|
Environmental Project no. 902, 2004 The Influence of Sorption on the Degradation of Pesticides and other Chemicals in SoilContents6 The influence of sorption on the degradation described by mathematical models 8 Methods for measuring the influence of sorption on the degradation 9 Soil treatment as a factor influencing the relation between binding and degradation 10 Pesticides mainly bound to clay minerals: glyphosate, paraquat, and diquat PrefaceThis report is the result of a fact-finding project initiated at the Danish Institute of Agricultural Sciences (DIAS) on the initiative of the Danish Environmental Protection Agency. The aim of the project was – based on an examination of literature – to throw light upon the connection between degradation and sorption of chemicals in soil and to assess the influence of the sorption on the fate of pesticides. In the examination of literature and the compilation of material, fOC us has been on 1) summing up data from a broad range of published studies, 2) comparing the methods applied for studying simultaneous sorption and degradation, and 3) identifying mathematical models applied for describing the connection between sorption and degradation. Finally, information about ongoing experiments at DIAS has been included. The report has been prepared on the basis of a systematic examination of all the papers that have been identified in the Agricola and Chemical Abstract databases and that describe the connection between sorption and degradation. Where relevant, papers dealing with methods of analysis or isolated degradation and sorption experiments have also been studied. As regards the latter, the examination is not complete as the number of separate papers dealing with degradation or sorption is numbering thousands. Initially, the inclusion of all types of xenobiotic chemicals in the assessment was under discussion. The examination has, however, turned out to be dominated by studies concerning pesticides, which are the substances that have primarily aroused interest when the influence of sorption on the degradation was to be described. The project was financed by the Danish Environmental Protection Agency. The steering committee for the project consisted of Christian Dejbjerg Hansen, the Danish Environmental Protection Agency, and Inge S. Fomsgaard, DIAS. Inge S. Fomsgaard has been in charge of the examination of literature and the compilation of material. Our thanks to Kirsten Jensen, DIAS, for a large effort in translating the report into English and to Sonja Graugaard and Henny Rasmussen for the lay-out of text and figures. Sammenfatning og konklusionerDet er ofte antaget at kemiske stoffer i jord nedbrydes efter en simpel 1. ordens kinetik, hvoraf udledes en halveringstid, DT50-værdi. Det har dog i mange tilfælde vist sig, at nedbrydningen ikke følger en simpel 1. ordens kinetik. Årsagen til disse rapporterede afvigelser fra en simpel 1. ordens kinetik kan formodentlig findes dels i det mikrobielle miljø i jorden, dels i, at sorptionen over længere tid (og dannelsen af bound residues) foregår efter mere komplicerede processer end almindeligvis antaget. Alle kendte modeller, der anvendes til at beskrive skæbne, transport eller risikovurdering af kemiske stoffer i jord indeholder termer, der beskriver henholdsvis binding og nedbrydning. En forståelse af disse parametres dynamik og den indbyrdes sammenhæng mellem dem er således uomgængelig, hvis modellerne skal anvendes med succes. Selvom det hidtil ikke har været muligt at give en komplet beskrivelse af bindingen af miljøfremmede kemiske stoffer til jord, så er der udført et utal af undersøgelser på området og beskrevet et stort antal processer, idet stoffernes binding i jorden er af stor betydning for deres videre skæbne. Sorption af kemiske stoffer til jord kan variere fra at være total reversibel til at være total irreversibel. En meget kompleks sammenhæng mellem sorption af miljøfremmede kemiske stoffer til jordbundens organiske materiale og den mikrobielle nedbrydning af stofferne i jordmiljøet er mere reglen end undtagelsen. For at kunne belyse sammenhængen er udviklingen de sidste år således gået i retning af at foretage undersøgelser af den mikrobielle nedbrydning af kemiske stoffer i jord under omstændigheder, der er så tæt på det virkelige miljø som muligt. Det er hyppigt set, at sorptionen har en begrænsende effekt på nedbrydningshastigheden af kemiske stoffer, hvilket forklares med, at det først og fremmest er de mængder af det kemiske stof, der er tilgængelig i jordens vandfase, der kan nedbrydes mikrobielt. Selvom dette forhold gør sig gældende, vil der dog stadig til en vis grad kunne foregå en nedbrydning af sorberet kemisk stof. Hvis desorptionen er afhængig af tid og foregår med en hastighed, der er på niveau med, eller lavere end nedbrydningshastigheden, så leder nedbrydningens afhængighed af sorptionen til en komplex kinetik, selvom selve nedbrydningskinetikken er en simpel 1. ordens. Nyere undersøgelser er domineret af den holdning, at en to-compartments 1. orden kinetik giver en bedre beskrivelse af kemiske stoffers nedbrydning i jord, når der arbejdes med systemer, der simulerer de naturlige omstændigheder. To-compartmentskinetikken udtrykker en fordeling af det tilførte kemiske stof mellem vandfasen og jordfasen og angiver en 1. ordens hastighedskonstant for nedbrydningen i henholdsvis vandfasen og jordfasen. To-compartmentskinetikken er ofte foretrukket uanset om der er tale om kemiske stoffer, der fortrinsvis bindes til jordens organiske materiale eller til lermineralerne. Af to-compartmentsmodellen kan der ikke udledes en halveringstid, hvorfor den ikke uden tilpasning kan anvendes i de almindeligt anvendte dynamiske udvaskningsmodeller som PRZM-2, PRZM, PELMO, GLEAMS, PESTLA, VARLEACH, LEACHM, MACRO, PLM og MIKE-SHE, der anvendes til at forudsige udvaskning til grundvandet af pesticider. Både i godkendelsessammenhænge og i modelkørsler ville det være relevant at tage højde for at nedbrydningen følger en mere komplex kinetik end den simple 1. ordens, ligesom det ville være relevant at tage højde for problematikken omkring bound residues, som der har været overordentlig meget fokus på inden for de sidste 10 år. Der findes allerede en lang række undersøgelser af bindingsmekanismer både for desorbare miljøfremmede stoffer og for bound residues. Det er dog nødvendigt at koble disse undersøgelser op på undersøglser af biotilgængelighed for at afgøre, i hvor stor en grad stofferne vil kunne reaktiveres i det naturlige system. Det er vigtigt at understrege, at laboratorieforsøg, der ønskes anvendt til at beskrive den komplexe sammenhæng der er mellem bindingen og nedbrydningen må tage højde for, at disse processer varierer overordentlig meget. Der er i forvejen en stor variation, som blot skyldes jordens heterogenitet. Den del af variationen, der kan skyldes metoden, må elimineres. Det første skridt her må være at udføre undersøgelserne under betingelser, der ligner de naturlige betingelser så meget som muligt. Det er desuden nødvendigt at udvikle kemiske analysemetoder, der kan ekstrahere mængder af de miljøfremmede stoffer fra jorden, som kan korreleres med de biotilgængelige mængder der er resultaterne af biologiske undersøgelser af biotilgængelighed. Både fra et lovgivningsmæssigt synspunkt og fra et videnskabeligt synspunkt er det vigtigt at differentiere mellem frie pesticidrester og bundne pesticidrester, ligesom det er vigtigt at skelne mellem biologisk persistens og kemisk persistens, når man taler om persistens, da disse ikke nødvendigvis følges ad. Summary and conclusionsIt has often been assumed that chemicals in soil are degraded according to simple first-order kinetics from which a half-life, a DT50-value, is deduced. However, it has emerged from many cases that the degradation does not follow simple first-order kinetics. The cause for these reported deviations from simple first-order kinetics may probably be found partly in the microbial environment in the soil, partly in the fact that the sorption over longer time (and the formation of bound residues) take place according to more complicated processes than generally assumed. All known models used for describing the fate, transport or risk evaluation of chemicals in soil include terms describing binding and degradation, respectively. Thus, an understanding of the dynamics of these parameters and the mutual connection between them is unavoidable if the models are to be used successfully. Even though it till now has not been possible to give a complete description of the binding of xenobiotic chemicals to soil, a vast number of studies of this subject have been carried out just as a large number of processes have been described because the binding of the substances in the soil is of great importance to their fate. The sorption of chemicals to soil may vary from being completely reversible to being completely irreversible. A very complex connection between the sorption of xenobiotic chemicals to the soil organic matter and the microbial degradation of the substances in the soil environment is more the rule than the exception. In order to throw light upon this connection, the development in the past few years has been in the direction of investigating the microbial degradation of chemicals in soil under circumstances as close to the real environment as possible. It has often been seen that the sorption has a limiting effect on the degradation rate of pesticides, which can be explained by the fact that first of all it is the amounts of pesticide available in the aqueous phase of the soil that can be microbially degraded. Even though this factor plays a part, a degradation of sorbed chemicals may still take place to a certain extent. If the desorption is dependent on time and takes place at a rate that is on a level with or lower than the degradation rate, the dependence of the degradation on the sorption leads to complex kinetics even though the degradation kinetics itself is simple first-order kinetics. Recent studies are dominated by the view that two-compartment first-order kinetics provides a better description of the degradation of chemicals in soil when systems are used that simulate the natural circumstances. The two-compartment kinetics expresses a distribution of the added chemical between the aqeous phase and the soil phase and indicates a first-order rate constant for the degradation in the fluid phase and the soil phase, respectively. The two-compartment kinetics is often preferred, both for chemicals that are mainly bound to the soil organic matter and for chemicals that are mainly bound to the clay minerals. A half-life cannot be deduced from the two-compartment model for which reason it cannot be used without adaptation in the commonly applied dynamic leaching models, such as PRZM-2, PRZM, PELMO, GLEAMS, PESTLA, VARLEACH, LEACHM, MACRO, PLM, and MIKE-SHE, which are used for predicting leaching of pesticides to the groundwater. Both in connection with approvals and in model runs, it would be relevant to take into account that the degradation follows more complex kinetics than simple first-order kinetics, just as it would be relevant to take into account the problem concerning bound residues, which has been extremely much in fOC us in the last 10 years. There are already a large number of studies of binding mechanisms for both desorbable xenobiotics and bound residues. However, it is necessary to couple these studies to studies of bioavailability to determine to which extent the substances can be reactivated in the natural system. It is important to emphasize that laboratory experiments that are intended to be used for describing the complex connection between the binding and the degradation must allow for the extreme variation of the processes. There is already a large variation, which is merely caused by the heterogeneity of the soil. The part of the variation, which may be caused by the method, must be eliminated. Here, the first step must be to carry out the studies under conditions that resemble the natural conditions as much as possible. Furthermore, it is necessary to develop chemical methods of analysis able to extract the amounts of the xenobiotics from the soil that can be correlated with the bioavailable amounts that are the results of biological studies of bioavailability. Both from a legal and a scientific point of view it is important to differentiate between free pesticide residues and bound pesticide residues just as it is important to distinguish between biological persistence and chemical persistence when persistence is in question, as these do not always go together. 1 IntroductionMany reported studies in which the fate of pesticides or other chemicals in soil have been studied have shown that it is exceedingly difficult to draw general conclusions about the degradation of these substances in soil compared with the soil parameters. A number of indexing methods have been developed that from few parameters for each substances aim to rank the pesticides according to the risk of leaching to the groundwater. E.g. the GUS-index ranks the pesticides solely according to inherent properties, degradability (measured as DT50-values), and sorption (measured as KOC) and in this way provides a measure of the potential of leaching. Lindhardt et al. (1998) showed that a ranking according to the GUS-index with a starting point in the DT50 and KOC –values of 12 pesticides that had been reported to the Danish Environmental Protection Agency involved great unreliability stemming from the large variation in the data material. For 12 pesticides (atrazine, bentazone, diuron, glyphosate, isoproturon, MCPA, mecoprop, metamitron, metsulfuron-methyl, phenmedipham, propiconazole, and triasulfuron), data of degradability DT50 and sorption KOC were collected in order to evaluate the dispersion on these parameters. The calculated GUSmin and GUSmax show that 11 of 12 substances (glyphosate left out) for extreme combinations of degradability and sorption conditions, based on information in the material that underlay the approval of the substances by the Danish Environmental Protection Agency, must be rated as "probably leachable" and that 8 of the 11 pesticides under other circumstances must be rated "probably not leachable". It has often been assumed that chemicals in soil are degraded according to simple first-order kinetics from which a half-life is deduced. However, it has emerged from many cases that the degradation does not follow simple first-order kinetics. The cause for these reported deviations from simple first-order kinetics may probably be found partly in the microbial environment in the soil, partly in the fact that the sorption over longer time (and the formation of bound residues) take place according to more complicated processes than generally assumed. In some connections authors choose to distinguish between the meanings of the expressions "half-life" and "DT50-value". "Half-life" is used when it is a question of first-order kinetics where the time for a number of halvings is the same, whereas "DT50-value" is used about the time that can be stated for a single halving (frequently the first one) but where the time varies for the following halvings. The present report does not distinguish between the meanings of these two expressions. According to the Uniform Principles Directive, 94/43 (OJ L227 31-55), a pesticide - if it is to be approved - must have a half-life of less than 90 days unless it can be shown that it is very probable that there are no harmful effects. The Danish Environmental Protection Agency generally accepts half-lives of 3-6 months if supplementary ecotox studies are carried out showing that the substance has no undesirable adverse effects. Glyphosate is an example of a substance which is bound extremely strongly to soil and which therefore has a very variable half-life. The assessment whether pesticides can be approved in spite of their long half-lives may depend on e.g. the type of binding. The pesticides are the group of substances of which by far the most information is available as to studies of the fate of chemicals in soil. This goes for both earlier applied hydrophobic substances such as DDT and similar substances and all the later developed substance groups of pesticides. In order that pesticides may be used, they must pass the approval system of the Danish Ministry of the Environment. Even though the pesticides have been approved for use in practical farming, there has generally been large interest within the research community in carrying out supplementary studies of the fate of the substances in the environment. And, indeed, a number of cases are known where the approvals have been withdrawn because of published studies. Within the last 10 years, there has been an interest in studying the fate of other chemicals in soil as the widespread practice of spreading sludge from sewage treatment works on to the agricultural soil has made it necessary not just to introduce limit values for the presence of chemicals in sludge but also to study the fate of the permitted concentrations of these chemicals in agricultural soil. The chemicals in question may be antibiotics, other medicinal substances, biocides, and detergents. Finally, fOC us has been put on the fate in soil of other chemicals, which among other things are present in the soil as industrial pollutions, as it – in the last cases – frequently is a question of the need to throw light upon purification techniques such as bioremediation. Just as pesticides are the most studied substances, pesticides are also the substances for which the most specific legislative requirements exist as regards their presence in and effects on the environment. In drinking water the concentration of a pesticide must not exceed 0.1 µg/l. Studies of the fate of the substances in soil have thus always been carried out with the aim of predicting whether the limit value for the presence in drinking water can be exceeded. But there are no legislative requirements concerning either the quantities of pesticides that may be present in soil or the form of the pesticides. All known models used for describing the fate, transport or risk evaluation of chemicals in soil include terms describing binding and degradation, respectively. Thus, an understanding of the dynamics of these parameters is unavoidable if the models are to be used successfully. One has to distinguish between chemicals that normally resist biological degradation (recalcitrant), such as PCBs and PAHs (polychlorinated biphenyls and polyaromatic hydrocarbons) and more readily transformable substances such as modern pesticides. PCBs and PAHs are hydrophobic, and their binding to the soil is caused by van der Waals forces or interaction with lipophile constitutents in the soil. Thus, these substances are not particularly available to microbial degradation. For the same reason a large part of the literature on bioremediation of these substances fOC uses on how the substances can be made available. 2 The soil organic matterOrganic matter (humus) is a result of the biological and chemical degradation of dead plant residue or animal remains in the soil. After a number of transformations the matter no longer has any resemblance to plant residue or animal remains after which it is called organic matter, macromolecular matter or humus. The soil organic matter has a complex structure, and it can be classified into soluble organic matter (DOM, dissolved organic matter) and insoluble matter. A classification of the organic matter according to its chemical structure includes fulvic acids, humic acids, and humin, where fulvic acid is soluble in both acidic and alkaline solutions, humic acid is soluble only in alkaline solutions, and humin is not soluble. The soil organic matter is further composed of polysaccharides, lignin, simple carbohydrates, lipids, proteins, and organic acids (Lassen, 1995; Stevenson, 1992). The exact structure of the organic matter is not known, but most hypotheses agree that the organic matter is composed of amorphous polymers of a three-dimensional structure of which both aromatic and aliphatic hydrocarbons form part and where there are both hydrophobic and hydrophilic side chains. Thus, it is not possible to give a complete description of the binding of pesticides and other xenobiotic chemicals to the soil organic matter. 3 SorptionEven though it till now has not been possible to give a complete description of the binding of xenobiotic chemicals to soil, a vast number of studies of this subject have been carried out just as a large number of processes have been described because the binding of the substances in the soil is of great importance to their fate. The sorption of chemicals to soil can vary from being completely reversible to being completely irreversible. For certain combinations of substance and soil, the desorption rate can be considerably slower than the sorption rate. The sorption depends on the molecular structure of the chemical and the soil properties, such as the amount and the type of organic matter, the distribution of particle size, and the cation composition. The sorption may be purely physical or chemical by nature. Gevao et al. (2000) distinguished between ionic bonding, hydrogen bonding, van der Waals forces, ligand exchange, complexes formed by charge transfer, hydrophobic partitioning, covalent bonding, and sequestration. Figure 1 shows an outline from Gevao et al. (2000) of different types of pesticides.
Figure 1. Outline of pesticides classified according to their molecular structure (from Gevao et al., 2000). The figure is reproduced with the kind permission from Elsevier. Ionic bonding occurs when the chemicals occur in cationic form in the soil or can be protonated, and the substances are bound to hydroxyl groups in humus, either phenolic or carboxyl groups. Cationic substances are e.g. diquat and paraquat while e.g. the triazines, which are alkaline, can form ionic bonds if both the chemical and the hydroxyl groups on humus have been ionised by the present pH-value in the soil. Hydrogen bonds can be formed between functional groups in humus that contains oxygen or OH and similar groups in the pesticide molecules. Hydrogen bonds are supposed to be of great importance to the sorption of non-ionic pesticides. If the pH-value in the soil is below the pKa-value of acidic and anionic pesticides such as phenoxy acids, phenoxyacetic esters, and dicamba, these are brought on a non-ionic form by which they also can form part of hydrogen bonds. The triazines can also form part of hydrogen bonds with the parts of the molecule that are not ionised. E.g. hydrogen bonds can be formed between carbonyl groups in humic acids and secondary amine groups in the triazines. Glyphosate is often mentioned as a substance that does not bind to the soil organic matter. However, Piccolo and Celano (1994) showed that glyphosate [N-(phosphonomethyl)glycine] via its phosphono group formed hydrogen bonds with O-atoms in soluble humic acids. Van der Waals forces are weak dipolar attractions that occur frequently where non-ionic or non-polar chemicals come into contact with humic acid molecules. Van der Waals forces are thought to be the dominant type of binding for picloram and 2,4-D (Khan, 1973). In hydrophobic partitioning the soil organic matter is regarded as an organic phase that is immiscible with water whether the organic matter is solid or dissolved. When referring to hydrophobic partitioning, the part of the chemical that is found on the hydrophobic phase is regarded as being dissolved in this phase. Hydrophobic partitioning is an important element in the binding of the "old" chlorinated compounds such as DDT, but it also occurs as one of the binding processes for trazine and urea herbicides. Covalent bonds are chemically well-defined bonds, which most frequently are irreversible and which by definition result in the originally well-defined structure of the pesticide, metabolite or other chemicals no longer existing. The chemicals that most frequently form covalent bonds are substances that have functional groups similar to the functional groups in humus (Bollag & Myers, 1992; Bollag et al., 1992; Senesi, 1992). E.g. substances that have phenolic groups will often form covalent bonds with humus. The covalent bonds are often formed by oxidative coupling and catalysed chemically, photochemically or enzymatically (Bollag & Myers, 1992; Dec & Bollag, 1997). In some cases, the microbial activity is the cause of the formation of the covalent bonds while some groups of substances can form covalent bonds without the participation of microorganisms (Parris, 1980). The sorption is often described by a simple equilibrium reaction between the concentration of substance sorbed to the solid matrix Cs (mg/kg) and the concentration in the aqueous phase Cw (mg/l). Several empirical expressions for describing this relation exist; the most common is the Freundlich-isotherm or a simple linear isotherm:
where K is called the Freundlich constant and n is a constant describing the "non-linearity" of the isotherm. If n = 1, the isotherm will be linear. If it is assumed that the sorption isotherm is linear for the individual substance to a given soil, a sorption coefficient can be calculated:
As most of the pesticides and other foreign chemicals that are found in soil are bound to the organic matter of the soil, a distribution coefficient between organic carbon and water KOC is often calculated on the basis of the sorption coefficient:
or
where fOC is the fraction of organic carbon. This relation is expected to be valid for soils with more than 0.1% organic carbon. By far the majority of the published studies on measurements of sorption have used the OECD method (OECD, 2000) in a more or less adapted form. This is a batch method by which a given volume of water with a known concentration of the substance is added to a known amount of air-dried soil. This mixture is shaken until equilibrium after which the concentration in the aqueous phase is measured. Based on the difference between the initial – and the equilibrium concentrations in the aqueous phase, the sorbed amount is measured. In recent times, however, a number of attempts at applying techniques for measuring the sorption have been carried out by means of which a natural soil/water ratio is retained (Weber & Young, 1997; Rochette & Koskinen, 1997). In addition, studies have been published in which more complex models have shown to describe the sorption better than simple one-compartment models (Pignatello & Xing, 1996; Ma & Selim, 1994; Streck, 1995; de Jonge et al., 2000). 4 DegradationThe degradation of a chemical in soil can take place through either chemical or microbiological processes. Often one or more steps take place chemically (e.g. a hydrolysis) whereas the following steps are microbiological. The degradation processes that take place chemically come to an end relatively quickly whereby the microbiological processes become the more interesting subject to study. The degradation happens gradually through the formation of one or more metabolites. By a total degradation of a chemical, CO2, salts, and water are formed, and parts of the chemical are built into new molecular structures in the soil humus or in biomass. Thus, it will not be a question of all the carbon atoms added to the soil through a chemical becoming CO2 (mineralised) within a foreseeable time. This is illustrated in Figures 2 and 3. Figure 2 shows a general diagram of the degradation of the herbicide mecoprop whereas Figure 3 shows the measurements of collected 14CO2, developed from the incubation of 14C-labelled mecoprop in soil in the concentration of 5 µg g-1. At the time when the incubation has stopped, about 62% of the added substance has been converted to 14CO2, but at this time no extractable residues of mecoprop or metabolites are found. The remaining 38% of the added radioactivity is thus either built into the organic constitutents of the soil or bound very strongly to these constituents.
Figure 2. Diagram illustrating the degradation of the pesticide mecoprop. An inter-stage may occur in which the metabolite 2-methyl-4-chlorphenol is formed.
Figure 3. Mineralisation of mecoprop 5 µg g-1 in soil. Degradation experiments can be carried out either by measuring the disappearance over time of the added pesticide or the formation of the mineralisation product 14CO2 from 14C-labelled pesticides. The soil microorganisms manage most of the degradation processes, and the degradation can either take place metabolically – that is microorganisms making use of the substances that are being degraded for growing – or it can take place cometabolically where the substances are degraded by microorganisms without these being able to make use of the pesticide as a source of energy or nourishment. In the metabolic degradation process the degradation rate of the substance is increased as the microorganisms are growing. Figure 4 shows an example of the illustration of degradation/mineralisation experiments when the process takes place with no-growth kinetics and growth kinetics, respectively. Growth kinetics may take place for selected chemicals but will in plough layer soil typically not be seen until the concentration of the substance is relatively high (Fomsgaard, 1999).
Figure 4. Examples of illustrating degradation and mineralisation of chemicals in soil when the processes take place with and without growth, respectively. Many factors influence the degradation of chemicals in soil. Factors such as temperature, water content, and other climatic factors, soil texture, microbiological activity, the composition of the other organic matter of the soil and the soil microbial biomass as well as the biological diversity and plant coverage. The fact that the depth of the soil influences the degradation rate of pesticides because of the very variable chemical and biological conditions has been described in many publications (Dictor et al., 1992; Mueller et al., 1992; Minton et al., 1990; Moorman & Harper, 1989; Pothuluri et al., 1990). The effect of temperature on the degradation rate of pesticides is also well described (Helweg, 1993; Helweg, 1987; Matoba et al., 1995; Ismail & Lee, 1995; Walker et al., 1996; Jones, 1986; Eberbach, 1998). Walker et al. (1996) reviewed a large number of studies of pesticide degradation and calculated mean Q10 -values. The water content of the soil has also often been described as being of importance (Ismail & Lee, 1995; Helweg, 1993; Helweg, 1987) just as the initial concentration of the pesticide (Helweg, 1993; Helweg, 1987; Reffstrup et al., 1998; Jacobsen & Pedersen, 1992; Parker & Doxtader, 1982; Mueller et al., 1992; Alexander, 1985; Helweg, 1993; Fomsgaard & Kristensen, 1999b). Temperature, water content, and soil depth are factors taken into account as having an influence on the degradation rate of pesticides in the 9 dynamic leaching models PRZM-2, PRZM, PELMO, GLEAMS, PESTLA, VARLEACH, LEACHM, MACRO, and PLM used in the fOC US comparisons (Boesten et al., 1995). Biological activity/biomass have also often been measured just as it has been attempted to relate them to the degradation rate of pesticides, either directly or through the variation in soil depth (Anderson, 1984; Torstensson & Stenström, 1986; Monrozier et al., 1993; Dictor et al., 1992). The amount of organic matter is normally also related to the soil depth, and its influence on the degradation of xenobiotic chemicals has also often been studied (Reddy et al., 1995; Duah-Yentumi & Kuwatzuka, 1980; Greer & Shelton, 1992; Knaebel et al., 1994). The pH-value in the soil (Smelt et al., 1983; Smelt et al., 1978), the oxygen conditions in the soil (Sinclair & Lee, 1992; Pothuluri et al., 1990; Ou et al., 1988), the water content of the soil Helweg, 1987; Helweg, 1993; Konopka & Turco, 1991), and repeated sprayings with the same substance (Moorman, 1990; Rahima et al., 2000) influence the degradation rate as well. Koetler et al. (2001) described how wetting/drying cycles reduced the extractable amount of atrazine and phenanthrene. Finally, there is the influence of the factor on the degradation, which the present report deals with, viz. sorption. To a great extent, the sorption of the substance determines whether it is available to microbial degradation. 5 BioavailabilityBioavailability is a term frequently used when one describes the possibility of plants absorbing the chemical or the availability of the substance to soil organisms, the latter case often being coupled with toxicity. Bioavailability understood as availability to the microorganisms in the soil is very consistently used in literature fOC using on bioremediation in which an increase in the microbial degradation of a chemical is wanted. The fOC using on techniques for increasing bioavailibity in this context may probably be attributed to the fact that many bioremediation tasks have fOC used on very hydrophobic substances such as the PAHs. Literature that describes the natural microbial degradation of pesticides in the soil environment only rarely makes use of the expression "bioavailability". Most publications phrase the problem – that the substances may be not be able to establish contact with the microorganisms – in different ways. E.g. formation of bound residues, the influence of the binding on the degradation, and microcavities in the soil where the chemical molecule can penetrate but where the space is too small for the microorganisms. Thus, the expression "bioavailability" can in principle be understood as the availability of the chemicals to microorganisms. The amount of organic matter in the soil will generally increase the binding of the substances to soil particles unless it is a question of substances, such as glyphosate, which are chiefly bound to clay particles and where in several cases a reverse correlation has been shown between the binding of glyphosate and the amount of organic matter (Parfitt et al., 1995; Glass, 1987). The first reactions between foreign chemicals and soil are physically-chemically reversible interactions. However, it applies to many pesticides in soil that the sorption-desorption process is not completely reversible, and the possibility of desorption is often reduced the longer the chemical has been present in the soil (Lehmann et al., 1990; Barriuso et al., 1992). At the same time as the extractability of the pesticide decreases, the formation of bound residues increases. A large number of authors have described this connection in conceptual diagrams (Novak et al., 1995; Wauchope & Meyers, 1985; Scow & Hutson, 1992; Calderbank, 1989). The amount of the organic matter in the soil has also another effect, as the presence of a large amount of organic matter in soil may be attributed to higher biological activity, as a large amount of degradable organic matter has been present on which the microorganisms could be propagated. A higher biological activity will often increase the transformation rate of not just the organic matter found naturally but also the added foreign organic substances. With this the organic matter may have two opposite effects in the soil. Mueller et al. (1992) showed that there was a positive linear correlation between the degradation rate for fluometuron and the content of organic matter in the soil and the microbial biomass while Simon et al. (1992) showed that such a correlation could not be shown for fenamiphos. Veeh et al. (1996) found a positive correlation between the degradation rate for 2,4-D and the number of microorganisms and asserted that it would be possible to find such a correlation for most pesticides with a low binding to the soil organic matter. Torstensson and Stenström (1986) measured the biological activity as a basic respiration rate and also studied the degradation of 2,4-D but in contrast to Veeh et al. (1996) found no clear correlation between the respiration rate and the degradation rate for 2,4-D. They found this correlation for linuron and glyphosate, however. A very complex connection between the sorption of xenobiotic chemicals to the soil organic matter and the microbial degradation of the substances in the soil environment is more the rule than the exception. In order to throw light upon this connection, the development in the past few years has been in the direction of investigating the microbial degradation of chemicals in soil under circumstances as close to the real environment as possible. When the sorption is so significant as to whether the added chemical comes into contact with the degrading microorganisms, there will be a great difference in what is found when degradation experiments are carried out in soil that has a natural water content as opposed to experiments where the soil is suspended in water. Therefore, publications that describe degradation experiments with pesticides in aqueous suspensions are becoming very rare. At pesticide concentrations of more than 1 µg/g in soil, substances that can be degraded metabolically will often be degraded according to growth kinetics whereas at lower concentrations the degradation will often take place according to no-growth kinetics. A normal consumption of pesticides will lead to concentrations of 1 g/g or less in the upper soil layer. On the other hand, point source pollution with pesticides or industrial pollution with industrial chemicals may lead to higher concentrations. Therefore, one ought to be aware whether experiments are carried out at realistic concentrations. While the aim is to carry out experiments under circumstances that resemble the natural ones, development has also gone in the direction of making a complete description of the mechanisms that control the degradation of the chemicals so that the influence of the sorption on this can be described. 6 The influence of sorption on the degradation described by mathematical modelsIt has often been seen that the sorption has a limiting effect on the degradation rate of pesticides, which can be explained by the fact that first of all it is the amounts of pesticide available in the aqueous phase of the soil that can be microbially degraded. Even though this factor plays a part, a degradation of sorbed chemicals may still take place to a certain extent. If the desorption is dependent on time and takes place at a rate that is on a level with or lower than the degradation rate, the dependence of the degradation on the sorption leads to complex kinetics even though the degradation kinetics itself is simple first-order kinetics. Guo et al. (2000) studied the influence of the sorption on the degradation rate of 2,4-D in the presence of varying amounts of activated carbon and described the sorption by a two-compartment first + first-order degradation process which took place in the aqueous phase and in the sorbed phase, respectively. Guo concluded that the degradation process took place in the two phases at different rates. Hill and Schaalje (1985) described a two-compartment model for the degradation of deltamethrin in soil. Jones et al. (1996) and Reid et al. (2000) pointed out that curves (Figure 5) that describe the course of the degradation
Figure 5. A biphasic degradation process for a chemical in soil (Reid et al., 2000). The figure is reproduced with the kind permission from Elsevier. of chemicals in soil often consist of two phases, an initial phase with a quick degradation and a second phase with a much slower degradation, where they were of the opinion that the relative significance of each of these phases would be determined by the volatility and hydrofobicity of the chemical. If the degradation takes place according to such a biphasic process, it is not logical to determine the degradation of chemicals by means of half-lives. Using 14C-labelled substances and measurement of the formed 14CO2, Scow et al. (1986) analysed the kinetics of the mineralisation of chemicals in soil and pointed out that this, too, was a two-phase process (Figure 6). Fomsgaard and Kristensen (1999a, 1999b) and Fomsgaard (1999) described the biphasic mineralisation of ETU with mathematical models, which were able to describe processes both with and without microbial growth (Figure 7), and stated, just like Scow (1986) and Brunner and fOC ht (1984), that the second – considerably slower – phase of the mineralisation process had to be a mineralisation of the soil organic matter in which 14C from the pesticide had already been built in or to which it had already been strongly bound. Whether - in the second phase - it is in actual fact a question of 14C being first built into the soil organic matter, which subsequently is mineralised, or whether a slow mineralisation takes place of the foreign chemical molecule that is bound to the surface of the soil is difficult to tell. If measurable amounts can be extracted when the mineralisation experiment is interrupted, then the latter is the case. In the cases in the studies of Fomsgaard and Kristensen (1999a, 1999b) where the mineralisation in the first compartment took place with no-growth kinetics, a two-compartment first + first-order process was the model which resulted in the best fit of the mineralisation.
Figure 6. Biphasic mineralisation curve for aniline in varying concentrations (from Scow et al., 1986). The figure is reproduced with the kind permission from American Society for Microbiology.
Figure 7. Two-compartment mineralisation of 14C-ETU (Fomsgaard, 1999). A. Data points ......... and model ________: B. Data points ......... and the first term of the model______: C. Data points........ and the second term of the model_____: Figur 7. To-compartment mineralisering af 14C-ETU (Fomsgaard, 1999). A. Datapunkter ......... og model ________: B. Datapunkter ......... og første led af modellen______: C. Datapunkter........ og andet led af modellen_____: Sjelborg et al. (2002a) compared the use of a one-compartment first-order model and a two-compartment first + first-order model respectively for the description of the degradation of ioxynil: The models were described as
The comparison of the application of the two different models is seen in Figure 8. A first-order degradation process can be solved analytically, and a half-life (DT50) can be determined. A two-compartment first + first-order process cannot be solved analytically, and it was concluded that the use of a half-life, calculated from the simple first-order process, for a description of degradation rates is not adequate. Many mathematical models, which were formerly used to describe the degradation kinetics of a chemical, took as their starting points the degradation of a single soluble chemical by means of a single culture of bacteria in a suspended experimental system. One of the models, which were often used where bacterial growth occurs, is the Monod model. From this, various other models can be derived for describing the degradation of a substance, each of these models having its own relevance depending on the initial concentration of the chemical and the bacterial density – models such as first-order, zero-order, Michalis-Menten, logarithmic, and logistic equations (Simkins & Alexander, 1984). As to chemicals in soil, first and foremost pesticides, it is frequently assumed that the degradation takes place according to simple first-order kinetics from which a DT50 -value is derived. Scow et al. (1986) and Fomsgaard (1999) showed that the mathematical description of mineralisation curves, in which the 14CO2 development from 14C-labelled pesticide is measured, is best carried out by means of two-term models where the second term describes the slow degradation of 14C from the pesticide, which has been built into or strongly bound to the soil constituents. Many authors have been of the opinion that a degradation that was much slower than expected is due to a slow diffusion of the chemical through the soil matrix before it reaches the microorganisms (Gustafson & Holden, 1990; Firestone, 1982; fOC ht & Shelton, 1987; Myrold & Tiedje, 1985; Schmidt & Gier, 1989). Pignatello (1989), Harmon et al. (1989), Brusseau and Rao (1989), and Brusseau et al. (1991) showed that diffusion is the process that dominates sorption and desorption processes. Scow and Hutson (1992) and Scow and Alexander (1992) developed a diffusion-sorption-biodegradation model (DSB) and applied it to empirical experiments. The model is outlined schematically in Figure 9. The results show that in suspensions of soil in water or if the soil aggregates are very small or when the concentration in the solution is very large a simple first-order process can often be used to describe the degradation of chemicals in soil. But if systems are involved where the
Figure 8. Application of first-order and two-compartment first-order model, respectively, for description of the degradation of ionynil (0.5 mg/kg) in plough layer soil and soil from a depth of 80-100 cm from Faardrup. The points are the experimental values whereas the full-drawn line is the mathematical model (Sjelborg et al., 2002a). water content of the soil is natural, if large aggregates are present or when the concentration in the soil water is small, a two-compartment model must be used to describe the degradation, and the rate constant for the slow part of the two-compartment degradation and the distribution of the amount of pesticide on the two degradation processes are determined by the diffusion/sorption of the substance in the soil (Scow & Hutson, 1992). Shelton and Doherty (1997a, 1997b) described a combined sorption-degradation model, which involved growth kinetics of the degradation process and used it for describing the decomposition of 2,4-D in soil. Gamerdinger et al. (1990) and Beigel et al. (1999) developed models that described the simultaneous sorption and degradation of chemicals in soil that could be used for studies, which were carried out as leaching experiments in small columns.
Figure 9. Schematic diagram showing the relation in the diffusion-sorption-biodegradation model (Scow & Hutson, 1992). The figure is reproduced with the kind permission from Soil Science of American Journal. Sjelborg et al. (2002b) compared the degradation of fenpropimorph in three different soils that had been incubated with the substances in the concentration of 0.5 mg/kg and with a natural water content and described the degradation by a two-compartment first + first-order process and a simple first-order process respectively (Figure 10 – Figure 12). It was entirely consistent that the description by the two-compartment model resulted in a higher correlation coefficient than the description by the simple first-order model (see the values in Figures 10 – 12). As in the literature mentioned above, the two compartments can be regarded as one compartment where a relatively quick decomposition of freely available pesticide takes place and another compartment where the decomposition is slower because it is controlled by a desorption or diffusion process. The distribution of the amounts of pesticides a and b between the two compartments is apparently determined by the structure of the pesticide, the amount of present organic matter to which the substance can be bound, and the rate k1 at which some of the pesticide can be decomposed in the first compartment before the
Figure 10. Degradation of phenpropimorph Faardrup 0-20 cm. a) Two-compartment first + first-order model. b) First-order model. Points: data points. Full-drawn line: model. Correlation coefficient r2. Half-life according to the first-order model = 15 days. The first three half-lives according to the first + first-order model: 4.2 days, 21.8 days, and 35.8 days, respectively (Sjelborg et al., 2002b)
Figure 11. Degradation of phenpropimorph Jyndevad 0-20 cm. a) Two-compartment first + first-order model. b) First-order model. Points: data points. Full-drawn line: model. Correlation coefficient r2. Half-life according to the first-order model = 122.6 days. The first three half-lives according to the first + first-order model: 66.3 days. The following half-lives cannot be calculated (Sjelborg et al., 2002b).
Figure 12. Degradation of phenpropimorph Tylstrup 0-20 cm. a) Two-compartment first + first-order model. b) First-order model. Points: data points. Full-drawn line: model. Correlation coefficient r2. Half-life according to the first-order model = 379 days. The first three half-lives according to the first + first-order model: 483 days, 623 days, and 624 days, respectively (Sjelborg et al., 2002b). remaining amount is bound in the second compartment. The rate of the degradation process in the first compartment k1 is probably determined by the biological activity whereas the rate of process in the second compartment k2 is determined by the desorption/diffusion rate. A model describing these relations is being prepared. In a two-compartment first + first-order equation the DT50-value cannot be found analytically; half-lives can only be indicated from readings of the curve or from an iterative method. The simple first-order equation, on the other hand, can be solved analytically as DT50 = ln2/k. The half-life according to the first-order equation is therefore the same no matter where one is in the process whereas the half-life according to the two-compartment first + first-order process changes over time and typically lengthens as the rate constant for the degradation is low in the second compartment, which becomes the dominant one as time passes. In the leaching model, MACRO, a possibility is built in of inserting two DT50-values for the aqueous phase of the soil and the sorbed phase respectively. The normal procedure is to insert the same DT50-value in both phases, which has been calculated on the basis of a simple first-order process. However, on the basis of the two-compartment model a half-life for each of the two compartments can be calculated from ln2/k, and the distribution of the of substance can be taken into account. In this way, one can assess the application of the two degradation models. However, a developed version of MACRO of which a model description combining the sorption and the degradation processes forms part would be preferable by far. The error that is made by using a simple first-order model as the basis for calculations of half-lives varies according to the other circumstances in the soil. In Figure 10 is seen that the half-life is estimated at 15 days according to the simple first-order process whereas it is just 4.2 days for the first halving according to the two-compartment model. If one assumes that the part of the substance that is decomposed in the second compartment has been extracted only owing to the application of an organic extraction agent and that it will not otherwise be available to the biology of the soil or to leaching to the groundwater, then a more severe assessment of the substance has been made when using the simple first-order process than might be considered reasonable. On the other hand, if the amount that is decomposed in the second compartment in actual fact is bioavailable or can be leached to the groundwater, the substance is assessed too mildly when using the simple first-order process as three halvings are estimated at just 45 days whereas it takes 62 days according to the two-compartment process. Thus it is not just a question of a need to find the right model for describing the sorption-degradation process but also a question of throwing light upon the type of binding and the bioavailability of the amounts of the substance that are decomposed in the second compartment. Sorensen et al. (1998) ranked the results of a number of monitoring findings of leached pesticides, compared this ranking with the ranking of various descriptors for the pesticides, and showed that there was a close connection between the ranking according to monitoring data and the ranking according to the three descriptors, dosage, sprayed area, and KOC-value. On the other hand, the ranking in relation to the DT50-value was not nearly so usable in relation to the found monitoring data. The ranking in relation to the degradation rate might have been more usable if a two-compartment model had been used for the description of the degradation. 7 Bound residuesSeveral decades ago "aging" was frequently mentioned in the pesticide literature whereby was understood the phenomenon that chemicals – most frequently described for pesticides – were becoming less extractable and with this less bioavailable after a prolonged residence time in the soil. In recent years mention of this phenomenon has again become relevant where it now most frequently is linked to discussions of the formation of bound residues. On examination of literature on bound residues there seems to be some confusion concerning the use of this term. Therefore, IUPAC (the International Union of Pure and Applied Chemistry) has found it relevant to define the term bound residues as "chemical species (active ingredient, metabolites and fragments) originating from pesticides, used according to good agricultural practice, that are unextracted by methods which do not significantly change the chemical nature of these residues, but which remain in the soil. These non-extractable residues are considered to exclude fragments, recycled through metabolic pathways leading to naturally occurring products" as stated by Roberts (1984) and Kearney (1982). The ability of the chemicals to be bound (sorbed) to the soil organic or mineral particles and to be degraded by the soil microorganisms is key properties when the leaching risk of the substances is to be assessed. These characteristics are determined by the molecular structure of the substances, but the current binding and degradation taking place in a given environment are further determined by the environment, e.g. the soil structure, biological activity, and the amount of water running through. The binding is more or less reversible, which was illustrated by e.g. Barriuso (1994) (Figure 13).
Figure 13. Distribution of pesticide over time between liquid phase and solid phase (Barriuso, 1994). The figure is reproduced with the kind permission from Elsevier. The distribution of pesticides between the aqueous phase and the soil phase was also illustrated by Barriuso (1994) as in Figure 14, where there is a distinction between a water-extractable part, a solvent-extractable part, and a non-extractable part of bound residues. Barriuso mentions a level of biocide pollution, which is found at the time when a lot of pesticide and/or metabolite is still present in the soil environment, which may have a measurable toxic effect on e.g. fish, plants or microorganisms; a level of chemical pollution, which is measured by chemical extraction methods where the efficiency of these methods influence on the given result; and a level of ecological pollution, which arises because he considers everything that is not measurable as active bound residues of the pesticide remaining in the environment. Barriuso (1994) was of the opinion that a chemical analysis with classic analytical techniques underestimates the chemical pollution, as not all bound residues present are extracted. At the same time Barriuso (1994) wrote that if the interesting part is the size of the amount of pesticide present in the soil that may have a toxic effect on soil organisms, then a chemical analysis will result in a overestimated result, as the chemical analysis is traditionally carried out with extraction agents that extract as large a quantity of the substance as possible whereas a toxic effect on soil organisms only occurs if the substance is present in the aqueous phase. In the same way a traditional chemical method of analysis will often overestimate the amount of chemical that may be a threat to the groundwater.
Figure 14. Soil pollution levels over time (Barriuso, 1994). The figure is reproduced with the kind permission from Elsevier. When an extraction method for chemicals in soil is being developed experiments with "spiked samples" are always carried out, that is experiments where known amounts of the chemical is added to the soil to which the extraction method is then applied. It is normal practice to leave the spiked sample for 24 hours as it is a well-known phenomenon that an extraction that is carried out immediately after adding the substance will overestimate the recovery percentage, which then does not cover the recovery percentage that exists when a "genuine" soil sample is analysed as the chemical in such a sample has been present for far more than 24 hours. Here, one is in reality also a long way into the problem concerning the lag period, which influences the binding (which again influences the degradation). Figures 15 and 16 show selected examples of the change in the desorption over time of pesticides to soil (Walker et al., 1995; Johnson et al., 2000).
Figure 15. Changes in desorption over time for a) linuron, isoxaben, propyzamide and b) metsulfuron-methyl (Walker et al., 1995). The figure is reproduced with the kind permission from British Crop Protection Council.
Figure 16. Change of sorption over time of imazmethabenz-methyl in soil at varying pH (Johnson et al., 2000). The figure is reproduced with the kind permission from Weed Science Society of America. A number of previous studies have described how 14C-labelled bound residues have been activated. The addition of fresh soil and addition of glucose or manure increased the decomposition of 14C-parathion bound residue (Racke & Lichtenstein, 1985), and the addition of glucose increased the decomposition of 14C-atrazine bound residue (Khan & Bheki, 1990). Both studies explained the activation by an increased microbial activity. However, a chemical analysis of the mentioned 14C-labelled bound residues was not carried out. Therefore, it is important to be aware whether studies describing the activation of 14C bound residues may describe a naturally increased degradation of the organic matter where 14C from the pesticide in a molecular form that differs from the original has been built in. Fomsgaard and Kristensen (2002) and Christensen (2002) showed that there is a significant difference between water-extractable and solvent-extractable isoproturon. Figure 17 shows selected results from Fomsgaard and Kristensen (2002) where the difference between the water-extractable and the solvent-extractable parts of isoproturon in a degradation experiment is significant.
Figure 17. Degradation of 14C-isoproturon in soil 0.1 g/g. Water extractable and solvent extractable amounts of IPU and metabolites, calculated in % of added amount of 14C-isoproturon (Fomsgaard & Kristensen, 2002). When the amount of extractable substance in Fomsgaard and Kristensen (2002) decreases in the course of time, it is first and foremost due to the microbial degradation, which can also be seen from Figure 7 showing the formation of 14CO2 from the pesticide. Nevertheless, after about 750 days only about 18% of the added amount of 14C (calculated in equivalent amounts of 14C-isoproturon) is transformed to 14CO2. And 20% of the added amount can be extracted (4% with water and 16% with an organic solvent). The remaining amount of 14C (62%) will be built into the soil microorganisms and into the soil organic matter or will exist as bound residues so strongly bound that it cannot be extracted with a solvent. The fact that a formation of 14CO2 still takes place after 800 days is natural as there will still be a microbiological activity, which can transform the organic matter into/to which 14C now is built/strongly bound. A large pool of organic matter, which is decomposed in the course of one growing season, is constantly found in the upper soil layers. Bureaul et al. (1998) estimated that close on 8 t of organic carbon per hectare is decomposed in the course of one growing season.
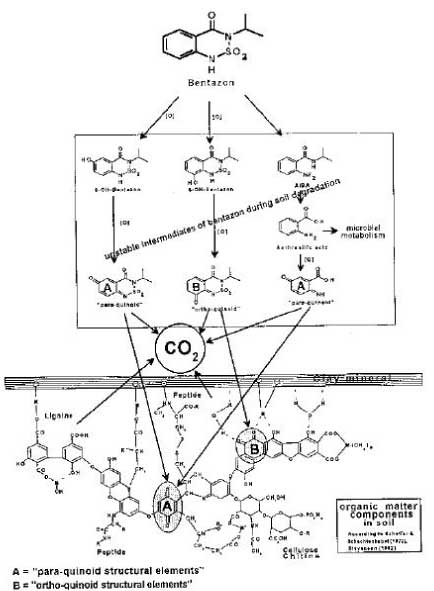
Figure 18. Outline of pesticides forming bound residues (Calderbank, 1989). The figure is reproduced with the kind permission from Springer Verlag. Bound residues are formed to a greater or lesser extent from ALL types of substances. There have been many reviews on bound residues (Klein & Scheunert, 1982; Khan, 1982; Roberts, 1984; Führ, 1987), and in 1996 a major workshop was held on this subject (Führ, 1996). Calderbank (1989) summed up the lists of Klein and Scheunert (1982), Khan (1982), and Roberts (1984) of pesticides that form bound residues (Figure 18). The size of the amounts of bound residues stated in Calderbank's (1989) list that has been identified as starting substance or metabolite varies. Bound residues of glyphosate have e.g. been quantitatively extracted from a volcanic ash soil with a strong alkaline solution (Nomura & Hilton, 1977). The bipyridilium herbicides diquat and paraquat are unstable in alkaline solutions but can be extracted from soil if the soil is opened with a strong sulphuric acid (Tucker et al., 1967). Such a procedure destroys the soil colloids whereby the herbicides can be quantitatively extracted from the soil. This procedure can only be applied to a very few pesticides as most of them will be destroyed at the same time as the soil colloids are destroyed. In an experiment with pirimicarb Hill (1976) showed that up to 70% of the added amount/quantity of pesticide was found as bound residues in the soil after 2 years. When extracted with 0.1M NaOH, 18% of this could be quantified as original pesticide. Khan (1982) showed that more than half the 14C-promethryn bound residues was present as the original molecule, and Capriel et al. (1985) showed that after 9 years 10% of the bound residues found in the soil from atrazine could be characterised as the original pesticide. As to substance groups such as ureas, anilides, phenoxy acids, nitroanilines, and pyrethroids, it has emerged that quantifiable amounts of the original substances cannot be extracted from the bound residues (Hsu & Bartha, 1976; Stott et al., 1983; Wheeler et al., 1979). Burauel et al. (1998) quantified bound residues from measurements of 14C in soil from 20 different outdoor lysimeter experiments where the experiments were carried out with 14C-labelled atrazine, terbuthylazine, chloridazon, dichlorprop-P, methabenzthiazuron, pyridate and anilazine. After two years' experiments, more than 80% of the remaining 14C was found in the upper 30 cm of the soil column of which the total length was 110 cm. Burauel et al. (1998) stated that 50-90% of the remaining radioactivity in the upper layer in such experiments normally can be characterised as "bound residues", that is non-extractable substance, metabolites or fragments of substance. Burauel et al. (1998) pointed out, however, that in many cases the structure of these bound residues has not been explained. In his lysimeter experiment with methabenzthiazuron, he found that 28.8% of the added radioactivity could be extracted from the soil in the form of the original substance after 127 days whereas < 1% of the added radioactivity could be extracted from the soil in the form of the original substance after 6.5 years. The bioavailability for the following crops was minimal with the highest amount of built-in radioactivity of 2% found in winter wheat – and it cannot be ruled out that the wheat has assimilated 14CO2 as a part of the incorporated 14C. In the lysimeter experiment with anilazine only about 10% of the radioactivity in the soil was found to be extractable after just 6 weeks. As a supplement Burauel et al. (1998) carried out 13C-NMR studies of humic and fulvic acids, which had been extracted from soil incubated with 13C-anilazine. The typical dialkoxy binding, which occurs when anilazine is built into humic and fulvic acids, was present whereas the free original substance was not. Fomsgaard et al. (2003a; 2003b) carried out lysimeter experiments with isoproturon and glyphosate. Experiments with 14C-labelled isoproturon were carried out as repeat determination in soil that had been treated conventionally whereas experiments with 14C-labelled glyphosate was carried out as repeat determination in soil that had been tilled with a reduced treatment. 48% and 54%, respectively, of the added radioactivity in the isoproturon experiments were found in the upper 10 cm of the soil while 17% and 14%, respectively, were found in soil from a depth of 10-20 cm. Extractions were carried out in the 0-10 cm soil, at first with an aqueous CaCl2 solution where 0.49% and 0.2%, respectively, of the added activity was extracted. By a subsequent extraction with acetonitril 1.19% and 0.56%, respectively, were found. Nothing extractable was found in the deeper layers. Because of the low extractable amounts no subsequent chemical analysis was carried out. In these lysimeter experiments, it must be noted that almost the whole amount of radioactivity left in the soil was bound residues. Further, it must be noted that some of the extractable amount (which may be assumed to be original matter or metabolites without it being known for certain) is more easily mobilisable than the rest, as it can be mobilised (extracted) with an aqueous solution while the rest needs acetonitrile. The possibility that the extractable substance may be mobilised under natural circumstances may be thought to be illustrated with the water extractable part. Whether the non-extractable part may be mobilised as an active pesticide or metabolite will need further investigation. It is very difficult to predict how large an amount of pesticide remains that the soil can bind and deactivate, but it must be assumed that the amount of pesticide remains is very small compared with the large amount of natural organic matter, which forms part of the soil cycle every year and that therefore no saturation of the sites where the pesticide remains can be bound will ever occur, especially when it is taken into consideration that a degradation also takes place. It is normally a matter of less than 1 kg of carbon, which comes from pesticides, out of a total amount of carbon, which constitutes between 20,000 and 40,000 kg/ha in an average plough layer depth in agricultural soil. Sequestration or entrapment in micropores is a phenomenon that has been studied and discussed very much in recent years. Waters and Oades (1991) and Bergström and Stenström (1998) described the differences in size between the soil pores, the chemical molecules, and the degrading organisms (Figure 19), and in their experiment with degradation of 2,4-D, Bergstöm and Stenström (1998) showed that after 7.7 days most of the chemical substance was found in micropores in the soil where there is no considerable exchange of water and where the bacteria cannot penetrate (Figure 20). Thus the availability of these parts of the substance is only possible if the substance diffuses into the major pores or if the degradation of the substances can take place by means of secreted enzymes from the microorganisms. Gevao et al. (2000) wrote that entrapment of chemicals in the soil could take place after a prolonged time or aging but did not define the prolonged time more closely. Dec and Bollag (1997) defined sequestration as a situation in which the substances could still be extracted from the soil. Sequestration differs from adsorption in that adsorption takes place relatively quickly whereas sequestration is a slower process; both processes can, however, take place at the same time (Huang et al., 1996; Pignatello & Xing, 1996). According to Alexander (1995), diffusion phenomena combined with sorption on available microsites, control the sequestration. What the chemical structure is like when bound residues are concerned is another matter. Barriuso (1994) mentioned chemical binding of the pesticides and their metabolites to the other soil constituents, incorporation in the phenolic polymers, incorporation in the soil microorganisms after these have been in control of a metabolic activity or binding in microcavities in the polycondensates of the soil. However, according to IUPAC's definition of < Figure 19. Size of soil particles compared with the size of pores and the size of chemical molecules and biota (from Bergström & Stenström, 1998). The figure is reproduced with the kind permission from The Royal Swedish Academy of Sciences. bound residues, chemical binding or incorporation in the soil microorganisms can only be included as examples of the formation of bound residues if the foreign substance in its original form or identifiable parts of it can be released again from the new complex molecule via a microbial or enzymatic decomposition. If one looks at degradation process for bentazone proposed by Huber and Otto (1994), which is illustrated in Figure 21, the degradation products from bentazone are covalently bound in the complicated molecular structure, which is a part of the soil organic matter. Whether the parts of the foreign organic molecule that originates from bentazone become identifiable and measurable in the process where the organic matter is decomposed by the microorganisms in the soil is doubtful, however. It is more likely that parts of < Figure 20. The influence of the soil structure on the availability of chemicals to degradation (Bergström & Stenström, 1998). The figure is reproduced with the kind permission from The Royal Swedish Academy of Sciences. that which originates from the bentazone molecule are degraded in the big pool in which the degradation of the whole amount of organic matter in the soil takes place. Only if e.g. the A or B structure is released again in the soil – and cannot be extracted with extraction agents that change the chemical structure – will it be bound residues. Measuring bound residues is often stated as best being carried out with 14C techniques (Barriuso, 1994; Calderbank, 1989). However, when using simple 14C techniques, it is not possible to distinguish whether parts of the original substance have formed part of new chemical structures or whether the chemical structure of the pesticide or of a well-known metabolite has been preserved and the substance is just bound to the soil particles.
Figure 21. Proposed structure of bentazone degradation products built into the soil macromolecular organic matter (Huber & Otto, 1994). The figure is reproduced with the kind permission from Springer Verlag. Barriuso and Koskinen (1996) discussed which methods might be the most careful for identification of where the bound residues are found in the soil. They referred to studies by Andreux et al. (1991), Khan (1982), Capriel et al. (1985), and Bertin and Scaviona (1989), who had chemically fractioned the soil organic matter and found that the soil content of fulvic acid played an important part in the initial quick immobilisation of pesticides while a higher extent of bound residues formed by association of pesticides with more polymeric humine links was observed over time. Barriuso and Koskinen (1996) questioned whether the chemical fractioning with alkaline extraction agents had modified the soil and the pesticides to an extent where it was no longer a matter of bound residues. Barriuso and Koskinen (1996) were therefore advocates of a physical fractioning of the soil particles. Barriuso and Koskinen (1996) added 14C-labelled atrazine to the soil in a number of lysimeters and studied how the binding of the substance over time was distributed on different fractions of the soil, which were grouped according to particle size (Figure 22). The formation of bound residues was increased over the first 4 months. Non-humus parts of the soil organic matter with particles sizes of more than 50 m showed the largest capacity for the formation of bound residues, and the ability of the organic matter to form bound residues with atrazine decreased with smaller particle size. This was explained by the fact that the access of the microorganisms to the substance was expected to decrease in the most difficultly accessible finest fractions of aggregates. The most stable bound residues, however, were formed in the fraction where the particle size was like that of clay. Depending on where bound residues are found, their subsequent fate varies. Barriuso and Koskinen (1996) were of the opinion that if they are found in the coarsest fractions and are bound to not fully transformed plant material then they may partly be released again. But if they are bound to the finest particles, they may be leached to the groundwater by particle-bound transport.
Figure 22. The distribution of bound residues from 14C-atrazine on different soil size fractions (Barriuso & Koskinen, 1996). The figure is reproduced with the kind permission from Soil Science Society of America. Northcott and Jones (2000) reviewed analytical techniques for measuring organic bound residues in soil and sediment and described the most dominating techniques as "release and identify" techniques (Dec & Bollag, 1997). Gevao et al. (2000) stated that the separation between free and bound chemicals in the soil is not always easy. Even though a substance can easily be extracted without its chemical structure being changed, it will not be completely free. Therefore, "extractability" is defined from the extraction agent and the other experimental conditions under which the extractions take place. Many methods of extraction have been used over the years for the extraction of chemicals from soil for subsequent analysis. Soxhlet extraction, shaking techniques with a solvent or ultrasound treatments are the more traditional methods of extraction. In addition to these, the following methods are now in use: supercritical fluid extraction (Khan, 1995; Khan, 1996, Barnabas et al., 1994; Weber & Young, 1997; Rochette & Koskinen, 1997), microwave extraction (Stout et al., 1996; Molins et al., 1996; Lopez-Avila et al., 1996), silylation previous to the extraction (Bolag & Stotsky, 2000), and accelerated solvent extraction. "Release and identify" techniques can only to a smaller extent tell something about the types of binding based on the solvents that extract the substances. The methods that extract different fractions of the organic matter, such as humic acid, fulvic acid, and humine, have a more elucidating effect concerning bindings. Methanolic saponification, alkaline extraction, silylation of humic compounds, and pyrolysis have been used to release chemicals that have been strongly bound to or sequestrated in the soil organic matter (Northcott & Jones, 2000; Eschenbach et al., 1994; Haider et al., 1992; Wais et al., 1995; Thorn et al., 1996; Schulten & Leinweber, 1996; Khan & Hamilton, 1980). However, with such methods, which are based on extraction, fractioning or destruction, there is a risk that the methods have also influenced the amount or the chemical form of the substance that is to be studied. In recent years a number of non-destructive spectroscopic methods have therefore been introduced, which now are preferred if one wishes to study the interaction of chemicals with the soil constituents. These spectroscopic methods can be ESR (Electron Spin Resonance spectroscopy), FTIR (Fourier Transform InfraRed Absorption spectroscopy), Fluorescence spectroscopy or NMR spectroscopy (Nuclear Magnetic Resonance spectroscopy). ESR is a good technique for studies of charge-transfer, which for example can be the mechanism for the binding of diquats and atrazine to humic acid (Sanchezpalacios et al., 1992; Senesi et al., 1987). Infrared absorption spectroscopy is very practicable for identifying the structure of organic compounds, as the infrared absorption spectrum is complex even for relatively simple compounds. FTIR is an advanced form of infrared spectroscopy with which the changes taking place in the adsorption spectrum of the organic matter because of bindings to the foreign chemicals can be detected. FTIR is particularly suitable for substances where hydrogen bondings and proton transfers occur (Aochi et al., 1992; Martin-Neto et al., 1996; Weissmahr, 1997). Fluorescence spectroscopy is most frequently used for measuring the association of hydrophobic substances such as PAH to dissolved humic matter (Backhus & Gschwend, 1990; Chin & Gschwend, 1992). NMR has been used very much for characterising the humus matter of the soil and is a well-tested non-destructive spectroscopic technique. 13C-NMR is the preferred method for studying covalent and non-covalent interactions between specific xenobiotic organic molecules and natural organic matter (Hatcher et al., 1993; Nanny et al., 1996; Kim et al., 1997; Haider et al., 1992; Haider et al., 1993; Wais et al., 1995). Roberts (1984) defined bound residues as "chemical species originating from pesticides, used according to good agricultural practice, that are unextracted by methods which do not significantly change the chemical nature of these residues". As mentioned above, this definition was used by IUPAC. Calderbank (1989) suggested a modification of this definition that took the bioavailability into consideration: "It may be that bound residues in soil could be defined as residues of the intact pesticide or degradation products derived from it that are no longer able to exert their original biological activity to any significant extent and/or cannot be extracted from the soil by extraction methods which do not degrade the compound unless such methods are able to destroy the soil structure without affecting the compound. Clearly the important matter is not so much how the residue is defined but the question of its biological availability". At a workshop held by the Deutsche Forschungsgemeinschaft, "Pesticide Bound Residues in Soil", a new definition of bound residues was suggested (Führ et al., 1996): "Bound residues represent compounds in soil, plant or animal which persist in the matrix in the form of the parent substance or its metabolite(s) after extractions. The extraction method must not substantially change the compounds themselves or the structure of the matrix. The nature of the bond can be clarified in part by matrix-altering extraction methods and sophisticated analytical techniques. To date, for example, covalent, ionic and sorptive bonds, as well as entrapments, have been identified in this way. In general the formation of bound residues reduces the bioaccessibility and the bioavailability significantly". The difference between the suggestion for a definition by Führ et al. (1996) and the existing IUPAC definition is important as Führ et al. (1996) incorporate a new condition, that the extraction agent must not change the chemical structure of either the pesticide or the soil matrix in order that it is possible to define the non-extractable part as bound residues, while IUPAC's definition only mentions that the extraction agent must not change the chemical structure of the pesticide in order that it is possible to define the non-extractable part as bound residues. In order to apply Führ's definition it would be necessary to define what it is meant by the soil matrix. If the soil structure is meant, that is the association of primary particles in secondary particles, then these can be destroyed by many substances, which would have as a result that, on the whole, practicable extraction agents for carrying out this separation could not be found. It must be assumed that by "matrix" is meant the soil colloids. The soil organic matter is to a most predominant extent bound to clay minerals and in that way forms an integrated part of the colloids. The clay minerals of the soil are affected by acids and bases, as they are more or less dissolved in these depending on the contact time (Borggaard, pers. comm.). It would therefore be obvious to regard organic solvents as extraction agents that can be used for distinguishing extractable residues from bound residues according to Führ's definition while to the extent that they do not destroy the foreign chemical, it will also be possible to use acids and bases as extraction agents according to IUPAC's definition. Whether IUPAC's or Führ's definition is used, it will nevertheless be necessary always to define the specific extraction procedure, but this limit will always solely be related to the chosen procedure and not to any naturally occurring clear-cut limit in the soil. The effect of the extraction agent on the soil colloids differs according to the given type of soil and perhaps even according to the different contents of iron compounds and organic matter in different soils (Christensen, pers. comm.). Alkaline and acid extractions as carried out by Nomura and Hilton (1977), Tucker et al. (1967), and Hill (1976) would thus yield different results in different types of soil when one is to assess whether the extracted substance has been bound residues. This discussion will be relevant when assessing whether glyphosate that is extracted from soil with a strong alkaline solution is to be regarded as bound residues or not. Jones et al. (2000) summed up the debate concerning bound residues: It has been shown for certain that bound residues are formed from potentially toxic/biologically active molecules in soil and that these residues are persistent. IF they can still be or again become bioavailable or biologically active although they cannot be extracted chemically – then they can still form part of the food chain or influence the fertility of the soil in the long run. Processes in the soil that result in the remobilization of bound residues may possibly also take place. If these assumptions are correct, bound residues are not to be desired. On the other hand, it is often pointed out that a reduced chemical extractability indicates that the substance has a weaker tendency to enter the soil water phase again and that lower extractability can be correlated with lower bioavailability. By this, the formation of bound residues must be seen as the first steps in a deactivation of the foreign organic molecule, which finally ends with a complete incorporation in the organic pool of the soil. Thus, increased binding and formation of bound residues can be understood as natural processes, which may even be used in the bioremediation as opposed to the manner in which bioremediation is most frequently carried out today, namely by increasing the bioavailability. "It seems likely that the significance, or otherwise, of bound residues in soils will ultimately be addressed by the bioavailability issue and this is likely to be the fOC us of ongoing research over the coming years" (Jones et al., 2000). 8 Methods for measuring the influence of sorption on the degradationAs it emerges from the chapters above, there has in recent times been a great deal of work going on carrying out studies where measurements of sorption are linked to measurements of the degradation, either through model descriptions of individual test readings of sorption and degradation, respectively, or through practical experiments where the sorption and the degradation are measured in the same series of experiments and a model is developed that describes the experimental results. The latter, by some called "reverse modelling", must be assumed to be the best solution, if the sorption- and degradation module in a leaching model such as MACRO is to be made more complex than it is in the present version, which uses Kd-value determinations for sorption specifications and simple first-order half-lives for specification of the degradation rate (Jarvis, pers. comm.). As mentioned in the part "Bound residues", spectroscopic methods that identify the type of binding between foreign chemicals and the soil organic matter are becoming wide-spread. A further development in this area is to be expected. Moreover, there have been attempts at studying the amount of dissolved substances in the soil water under circumstances where the ratio between soil and water was natural to be able in this manner to describe the amount of pesticide in the soil water over time. For this purpose a high-speed centrifugation technique (Ronday, 1997) and a technique using glass microfibre filters for collecting small amounts of soil water have been used (Gaillardon et al., 1991). The difficulties of deducing from chemical measurements on residues of pesticides and other chemicals whether there will be an undesirable effect on the soil biota (earthworms, insects, plants, and microorganisms) and presumably also on humans are obvious. Therefore, there is at present a large fOC us on developing methods and descriptions that can relate these to one another. Alexander (2000) stressed that, as chemical analytical methods used at present overestimate both exposure and risk of foreign chemicals in soil, it is important to develop methods for description of bioavailability. Such methods have so far predominantly been biological, but it is also necessary to develop chemical methods that describe bioavailability, as chemical methods are easier to handle than biological (Alexander, 2000). Kelsey et al. (1996) compared the amount of atrazine and phenanthrene that could be extracted by various methods of extraction with the amounts that could be absorbed by earthworms and/or degraded by bacteria. For one of the extraction agents, a good correlation was found. White et al. (1999) showed that in an aged soil with residues of phenanthrene the mineralisation curve and the desorption curve had the same slope, which indicated that the desorption was determinative for the mineralisation rate and thus for the bioavailability. Kelsey and Alexander (1997) compared the amounts of atrazine, phenanthrene, and naphthalene that were assimilated by earthworms with the amounts that could be extracted from soil with mild and strong extraction agents and found the best correlation when using a mild extraction agent. Kelsey and Alexander (1997) therefore stated that strong extractions should not be used if one wishes to estimate exposure and risk for selected organisms. Kelsey et al. (1997) presented selected chemical extraction agents to be used for predicting the bioaccessibility of chemicals in aged soil. Szmigielska et al. (1998) showed that anion interchange membranes could extract amounts of metsulfuron from soil that could be related to the biological effects on roots from lentil. At present Koskinen (pers. comm.) is working on studies of the application of genetically manipulated bacteria for determination of bioavailability of foreign chemicals in soil. The bacteria have had a gene incorporated, which means that they have a very well-developed ability to degrade a specific substance. Degradation experiments with the specific substance in a radioactive version will then provide a measure of the availability of the substance to degradation. For the present the National Academy of Science in the USA (2001) has set up a group of experts (the National Research Council Committee on Bioavailability of Contaminants in Soils and Sediments), which is to evaluate the scientific status concerning processes that influence whether chemical pollutions in soil and sediment are available to humans, animals, and plants. The group of experts will also evaluate the existing methods and methods which are being developed for the determination of bioavailability as well as assess how the bioavailability influences the effectiveness of bioremediation techniques. 9 Soil treatment as a factor influencing the relation between binding and degradationThe soil treatment influences many of the chemical, physical, and biological factors in the soil and is therefore of great importance to how quickly and how the pesticides are transported through the plough layer. The binding and the degradation of the pesticides and their metabolites are also dependent on whether the soil has been treated conventionally or not as the amount of organic matter and the microbial activity have a great influence on the binding and degradation of pesticides, which in their turn will influence whether the substances are leached. In the literature there are conflicting reports as regards degradation and leaching of pesticides in soil that had been subjected to conventional tillage (CT) and different degrees of reduced tillage (RT) or no tilllage (NT). Some publications proved that there was no difference between the leaching of pesticides when CT was compared with NT. Other publications proved that the leaching was greater from NT soil than from CT soil explaining the difference with the occurrence of a large number of macropores in the NT soil. Other publications showed that the leaching was less from NT soil than from CT soil explaining the difference with a larger binding and a more rapid degradation rate in the NT soil, which often has a higher content of organic matter and a higher biological activity than the CT soil. In Denmark the NT technique is used very infrequently. Some of the parameters that are different in soil that has been tilled differently are the amount of organic matter and the biological activity in the upper soil layers. Helling (1987) summed up the results of measurements of the amount of organic matter in the upper soil layers and found that the amount of organic matter in NT soil was between 1.5 and 2 times the amount in CT soil. Helling (1987) also quoted studies where the total number of aerobic microorganisms, the facultatively anaerobic microorganisms, and the denitrificating microorganisms were respectively 1.35, 1.57, and 7.31 times higher in NT soil than in the CT soil. In many of the known studies where the leaching or run-off of pesticides has been compared for different tillage systems, CT has been compared with NT (Fermanich et al., 1996; Isensee et al., 1990; Levanon et al., 1993; Fermanich & Daniel, 1991; Ritter et al., 1996; Sadeghi & Isensee, 1992; Isensee & Sadeghi, 1993; Afyuni et al., 1997). In most of these cases (Fermanich et al., 1996; Isensee et al., 1990; Fermanich & Daniel, 1991; Sadeghi & Isensee, 1992; Isensee & Sadeghi, 1993; Afyuni et al., 1997) it emerged that the leaching or the run-off was significantly greater in NT than in CT for the substances atrazine and deisopropylatrazine; atrazine, deethylatrazine, alachlor, cyanazine and carbofuran; carbofuran and chlorpyrifos; atrazine; atrazine, cyanazine and alachlor; chlorimuron and nicosulfuron whereas Levanon (1993) showed that the leaching was greater in CT than in NT for the substances atrazine, carbofuran, diazinon, and metolachlor. Ritter et al. (1996) showed that there was no important difference in the leaching of atrazine, simazine, cyanazine, and alachlor in CT and NT but that rain occurrences of more than 30 mm shortly after spraying was the important factor, which determined whether the substances were found in the groundwater. Comparisons between RT and CT were made by Cox et al. (1996), who studied the leaching of metamitron in disturbed columns from RT soil and CT soil and found that 9% was leached from CT soil while 5% was leached from RT soil. The difference was explained with a higher degradation rate in RT and a greater degree of preferential flow in CT. Gish et al. (1991) showed that the leaching of pesticides was more extensive in NT soil than in CT soil because of preferential flow, but they emphasized that it mainly applied to wettable powder and liquid formulations while encapsulated formulations reduced the significance of preferential flow. It has often been stated that earthworm channels in the upper layers of NT soil are a very important factor when a greater degree of preferential flow is seen in NT soil than in CT soil. However, it is important to note that Stehouwer et al. (1993) showed that there was about three times as large an amount of organic matter in the linings of the earthworm channels than in the matrix soil, which might increase the binding of atrazine. Zablotowicz et al. (2000) studied the degradation of fluometuron in CT soil and NT soil and found that the rate constant for the degradation in the upper two cm in CT was twice that in NT. 48% more organic matter was found in the upper two cm of the NT soil than in the upper two cm of the CT soil, and the microbial biomass and fluorescein diacetate hydrolysis activity was 106% and 127% higher, respectively, in the upper two cm of the NT soil compared with that of the CT soil. In this case the amount of organic matter has apparently increased the binding of the pesticide so much that it – despite the higher biological activity – is degraded more slowly in the NT soil. Reese et al. (1999) found no significant difference in the degradation rates of isoproturon and diflufenican when comparing CT and RT. Giupponi et al. (1996), on the other hand, found that soil tillage influences the degradation rate of isoproturon. In a prolonged study in Italy between 1993 and 1995, the results showed that the Kd-values were higher in RT soil than in CT soil. The degradation was faster in a ploughless ridge-cultivated soil than in both CT and NT soils. Modelling in VARLEACH, where the measured values for degradation rates and sorption were applied, showed that ploughless ridge cultivation was the form of tillage that had the least influence on the quality of the groundwater as regards leaching of pesticides. Fomsgaard and Kristensen (2002) carried out a number of studies of the degradation of isoproturon in three types of soil that had been tilled differently: a) newly ploughed: Soil from a field that for 20 years previous to the study had only been harrowed but was ploughed shortly before samples were taken (RT-CT), b) harrowed: Soil that for 20 years had not been ploughed but only harrowed (RT), and c) ploughed: Soil that had been ploughed for a number of years before sampling (CT). The studies were carried out in different concentrations (0.1 µg/g and 0.01 µg/g) in both plough layers and deeper soil layers. 14C-labelled pesticide was used so that the total mineralisation to 14CO2 could be followed. At the time samples were continuously interrupted during the incubation of some of the experiments so that the remaining amount of isoproturon and formed metabolites could be followed over time. Some of the samples were analysed by two extraction methods, an aqueous and an organic one, by means of which it was possible to a certain extent to distinguish between desorbable pesticide and non-desorbable pesticide. The mineralisation rate for isoproturon in plough layers at the concentration of 0.1 µg/g was highest in RT soil. The biological activity, measured as the mineralisation rate of Na-acetate, was also highest in this soil. Figure 23 shows the results of the measurements of the mineralisation where the rise in the first part of the curve is the measure of the rate that is calculated by the mathematical modelling. The mineralisation rate for isoproturon was higher in soil that normally had been ploughed (CT) than in soil that had been ploughed for the first time in many years (RT-CT). A similar difference between the mineralisation rates in the different types of soil was seen in the concentration of 0.01 µg/g. The recent ploughing of the soil that formerly had only been harrowed has also reduced the mineralisation rate considerably. The microcosmos in which the bacteria live has been decisive for their activity. And the activity has been so high that it has been able to cause a more rapid mineralisation even though the binding, measured by Kd-values, was not significantly different in the harrowed and the newly ploughed soil (Fomsgaard & Kristensen, 2002). The influence of the amount of organic matter on sorption and degradation, respectively, has been discussed in many publications. Generally, it will be expected that a larger amount of organic matter causes an increased binding which then is expected to reduce the degradation rate because the substance is less available to the microorganisms in the aqueous phase. However, the opposite – that a greater amount of organic matter is linked to a greater biological activity – is often seen to be the case, which then increases the degradation rate for the pesticides, as seen here in Fomsgaard & Kristensen (2002) where the binding was not influenced when the Kd-values for newly ploughed and harrowed soil are compared. A more detailed knowledge of the type of organic matter would in this case have been interesting. What the connection between the mineralisation presented in Figure 23 and the disappearance of the parent substance (which for one series of experiments is shown in Figure 17) is like has not been fully analysed yet. Only with these results can it be determined whether the lower mineralisation rates result in residues of extractable substance in the soil.
Figure 23. Mineralisation rates for isoproturon at 15°C and 5°C and mineralisation rates for Na-acetate. The trials/experiments have been carried out in CT soil (soil that has been ploughed normally for a number of years), RT (soil that has only been harrowed for 20-30 years), and RT-CT (soil that has only been harrowed in the previous 20-30 years but was ploughed for the first time shortly before sampling) (Fomsgaard & Kristensen, 2002). 10 Pesticides mainly bound to clay minerals: glyphosate, paraquat, and diquatBoth glyphosate and the metabolite of glyphosate, AMPA, are quickly and strongly adsorped in most soil types. Unlike most other pesticides glyphosate will not be very strongly bound to the soil organic matter, but on the other hand primarily to the soil minerals. The content of cations on the clay minerals is thought to be important for the binding. The binding is probably caused by metal cations (e.g. Fe3+ and Al3+) forming complexes with glyphosate and AMPA through the phosphonic acid part (Glass, 1987; Sprankle et al., 1975). The binding of glyphosate takes place in competition with free inorganic phosphate. The binding mechanisms have not been fully explained, however (Franz et al., 1997). Kd-values for glyphosate in a number of surface soils are reported to be between 33 and 377 (Jacobsen et al., 1998). In a study of the sorption of 14C-marked glyphosate to a number of Nordic soils (Tiberg, 1998), Kd-values above 100 were similarly found in most soils. Measurements of the sorption in subsurface soils showed an even greater sorption than the corresponding surface soils. Studies in Sweden (Torstensson & Lindholm, 1988) have similarly shown Kd-values of 26 to 83 in soil sampled below railways in Sweden. Similar values have been found in Danish soils and in samples taken at a depth of about 1 metre (Petersen et al., 1998). The degradation time for glyphosate can vary greatly from one soil to another. On measuring the degradation time on the basis of the liberation of 14CO2- from 14C-labelled glyphosate, half-lives from a few days to 175 days were found. In soil sampled under the plough layer the degradation was considerably slower (Jacobsen et al., 1998). In most cases the half-life seems to be relatively short in agricultural soils and has been shown to be similarly short in forest soils (Torstensson, 1985) even though a few half-lives of more than 10 years have been proved in 2 soils from Hawaii (Franz et al., 1998). In a number of studies, agricultural soils have been analysed for residues of glyphosate and AMPA between 3 and 8 months after being treated with glyphosate. The results showed (Torstensson, 1985) that residues of glyphosate and AMPA can be proved in the soils after 3 to 8 months. In some cases mainly AMPA, in others mainly glyphosate. No results have been found in the refereed studies showing an accumulation of glyphosate or AMPA after incubation for a long time. It has previously been shown that pesticides that are strongly bound to clay particles can be stabilised in the soil so that they get a very long half-life even though they are substances that in unbound state are relatively easily degradable. Especially long persistence after binding has been proved for the herbicide paraquat. Studies by Fryer et al. (1975) showed that almost the whole amount of paraquat, which was added in field experiments over 6 years, could be found again in the soil. Most remained in the upper 5 cm, but low contents were also found to a depth of 25-35 cm, which were attributed to the sampling method or to particle transport, cracks or earthworm activity. Soil milling did not influence the found residues of paraquat. Similarly, Smith and Mayfield (1978) found that under a number of different incubation conditions paraquat was not degraded in 16 months, and Riley et al. (1976) stated that bound residues of paraquat were not available to living organisms such as plants, earthworms or microorganisms. Riley et al. (1976) distinguished between "loosely bound" and "tightly bound" paraquat as they defined "loosely bound" paraquat as the part that could be extracted from the soil with 5M NH4Cl and "tightly bound" paraquat as the part that could be extracted by boiling with 18 N sulphuric acid. Riley et al. (1976) stated that it has been shown in greenhouses that "loosely bound" paraquat potentially might be available to plants again. Eberbach (1998) studied the mineralisation of 14C-labelled glyphosate and demonstrated a biphasic course in which a relatively rapid mineralisation of available glyphosate and a slower mineralisation of bound glyphosate took place. The biphasic course of a degradation curve is thus not reserved for chemicals that are bound to the soil organic matter but occurs also for substances mainly bound to the clay particles of the soil. Nomura and Hilton (1977) and Rueppel et al. (1977) also mentioned the influence of sorption on the course of the degradation of glyphosate. Eberbach found that glyphosate is most strongly bound in soil with acid pH. Heinonen-Tanski (1989) found that liming increases the degradation rate for glyphosate and de Jonge et al. (2001) showed that the binding of glyphosate decreased with an increased presence of phosphate. Despite the biphasic course proved by Eberbach, there are many proofs that a higher biological activity in the soil increases the decomposition rate of glyphosate. Thus, it is to be expected that the decomposition will be quicker in a reduced tillage soil than in a conventionally tilled soil. Fomsgaard et al. (2003a; 2003b) carried out lysimeter experiments with isoproturon and glyphosate. Experiments with 14C-labelled isoproturon and unlabelled glyphosate were carried out as repeat determination in soil that had been tilled conventionally while experiments with 14C-glyphosate and unlabelled isoproturon were carried out as repeat determination in soil that had been tilled with a reduced tillage technique. In the reduced tillage soil, glyphosate had been applied regularly until 3 years before sampling while glyphosate had not been applied in the last 5 years in the conventionally tilled soil. 16% and 14%, respectively, of the added radioactivity in the 14C-glyphosate lysimeters were found in the top 10 cm of the soil while 0.2% and 0.7%, respectively, were found in soil from 10-20 cm. Alkaline extractions were carried out in 0-10 cm depth and specific analysed on LC/MS of the residues of glyphosate and AMPA in the entire soil profile in both repeat determinations. The amounts of 14C found in the upper soil layers were on a level with findings of non-extractable radioactivity from 14C-mecoprop in Felding et al. (1996) and considerably lower than the findings of Burauel et al. (1998) of more than 80% of the added radioactivity in the upper 30 cm in 20 different outdoor lysimeter experiments carried out with 14C-labelled atrazine, terbuthylazine, chloridazon, dichlorprop-P, methabenzthiazuron, pyridate, and anilazine and considerably lower than the abovementioned results of 14C from 14C-isoproturon in lysimeter plough layers from the two lysimeters taken from conventionally tilled soil. With the specific analysis significantly greater amounts of AMPA residues were found in the soil that had been cultivated with reduced tillage than in soil that had been conventionally tilled. Decomposition has either been slower in the soil that has been cultivated with reduced tillage than in the soil that has been tilled conventionally or the decomposition of glyphosate and AMPA has possibly not been sufficiently rapid for the residues of the previous years to disappear completely from the soil given the actual pattern of pesticide consumption. As glyphosate according to many studies is strongly bound, the second part of the two-compartment process can be expected to pass off exceedingly slowly. No direct comparisons can be made between the amount of 14C and the amounts of the substance, which were proved in the specific analysis, as the 14C-analyses do not measure the specific substances. It would be highly interesting to know how glyphosate was bound to the soil particles of different types and sizes. Gimsing et al. (2001) showed that the soil oxides are of great importance as regards the ability of the soil to adsorb glyphosate while the content and the type of clay are of small importance. It was shown by deJonge and deJonge (1998) that a larger dispersion of clay did not increase the binding of glyphosate to the clay minerals. 11 DiscussionWhether it is a matter of chemicals mainly bound to the soil organic matter or to the clay minerals, it is apparently a matter of basically the same model for the influence of sorption on the degradation as illustrated in Figure 24: Sorption and degradation take place in two dominating compartments, the aqueous phase and the soil phase. A part of the xenobiotic chemical may be degraded relatively rapidly in the first compartment, the aqueous phase, possibly through the formation of identifiable metabolites, and can be completely mineralised to CO2. If the substance is not transformed sufficiently rapidly, it may be leached. Both the xenobiotic chemical or metabolites from this may be adsorbed to soil particles through a number of different binding mechanisms, and the substances may partly be desorbed from this again and go back into the aqueous phase where they may be degraded or leached. From being more "loosely" bound to the soil particles, the substances may be more "firmly" bound whereby it becomes a matter of "bound residues". "Bound residues" appear in various forms: The entire molecule of the xenobiotic or parts of it may have entered a new molecular structure as a part of the organic matter, it may be sequestrated in inaccessible places in the soil aggregates or it may be strongly sorbed. There is a difference on what can be extracted by chemical methods and what can be activated purely biologically. The dotted lines (A) in Figure 24 represent substances that can be extracted with extraction agents that do not change the chemical structure of the substances, the soild lines (B) represent the bound residues that can be extracted with stronger extraction agents, and the dashed lines (C) represent the bound residues that cannot be extracted chemically. In each of the three groups, there may be subgroups that may be/become bioavailable to both plants, animals, and degrading microorganisms, as these subgroups are substances that can be reactivated as the original molecule or a metabolite from this after which it can be degraded or leached (process (1)). Other parts of the substances may, in the strongly bound structure in which they form part, be directly transformed to CO2 by the microorganisms as a constant transformation of the organic pool takes place (process (2) in Figure 24). The fundamental principles – the model according to which the substances are degraded – are as mentioned before the same for all types of substances. The variations that are caused by the chemical structure of the substances and the soil texture are seen in the distribution of the amounts of substance in the two compartments, in the distribution of the amounts of substance in the individual subgroups A, B, and C, and in how much of the amounts in A, B, and C that can be transformed according to processes 1 and 2, respectively. When the substances are bound to the clay minerals, they will not be transformed according to process 2 but only according to process 1. If the part that is transformed according to process 1 is small it will result in larger amounts remaining in the soil as bound residues. The crucial question here is whether there may be changes in the soil that can lead to an increased reactivation and whether this reactivation may lead to the occurrence of biologically active reactivated amounts of the substance that may have an adverse effect on the soil environment or may be leached before it is degraded.
Figure 24. Outline of the problems concerning the influence of sorption on the degradation of pesticides and other chemical in soil. The degradation in the second slow compartment is considerably slower than the average degradation rate that usually can be calculated from a simple first-order process, but at the same time the degradation is also considerably quicker in the first compartment than it is when a simple first-order process is used. The choice of model may not be particularly crucial for some substances, but for others where the course is special in one compartment it may be crucial. As far back as 25 years ago, Hamaker and Goring (1976) described that the degradation of pesticides depends very much on the extent to which the substance is bound to the soil particles and that the degradation therefore can be described as taking place in two compartments. At that time, results had to be marked and calculated on semilogarithmic paper, so the simple first-order kinetics has often been assumed for the calculation of degradation rates. The parameters in more complex models can now be calculated by means of a computer, so the time should have come where the importance of using one or the other model for the evaluation of the tendency towards leaching of substances is assessed. It is therefore highly relevant that a number of comparisons are made of the calculated remaining amounts of substances after certain intervals during the application of the relevant simple first-order model and the two-compartment model, so that there is a basis for assessing whether the approval system for pesticides should be changed to allow for this fact, and that the assessment of the persistence of the substances according to a new approval system is carried out on the basis of calculations in which the two-compartment model has been used. It is necessary to take a position on the biphasic system for degradation and identify the different results of the most frequently used models in leaching scenarios. Such model runs will, however, only be a step on the way to a better description. Soil that has a higher content of organic matter will in some cases delay the degradation and the leaching of chemicals due to the stronger binding. At the same time the larger amount of organic matter may result in a higher microbiological activity, which increases the degradation rate for pesticides. It is imperative that more studies are carried out in which measurements of sorption and measurements of the degradation are linked, e.g. through model descriptions of individual measuring results of sorption and degradation, respectively, or through practical experiments, in which the sorption and the degradation are measured in the same experiment process and a model is developed that describes the results. The latter, so-called reverse modelling, must be assumed to be the best solution. The published examples, in which such models have been developed, have naturally concentrated on individual substances. A test of these different models on a large and varied data material would provide the best basis for assessing which of these models allow for the varying soil conditions to the greatest extent. Such a model might then be built into the most frequently used leaching models as a substitute for the existing sorption and degradation modules, which use Kd-value determinations for sorption specifications and simple first-order half-lives for specification of degradation rates. The error that is made by using a simple first-order model as the basis for calculations of half-lives varies according to the other circumstances in the soil. From Figure 10 is seen that the half-life is estimated at 15 days according to the simple first-order process whereas it is just 4.2 days for the first halving according the two-compartment model. If the part of the substance that is transformed in the second compartment is assumed only to have been extracted due to the application of an organic extraction agent and otherwise will not be available to the biology of the soil or to leaching to the groundwater, then the substance has been assessed harder than what could be regarded as reasonable at the application of the simple first-order process. If, on the other hand, the amount that is transformed in the second compartment in reality is bioavailable or can be leached to the groundwater, the substance is judged too mildly at the application of the simple first-order process as three halvings are estimated at just 45 days while it takes 62 days according to the two-compartment process. Thus, it is not just a question of a need to find the right model for describing the sorption-degradation process but also the question of a need to throw light upon the type of binding and the bioavailability of the amounts of substance that are transformed in the second compartment. If the interesting factor is how much of the pesticide present in the soil that may have a toxic effect on soil organisms, a traditional chemical analysis will provide an overestimated result because the chemical analysis is traditionally carried out with extraction agents that extract as much of the substance as possible while after all a toxic effect on soil organisms only occurs if the substance is present in the aqueous phase. In the same way a traditional chemical method of analysis will often overestimate the amount of chemical that might be a threat to the groundwater. If the two-compartment model is taken into account in connection with approvals and in model runs, the problem concerning bound residues must also be taken into account as it has been extremely much in fOC us during the last 10 years. There are already a large number of studies of binding mechanisms for both desorbable xenobiotics and bound residues. However, it is necessary to couple these studies to studies of bioavailability to determine to which extent the substances can be reactivated in the natural system. Adsorption/desorption is a balance that over time may be disturbed and adjusted in a new balance where parts of bound substance will possibly be released. It would be very appropriate if there were chemical methods of analysis able to extract the amounts of the xenobiotics from the soil that could be correlated with the bioavailable amounts that are the results of biological studies of bioavailability. For the time being the biological studies are necessary to measure bioavailability, but they are expensive and difficult. The work on developing such methods should be intensified. There has been much fOC us on developing chemical methods of analysis that can extract everything, so the chemical laboratories needs to adapt themselves to fOC us on measurements of bioavailable amounts. It is important to emphasize that laboratory experiments that are intended to be used for describing the complex connection between the binding and the degradation must allow for the extreme variation of the processes. There is already a large variation, which is merely caused by the heterogeneity of the soil. The part of the variation, which may be caused by the method, must be eliminated. Here, the first step must be to carry out the studies under conditions that resemble the natural conditions as much as possible, that is by application of the concentrations of pesticides that will be relevant according to normal farming practice, incubation at temperatures, water content, and oxygen conditions that conform to the climate and the soil where the pesticides are used. Further, it is important to establish to which extent the degrading microorganisms are influenced by the sieving and mixing of the soil that is frequently carried out before the experiments. If the microorganisms and with them the degradation process are significantly affected by this, experiments can be carried out in microcosmos setups with undisturbed soil columns where the conditions of the microorganisms are not changed. In theory, one might imagine that the evaluation of the persistence of pesticides by the approval system required studies of degradation and sorption by methods closer to reality than the ones used at present and that these studies had to be carried out in a number of different soils with a varying content of organic matter and clay. At the same time it might be required that the degradation process was described by the two-compartment model. Different limit values for the degradation rate of the pesticides in the two compartments might be introduced. It may be accepted that the degradation rate in the second compartment (where the substance is bound) has a lower value, but this value must be dependent on the quantitative distribution of the pesticide in the two compartments. A demand for supplementary measurements of microbiological activity in the soil could also be attached to the system to ensure that the microbiological activity in the used soil samples was within a standard area. Both from a legal and a scientific point of view it is important to differentiate between free pesticide residues and bound pesticide residues just as it is important to distinguish between biological persistence and chemical persistence when persistence is in question, as these do not always go together. Therefore, it might be assumed that the approval system required a distinction between different types of extraction agents in the analysis of degradation and sorption measurements, so it was possible to predict the possibility of reactivation of the remaining amounts of substances. 12 ReferencesAfyuni, M., Wagger, M. & Leidy, R. (1997) Runoff of two sulfonylurea herbicides in relation to tillage system and rainfall intensity. Journal of Environmental Quality, 26, 1318-1326. Alexander, M. (2000) Aging, Bioavailability, and Overestimation of Risk from Environmental Pollutants. Environmental Science and Technology, 34, 4259-4265. Alexander, M. (1995) How toxic are toxic chemcials in soil? Environmental Science and Technology 29 2713-2717. Alexander, M. (1985) Biodegradation of organic chemicals. Environ. Sci. Technol., 18, 106-111. Alexander, M. Oral presentation, SETAC Europe Conference, Organic Soil Contaminants 2001, 2-5 Sept 2001, Copenhagen, Denmark. Anderson, J.P.E. (1984) Herbicide degradation in soil: influence of microbial biomass. Soil Biol. Biochem., 16, 483-489. Andreux, F., Portal, J.M., Schiavon, M., Bertin, G. & Barriuso, E. (1991) The usefulness of humus fractionation methods in studies about the behaviour of pollutants in soils. Toxicol-Environ-Chem 31/32 29-38. Anonymous (1991) EC Directive 91/414/EEC concerning the placing of plant protection products on the market (Authorisations Directive). Commission of the European Communities - Directorate-General for Agriculture - DG VI B II-I, Brussels, Belgium. Anonymous (1995) EC Directive 95/36 establishing the data requirements for environmental fate and behaviour in Annex II (chapter 7) and Annex III (chapter 9) of directive 91/414. Commission of the European Communities - Directorate-General for Agriculture - DG VI B II-I, Brussels, Belgium. Anonymous (1997) EC Directive 97/57 establishing Annex VI (Uniform Principles) to directive 91/414. Commission of the European Communities - Directorate-General for Agriculture - DG VI B II-I, Brussels, Belgium. Anonymous (1997) EC Guidance Document 9188/VI/97, Persistence in Soil. Commission of the European Communities - Directorate-General for Agriculture - DG VI B II-I, Brussels, Belgium. Aochi, Y., Farmer, W. & Sawhney, B. (1992) In situ investigation of 1,2-dibromoethane sorption/desorption processes on clay mineral surfaces by diffuse reflectance infrared spectroscopy. Environmental Science and Technology, 26, 329-335. Backhus, D. & Gschwend, P. (1990) Fluorescent polycyclic aromatic hydrocarbons as probes for studying the impact of colloids on pollutant transport in groundwater. Environmental Science and Technology, 24, 1214-1223. Barnabas, I. J., Dean, J. R. & Owen, S. P. (1994) Critical review: Supercritical fluid extraction of analytes from environment samples - A review. Analyst 119[11], 2381-2394. Barriuso,E (1994). Pesticide Pollution of Soils – Analytical Aspects. Analusis Magazine, 22, M13-M15. Barriuso, E., Koskinen, W. & Sorenson, B. (1992) Modification of atrazine desorption during field incubation experiments. The Science of the total Environment, 123/124, 333-344. Barriuso, E. & Koskinen, W. (1996) Incorporating nonextractable atrazine residues into soil size fractions as a function of time. Soil Science Society of America Journal, 60, 150-157. Beigel, C. & diPietro, L. (1999) Transport of triticonazole in homogeneous soil columns: influence of nonequilibrium sorption. Soil Science Society of America Journal, 63, 1077-1086. Bergström, L. & Stenström, J. (1998) Environmental Fate of Chemicals in Soil. Ambio, 27, 16-23. Bertin, G., Schiavon, M. & Pottier, C. (1990) Plant bioavailability of "natural" and "model" humic acid-bound [14C] atrazine residues. Toxicol-Environ-Chem 26 203-210. Boesten, J., Businelli, M., Delmas, A., Edwards, E., Helweg, A., Jones, R., Klein, M., Kloskowski, R., Layton, J., Marcher, S., Schäfer, H., Smeets, L., Styzcen, M., Russel, M., Travis, K., Walker, A. & Yon, D. Leaching models and EU registration. The final report of the work group of the Regulatory Modelling Work Group of fOC US. 1995. European Commision. Bollag, J. & Stotsky, G. (2000) Soil biochemistry Vol. 10. Marcel Dekker Inc.; New York; USA. Bollag, J. & Myers, C. (1992) Detoxification of aquatic and terrestrial sites through binding of pollutants to humic substances. Science of the Total Environment, 117/118, 357-366. Bollag, J., Myers, C.J. & Minard, R.D. (1992) Biological and chemical interactions of pesticides with soil organic matter. The Science of the Total Environment, 123/124, 205-217. Brunner, W. & fOC ht, D.D. (1984) Deterministic three-half-order kinetic model for microbial degradation of added carbon substrates in soil. Appl. Environ. Microbiol., 47, 167-172. Brusseau & M.L. (1993) The influence of solute size, pore water velocity, and intraparticle porosity on solute disperion and transport in soil. Water-resour-res 29 1071-1080. Burauel, P. & Fuhr, F. (2000) Formation and long-term fate of non-extractable residues in outdoor lysimeter studies. Environmental Pollution, 108, 45-52. Burauel, P., Wais, A. & Führ, F. (1998) Soil-bound residues in Führ, F, Hance, RJ, Plimmer, JR, and Nelson, JO. (Eds). The lysimeter concept. 177-188. American Chemical Society. ACS Symposium Series. Calderbank, A. (1989) The occurrence and significance of bound pesticide residues in soil. Reviews of Environmental Contamination and Toxicology, 108, 71-103. Capriel, P., Haisch, A. & Kahn, S. Distribution and nature of bound (nonextractable) residues of atrazine in a mineral soil nine years after herbicide application. Journal of Agricultural and Food Chemistry, 33. Cerniglia, C. (1992) Biodegradation of polycyclic aromatic hydrocarbons. Biodegradation, 3, 351-368. Chin, Y. & Gschwend, P. (1992) Partitioning of polycyclic aromatic hydrocarbons in marine porewater organic colloids. Environmental Science and Technology, 26, 1621-1626. Cox, L., Calderon, M., Celis, R., Hermosin, M., Moreno, F., & Cornejo, J. (1996). Mobility of metamitron in soils under conventional and reduced tillage., Fresenius Environmental Bulletin 5: 528-533. Christensen, P. (2002) Modelling the mineralisation and decay of isoproturon and glyphosate in conventional and reduced tillage soil from an experimental approach. Master thesis. University of Copenhagen and Danish Institute of Agricultural Sciences. 173 pp. Craven, A. (2000) Bound residues of organic compounds in the soil: the significance of pesticide persistence in soil and water: a European regulatory view. Environmental Pollution, 108, 15-18. Dec, J. & Bollag, J. (1997) Determination of covalent and non-covalent binding interactions between xenobiotic chemicals and soil. Soil Science, 162, 858-874. deJonge, H. & deJonge, L. (1999) Influence of pH and solution composition on the sorption of glyphosate and prochloraz to a sandy loam soil. Chemosphere, 39, 753-763. deJonge, H., deJonge, L., Jacobsen, O., Yamaguchi, T. & Moldrup, P. (2001) Glyphosate sorption in soils with varying phosphorous and lime application rates. Soil Science, 166, 230-238. deJonge, L., deJOnge, H., Moldrup, P., Jacobsen, O. & Christensen, B. (2000) Sorption of prochloraz on primary soil organomineral size separates. Journal of Environmental Quality, 29, 206-213. Dictor, M. C., Soulas, G., Takagi, K., Anderson, J. P. E., Lewis, K., & Lewis, F. (1992) Subsoil microbiology: Microbial activity and potential for pesticide degradation in the unsaturated zone. Proceedings of the International Symposium on Environmental Aspects of Pesticide Microbiology. 284-290. 1992 Eberbach, P. (1998) Applying non-steady-state compartmental analysis to investigate the simultaneous degradation of soluble and sorbed glyphosate (N-(phosphonomethyl)glycine) in four soils. Pesticide Science, 52, 229-240. Eschenbach, A., Kastner, M., Bieri, R., Schaefer, G. & Mahro, B. (1994) Evaluation of a new effective method to extract polycyclic aromatic hydrocarbons from soil samples. Chemosphere, 28, 683-692. Felding, G., Odgaard, P. & Helweg, A. (1996) Leaching of mecoprop determined in lysimeters containing undisturbed soils. Brown, H et al. Proceedings of the Second International Weed Control Congress. I, 317-324. Fermanich, K.J. & Daniel, T.C. (1991) Pesticide mobility and persistence in microlysimeter soil columns from a tilled and no-tilled plot. Journal of Environmental Quality, 20, 195-202. Fermanich, K., Bland, W., Lowery, B. & McSweeney, K. (1996) Irrigation and tillage effects on atrazine and metabolite leaching from a sandy soil. Journal of Environemental Quality, 25, 1291-1299. Firestone, M.K. (1982) Biological denitrification. Stevenson, FJ. Nitrogen in agricutural soils. Agron. Monograph 22. Madison, WI, ASA, CSSA and SSSA. fOC ht, D.D. & Shelton, D. (1987) Growth kinetics of Pseudomonas alcaligenes C-0 relative to inoculation and 3-chlorobenzoate metabolism in soil. Applied and Environmental Microbiology, 53, 1846-1849. Fomsgaard, I.S. (1999) The mineralisation of pesticides in surface and subsurface soil - in relation to temperature, soil texture, biological activity and initial pesticide concentration. Denmark, Danish Institute of Agricultural Sciences. DIAS Report Plant Production. Fomsgaard, I.S. & Kristensen, K. (1999a) ETU mineralisation in soil under influence of organic carbon content, temperature, concentration, and depth. Toxicological and Environmental Chemistry, 70, 195-220. Fomsgaard, I.S. & Kristensen, K. (1999b) Influence of microbial activity, organic carbon content, soil texture and soil depth on mineralisation rates of low concentrations of 14C-mecoprop - development of a predictive model. Ecological Modelling, 122, 45-68. Fomsgaard, I.S. & Kristensen, K. (2002) Modelling the degradation of isoproturon in reduced tillage and normal tillage loamy soil. In preparation. Fomsgaard, I.S., Spliid, N. & Felding, G. (2003a) Leaching of pesticides through normal-tillage and low-tillage soil - a lysimeter study. II. Glyphosate.Journal of Environmental Science and Health B. In press Fomsgaard, I.S., Spliid, N. & Felding, G. (2003b) Leaching of pesticides through normal-tillage and low-tillage soil - a lysimeter study. I. Isoproturon. Journal of Environmental Science and Health B. In press. Franz, J., Mao, M. & Sikorski, J. (1996) Glyphosate: A Unique Global Herbicide. American Chemical Society. Fryer, J., Hance, R. & Ludwig, J. (1975) Long-term persistence of paraquat in a sandy loam soil. Weed Research, 15, 189-194. Führ, F. Non-extractable pesticide residues in soil. (1987) Greenhalgh, R. & Roberts, T.R. Pesticides Science and Biotechnology. Proc. 6th Int. Congr. Pestic. Chemistry, IUPAC. 381-389. London, Blackwell Scientific Publications. Führ, F. & Ophoff, H. (1998) Pesticide Bound Residues - Workshop- Senate Commission for the Assesment of Chemicals Used in Agriculture. Wiley-VCH Verlag, Weinheim. Führ, F., Ophoff, H., Burauel, P., Wanner, U. & Haider, K. (1998) Modification of the definition of bound residues. Pesticide Bound Residues - Workshop- Senate Commission for the Assessment of Chemicals Used in Agriculture. 175-176. 1998. Wiley-VCH Verlag, Weinheim. Gaillardon P., Fauconnet F., Jamet P., Soulas G. & Calvet, R. (1991) Study of diuron in soil solution by means of a novel simple technique using glass microfibre filters. Weed Research 31[6], 357-366. Gamerdinger, A.P., Wagenet, R.J. & van Genuchten, M.T. (1990) Application of two-site/two-region models for studying simultaneous nonequilibrium transport and degradation of pesticides. Soil Science Society of America Journal, 54, 957-963. Gevao, B., Semple, K. & Jones, K. (2000) Bound pesticide residues in soils: a review. Environmental Pollution , 108, 3-14. Gish, T., Isensee, A., Nash, R. & Helling, C. (1991) Impact of pesticides on shallow groundwater quality. Transactions of the ASAE, 34, 1745-1753. Giupponi, C., Bonaiuti, G., Capri, E., Errera, G. & Trevisan, M. (1996) Effects of alternative soil tillage systems on the degradation and sorption of herbicides. Re-AAM-del, Capri-E, Evans-SP, and Trevisan-M. The environmental fate of xenobiotics. Proceedings of the 10th Symposium Pesticide Chemistry, 151-161. 1996. Milan; Italy, Facoltá di Agraria, Universitá Cattolica del Sacro Cuore. Glass, R. (1987) Adsorption og glyphosate by soil and clay minerals. Journal of Agricultural and Food Chemistry , 35, 497-500. Greenhalgh, R. & Roberts, T.R. Pesticide Science and Biotechnology. 405-410. Oxford, Blackwell Scientific. Guo, L., Jury, W., Wagenet, R. & Flury, M. (2000) Dependence of pesticide degradation on sorption: nonequilibrium model and application to soil reactors. Journal of Contaminant Hydrology, 43, 45-62. Gustafson, D.I. & Holden, L.R. (1990) Nonlinear Pesticide Dissipation in Soil: A New Model Based on Spatial Variability. Environ. Sci. Technol., 24, 1032-1038. Haider, K., Spiteller, M., Reichert, K. & Fild, M. (1992) Derivatization of humic compounds: an analytical approach for bound organic residues. International Journal of Environmental Analytical Chemistry, 46, 201-211. Haider, K., Spiteller, M., Wais, A. & Fild, M. (1993) Evaluation of the binding mechanism of anilazine and its metabolites in SOM. International Journal of Environmental Analytical Chemistry, 46, 125-137. Harmon, T.C., Ball, W.P. & Roberts, P.V. (1989) Nonequilibrium transport of organic contaminants in ground water. Sawhney, BL and Brown, K. Reactions and movement of organic chemicals in soils. SSSA Special Publication 22. 45-80. Soil Science Society of America. SSSA Special Publication. Hatcher, P., Bortlalynski, J., Minard, R., Dec, J. & Bollag, J. (1993) Use of high resolution 13C NMR to examine the enzymatic covalent binding of 13C-labelled 2,4-dichlorophenol to humic substances. Environmental Science and Technology, 27, 2098-2103. Heinonen-Tanski, H. (1989) The effect of temperature and liming on the degradation of glyphosate in two arctic forest soils. Soil Biology Biochemistry, 21, 313-317. Helling, C.S. (1987) Effect of conservation tillage on pesticide dissipation. Effects of Conservation Tillage on Groundwater Quality: Nitrates and Pesticides. 179-187. USA, Lewis Publishers. Helweg, A. (1987) Degradation and adsorption of 14C-MCPA in soil - influence of concentration, temperature and moisture content on degradation. Weed Research, 27, 287-296. Helweg, A. (1993) Degradation and adsorption of 14C-mecoprop (MCPP) in surface soils and in subsoil. Influence of temperature, moisture content, sterilization and concentration on degradation. The Science of the Total Environment, 132, 229-241. Helweg, A., Fomsgaard, I., Reffstrup, T. & Sørensen, H. (1997) Degradation of mecoprop and isoproturon in soil. International Journal of Environmental Analytical Chemistry, 4, 1-17. Hill, I.R. (1976) Degradation of the insecticide pirimicarb in soil - characterization of "bound" residues. Kaufmann.D.D., Still, G.G., Paulson, G.D. & Bandal, S.K. Bound and Conjugated Pesticide Residues. 358-361. ACS Symposium Series. Hill, B.D & Schaalje, G.B. (1985) A Two-Compartment Model for the Dissipation of Deltamethrin in Soil. J. Agric. Food Chem. 33, 1001-1006. Hsu, T. & Bartha, R. (1976) Hydrolyzable and nonhydrolyzable 3,4-dicholoaniline-humus complexes and their respective rates of degradation. Journal of Agricultural and Food Chemistry, 118-122. Huang, W., Schlautman, M. & Weber, W. (1996) A distributed reactivity model for sorption by soils and sediments 5: The influence of near-surface characteristics in mineral domains. Environmental Science and Technology, 30, 2993-3000. Huber, R. & Otto, S. (1994) Environmental behavior of bentazon herbicide. Rev. Environ. Contam. Toxicol., 137, 111-134. Inderjit & Dakshini, K. (1994) Allelopathic potential of the phenolics from the roots of Pluchea lanceolata. Physiologia Plantarum 92 571-576. Isensee, A., Nash, R. & Helling, C. (1990) Effect of conventional versus no-tillage on pesticide leaching to shallow groundwater. Journal of Environmental Quality 19 434-440. Isensee, A. & Sadeghi, A. (1993) Impact of tillage practice on runoff and pesticide transport. Journal of Soil and Water Conservation, 48, 523-527. Ismail, B. S. & Lee, H. J. (1995) Persistence of metsulfuron-methyl in two soils. J. Environ. Science Health B 30[4], 485-497. Jacobsen, C.S. & Pedersen, J.C. (1992) Mineralization of 2,4-dichlorophenoxyacetic acid (2,4-D) in soil inoculated with Pseudomonas cepacia DBO1 (pRO101), Alcaligenes eutrophus AEO106(pRO101) amd Alcaligenes eutrophus JMP134(pJP4): effects of inoculation level and substrate concentration. Biodegradation, 2, 253-263. Jacobsen, C., Helweg, A. & Spliid, N. (1998) Glyphosat og AMPA i jord. 15 Danske Planteværnskonference. Pesticider og Miljø, 17-25. Johnson, D., Shaner, D. L., Deane, J, Mackersie, L.A. & Tuxhorn, G. (2000) Time-dependent adsorption of imazethapyr to soil. Weed Science, 48, 769-775. Jones, K., Gevao, B., Mordaunt, C., Northcott, G. & Semple, K. (2000) Introductory remarks to the Special Issue . Environmental Pollution, 108, 1-2. Jones, R.L. Field, laboratory, and modeling studies on the degradation and transport of aldicarb residues in soil and ground water. (1986) Garner, W. Y., Honeycutt, R. C. & Nigg, H. N. Evaluation of Pesticides in Ground Water. 197-218. American Chemical Society. ACS Symposium Series. Kearney, P. (1982) IUPAC pesticide commission report. Journal of the Association of Official Analytical Chemists, 65, 1030-1032. Kelsey, J. & Alexander, M. (1997) Declining bioavailability and inappropriate estimation of risk of persistent compounds. Environmental Toxicology and Chemistry, 16, 582-585. Kelsey, J., Kottler, B. & Alexander, M. (1996) Selective Chemical Extractants To Predict Bioavailability of Soil-Aged Organic Chemicals. Environmental Science and Technology, 31, 214-217. Khan, S. (1995) Supercritical fluid extraction of bound pesticide residues from soil and food commodities. Journal of Agricultural and Food Chemistry, 43, 1718-1723. Khan, S. (1982) Bound pesticide residues in soil and plants. Residue Reviews, 84, 1-24. Khan, S. (1973) Equilibrium and kinetic studies of the adsorption of 2,4-D and picloram on humic acid. Canadian Journal of Soil Science, 53, 429-434. Khan, S. (1996) Supercritical fluid extraction of bound pesticide residues from soil in Re, A.A.M. del, Capri, E., Evans, S.P. & Trevisan, M. (Eds). The environmental fate of xenobiotics. Proceedings of the 10th Symposium Pesticide Chemistry. 533-541. Khan, S. & Bheki, R. (1990) Effects of Pseudomonas species on the release of bound 14C-residues from soil treated with 14C-atrazine. Journal of Agricultural and Food Chemistry, 38, 2090-2093. Khan, S. & Hamilton, H. (1980) Extractable and bound (unextractable) residues of prometryn and its metabolites in an organic soil. Journal of Agricultural and Food Chemistry, 28, 126-132. Kim, J., Fernandes, E. & Bollag, J. (1997) Enzymatic coupling of the herbicide bentazon with humus monomers and characterization of reaction products. Environmental Science and Technology, 31, 2392-2398. Kloskowski, R., Führ, F. & Mittelstaedt, W. (1986) Plant availability of bound anilazine residues in a degraded loeww soil. Journal of Environmental Science and Health, 21, 487-505. Kloskowski, R., Führ, F. & Mittelstaedt, W. (1987) The uptake of non-extractable soil-bound pesticide residue by roots - standardized experiments with four pesticides. Konopka, A. & Turco, R. (1991) Biodegradation of organic compounds in vadose zone and aquifer sediments. Applied and Environmental Microbiology, 57, 2260-2268. Kottler, B., White, J. & Kelsey, J. (2001) Influence of soil moisture on the sequestration of organic compounds in soil. Chemosphere, 42, 893-898. Lassen, P. (1995) On the possible role of humic substances in the environmental fate of pollutants. Ph.D. thesis. National Environmental Research Institute, Roskilde, Denmark. Lehmann, R., Miller, J. & Laskowski, D. (1990) Fate of fluroxypyr in soil. II Desorption as a fundtion of incubating time. Weed Research, 30, 383-388. Levanon, D., Codling, E., Meisinger, J. & Starr, J. (1993) Mobility of agrochemicals through soil from two tillage systems. Journal of Environmental Quality, 22, 155-161. Lopez-Avila, V., Young, R. & Teplitsky, N. (1996) Microwave-assisted extraction as an alternative to soxhlet, sonication and supercritical fluid extraction. Journal of AOAC International 79 142-156. Ma, L. & Selim, H.M. (1994) Predicting atrazine adsorption-desorption in soils: A modified second-order kinetic model. Water Resources Research, 30, 447-456. Martin-Neto, L., Vieira, E. & Sposito, G. (1994) Mechanisms of atrazine sorption by HA: A spectroscopic study. Environmental Science and Technology, 28, 1867-1873. Matoba, Y., Ohnishi, J.I. & Matsou, M. (1995) Temperature- and humidity-dependency og pesticide behaviour in indoor simulation. Chemosphere, 30, 933-952. Miljøstyrelsen. Miljø- og Energiministeriets bekendtgørelse nr. 241 af 27. april 1998 om bekæmpelsesmidler. 1998. Miljøstyrelsen, Miljø- og Energiministeriet. Miljøstyrelsen. Rammer for vurdering af plantebeskyttelsesmidler. 1994. Minton, N.A., Leonard, R.A. & Parker, M.B. (1990) Concentration of total fenamiphos residue over time in the profile of two sandy soils. Appl. Agric. Res. 5 127-133. Molins, C., Hogendoorn, E.A., Heusinkveld, H.A.G., van Harten, D.C., van Zoonen, P. & Baumann, R.A. (1996) Microwave assisted solvent extraction (MASE) for the efficient determination of triazines in soil samples with aged residues. Chromatographia 43 527-532. Moorman, T.B. & Harper, S.S. (1989) Transformation and mineralization of metribuzin in surface and subsurface horizons of a Mississippi delta soil. J. Environ. Qual. 18 302-306. Monrozier, L. J., Guez, P., Chalamet, A., Bardin, R., Martins, J. & Gaudet, J..P. (1993) Distribution of Microorganisms and Fate of Xenobiotic Molecules in Unsaturated Soil Environments. Sci.Tot. Environ. 136[1-2], 121-133. Moorman, T.B. Adaptation of microorganisms in subsurface environments. (1990) Racke, K.D. & Coats, J.R. Enhanced biodegradation of pesticides in the environment. [426], 167-180. American Chemical Society. ACS Symposium Series. Mueller, T.C., Moorman, T.B. & Snipes, C.E. (1992) Effect of Concentration, Sorption, and Microbial Biomass on Degradation of the Herbicide Fluometuron in Surface and Subsurface Soils. J. Agric. Food Chem. 40[12], 2517-2522. Myrold, D. & Tiedje, J. (1985) Diffusional constraints on denitrification in soil. Soil Science Society America Journal, 49, 651-657. Nanny, M., Bortiatynski, J., Tien, M. & Hatcher, P. (1996) Investigations of enzymatic alterations of 2,4-dichlorophenol using 13C-nuclear magnetic resonance in combination with site specific 13C-labelling: understanding the environmental fate of this pollutant. Environmental Toxicology and Chemistry, 15, 1857-1864. National Academy of Science, USA. (2000) Bioavailability of Contaminants in Soils and Sediments, Project Identification Number WSTB-U-99-05-A. Nomura, N. & Hilton, H. (1977) The adsorption and degradation of glyphosate in five Hawaiian sugarcane soils. Weed Research, 17, 113-121. Northcott, G. & Jones, K. (2000) Experimental approaches and analytical techniques for determining organic compound bound residues in soil and sediment. Environmental Pollution , 108, 19-43. Novak, J.M., Jayachandran, K. & Moorman. T.B. (1995) Sorption and binding of organic compounds in soils and their relation to bioavailability. Soil Science Society of America. Bioremediation: Science and Application. Madison, USA, Soil Science Society of America. OECD. (2000) Guideline for Testing of Chemicals No 106, Adsorption - Desorption Using a Batch Equilibrium Method in the distribution of the amounts of substance, (Updated Guideline, adopted 21st January 2000). Ou, L., Rao, P., Edvardsson, K., Jessup, R., Hornsby, A. & Jones, R. (1988) Aldicarb degradation in sandy soils from different depths. Pestic. Sci., 23, 1-12. Parker, L.W. & Doxtader, K.G. (1982) Kinetics of microbial decomposition of 2,4-D in soil: effects of herbicide concentration. J. Environ. Qual., 11, 679-684. Parris, G. (1980) Covalent binding of aromatic amines to humates. I: Reactions with carbonyls and ketones. Environmental Science and Technology, 14, 1099-1106. Petersen, L.J.J., Rabølle, M. & Felding, G. (1998) Binding og mobilitet af glyphosat i jord. 15. Danske Planteværnskonference, 7-15. Piccolo, A. & Celano, G. (1994) Hydrogen-bonding interactions between the herbicide glyphosate and water-soluble humic substances. Environmental Toxicology and Chemistry, 13, 1737-1741. Pignatello, J. & Xing, B. (1996) Mechanisms of slow sorption of organic chemicals to natural particles. Environmental-Science-and-Technology, 30, 1-11. Pothuluri, J., Moorman, T., Obenhuber, D. & Wauchope, R. (1990) Aerobic and anaerobic degradation of alachlor in samples from a surface-to-groundwater profile. J. Environ. Qual., 19, 525-530. Racke, D. & Lichtenstein, E. (1985) Effects of soil microorganisms on the release of bound 14C-residues from soil previously treated with 14C-parathion. Journal of Agricultural and Food Chemistry, 33, 938-943. Rahima, A., Houot, S. & Barriuso, E. (2000) Dependence of atrazine degradation on C and N availability in adapted and non-adapted soils. Soil Biology and Biochemistry, 32, 389-401. Reese-Stahler, G., Pestemer, W. & Pallutt, B. (1999) Okotoxische Auswirkungen der Applikation von Fenikan (Diflufenican + Isoproturon) in Wintergerste bei unterschiedlicher Bodenbearbeitung. Teil 1: Allgemeine Angaben zur Versuchsdurchfuhrung und das Ruckstandsverhalten von Diflufenican und Isoproturon unter dem Einfluss verschiedener Bodenbearbeitungsmassnahmen. Nachrichtenblatt-des-Deutschen-Pflanzenschutzdienstes, 51, 176-184. Reffstrup, T.K., Sørensen, H. & Helweg, A. (1998) Degradation of mecoprop (MCPP) in a simulated concentration gradient from a pointsource in surface and subsurface soil. Pestic. Sci., 52, 126-132. Reid, B., Jones, K. & Semple, K. (2000) Bioavailability of persistent organic pollutants in soils and sediments-a perspective on mechanisms, consequences and assessment. Environmental Pollution, 108, 103-112. Riley, D., Wilkinson, W., & Tucker, B.V. (1976) Biological unavailability of bound paraquat residues in soil. In: Kaufman, D.P., Still, G.G., Paulson, G.D., & Bandal, S.K., ed. Bound and conjugated pesticide residues, pp. 301-353 (ACS Symposium Series No. 29). Ritter, W., Chirnside & Scarborough, R. (1996) Movement and degradation of triazines, alachlor, and metolachlor in sandy soils. Journal of Environmental Science and Health. Part A, 31, 2699-2721. Roberts, T. (1984) Non-extractable pesticide residues in soils and plants. Pure and Applied Chemistry, 56, 945-956. Rochette, E.A. & Koskinen, W.C. (1996) Supercritical carbon dioxide for determining atrazine sorption by field-moist soils. Soil Sci. Soc. Am. J., 60, 453-460. Ronday, R. (1997) Centrifugation method for soil pore water assessment of the bioavailability of organic chemicals in soil. Communications of Soil Acience and Plant Analysis, 28, 777-785. Rueppel, M.L., Brightwell B.B., Scaefer, J. & Marvel, J.T. (1977) Metabolism and degradation of glyphosate in soil and water. J. Agric. Food Chem., 25, 517-528. Sadeghi, A. & Isensee, A. (1992) Effect of tillage systems and rainfall patterns on atrazine distribution in soil. Journal of Environmental Quality, 21, 464-469. Sanchez-Palacios, A., Perez-Ruiz, T. & Martinez-Lozano, C. (1992) Determination of diquat in real samples by electron-spin-resonance spectrometry. Analytica chimica Acta, 268, 217-223. Schlebaum, W. (1998) Organic contaminants in soil: desorption kinetics and microbial degradation. Landbouwuniversiteit Wageningen (Wageningen Agricultural University); Netherlands. Schmidt, S. & Gier, M. (1989) Dynamics of microbial populations in soil: Indigenous microorganisms degrading 2,4-dinitrophenol. Microbial Ecology, 18, 61-114. Schulten, H. & Leinweber, P. (1996) Characterisation of humic and soil particles by analytical pyrolysis and computer modelling. Journal of Analytical and Applied Pyrolysis, 38, 1-53. Scow, K.M. & Alexander, M. (1992) Effect of diffusion on the kinetics of biodegradation: experimental results with synthetic aggregates. Soil Sci. Soc. Am.J., 56, 128-134. Scow, K. & Hutson, J. (1992) Effect of diffusion and sorption on the kinetics of biodegradation: theoretical considerations. Soil Science Society of America Journal, 56, 119-127. Scow, K., Simkins, S. & Alexander, M. (1986) Kinetics of mineralization of organic compounds at low concentrations in soil. Appl. Environ. Microbiol., 51, 1028-1035. Senesi, N. (1992) Binding mechanisms of pesticides to soil humic substances. Science of the total Environment, 123/124 , 63-76. Senesi, N., Testini, C. & Miano, T. (1987) Interaction mechanisms between HA's of different origin and nature and electron-donor herbicides - a comparative IR and electron-spin-resonance study. Organic Geochemistry, 11, 25-30. Shelton, D.R. & Doherty, M.A. (1997) Estimating losses of efficacy due to pesticide biodegradation in soil: model simulations. Soil-Sci-Soc-Am-J., 61, 1085-1090. Shelton, D.R. & Doherty, M.A. (1997) A model describing peesticide bioavailability and biodegradation in soil . Soil Science Society America Journal., 61, 1078-1084. Simkins, S. & Alexander, M. (1984) Models for mineralization kinetics with the variables of substrate concentration and population density. Appl. Environ. Microbiol., 47, 1299-1306. Simon, L., Spiteller, M., Haisch, A. & Wallnofer, P.R. (1992) Influence of Soil Properties on the Degradation of the Nematocide Fenamiphos. Soil Biol. Biochem. 24[8], 769-773. Sinclair, J. & Lee, T. (1992) Biodegradation of atrazine in subsurface environments. EPA/600/S-92/001, 1-8. Sjelborg, P., Fomsgaard,I.S. & Helweg, A (2002a) Degradation and sorption parameters. In: Kjær, J., (ed). The Danish Pesticide Leaching Assessment Programme. Monitoring Results May 1999 - July 2000. Copenhagen, Geological Survey of Denmark and Greenland (GEUS). Sjelborg, P., Fomsgaard, I.S. & Helweg, A (2002b). Degradation and sorption parameters. In: Kjær,J. (ed.). The Danish Pesticide Leaching Assessment Programme. Monitoring Results May 1999 - June 2001. Second report. Copenhagen, Geological Survey of Denmark and Greenland (GEUS). Smelt, J.H., Leistra, M., Houx, N.W.H. & Dekker, A. (1978) Conversion rates of aldicarb and its oxidation products in soils. III. Aldicarb. Pestic. Sci., 9, 293-300. Smelt, J., Dekker, A., Leistra, M. & Houx, N. (1983) Conversion of four carbamoyloximes in soil samples from above and below the soil water table. Pestic. Sci., 14, 173-181. Smith, A. & Mayfield, C. (1978) Paraquat:determination, degradation and mobility in soil. Water, Air and Soil Pollution, 9, 439-452. Sorensen, P.B., Mogensen, B.B., Gyldenkaerne, S. & Rasmussen, A.G. (1998) Pesticide leaching assessment method for ranking both single substances and scenarios of multible substance use. Chemosphere, 36, 2251-2276. Sprankle, P. & Meggit, W.P.D. (1975) Adsorption, mobility and microbial degradation of glyphosate in soil. Weed Science, 23, 229-234. Stehouwer, R., Dick, W. & Traina, S. (1993) Characteristics of earthworm burrow lining affecting atrazine sorption. Journal of Environmental Quality, 22, 181-185. Stevenson, F. (1982) Humus chemistry: Genesis, composition, and reactions. John Wiley and Sons, New York. Stott, D., Martin, J., fOC ht, D. & Haider, K. (1983) Biodegradation, stabilization in humus and incorporation into soil biomass of 2,4-D and chlorocathechol carbons. Soil Science Society American Journal, 47, 66-70. Stout, S.J., da Cunha, A. & Allardice, D.G. (1996) Microwave-assisted extraction coupled with gas chromatography/electron capture negative chemical ionization mass spectrometry for the simplified determination of imidazolinone herbicides in soil at the ppb level. Analytical Chemistry 68 653-658. Streck, T., Poletika, N., Jury, W. & Farmer, W. (1995) Description of simazine transport with rate-limited, two-stage, linear and non-linear sorption. Water Resources Research, 31, 811-822. Szmigielska, A.M., Schoenau, J. & Greer, K. (1998) Comparison of chemical extraction and bioassay for measurement of metsulfuron in soil. Weed Science, 46, 487-493. Sørensen, P., Brüggemann, R., Carlsen, L., Mogensen, B., Kreuger, J. & Pudenz, S. (2003) Analysis of monitoring data of pesticide residues in surface waters using partial order ranking theory. Journal of Environmental Toxicology and Chemistry accepted for publication. Thorn, K., Pettigrew, P., Goldenberg, W. & Weber, E. (1996) Covalent binding of aniline to humic substances. 2. 15NMR studies of nucleophilic addition reacitions. Environmental Science and Technology, 30, 2764-2775. Tiberg, E., Greve, M.H., Helweg, A., Yli-Halli, M., Ekol, M., Nyborg, Å.A., Solbakken, E., Öborn, I. & Stenström, J. (1998) Nordic Reference Soils. Torstensson, L. Behaviour of glyphosate in soils and its degradation.(1985) Grossbard, Atkinson. The Herbicide Glyphosate. 137-150. London, Butterworth. Torstensson, L. & Lindholm, O. (1988) Ogräsbekämpning på banvallar. 29. Svenska Ogräskonferensen, 27.-28. Jan, 1988, Uppsala. 203-212. Torstensson, L. & Stenstrom, J. (1986) "Basic" respiration rate as a tool for prediction of pesticide persistence in soil. Toxicity Assessment: An International Quarterly, 1, 57-72. Tucker, B., Pack, D. & Ospenson, J. (1967) Adsorption of the bipyridilium herbicides in soil. Journal of Agricultural and Food Chemistry, 15, 1005-1008. Veeh, R., Inskeep, W. & Camper, A. (1996) Soil depth and temperature effects on microbial degradation of 2,4-D. J. Environ. Qual, 25, 5-12. Wais, A., Haider, K., Spiteller, M., deGraaf, A., Burauel, P. & Führ, F. (1995) Using 13C-NMR spectroscopy to evaluate the binding mechanisms of bound pesticide residue in soils. 1. solution high resolution NMR spectroscopy. Journal of Environmental Science and Health B, 30 , 1-25. Walker, A., Helweg, A. & Jacobsen, O. S. Temperature and pesticide transformation. (1996) Boesten, J., Helweg, A., Businelli, M., Bergstrøm, L., Schäfer, H., Delmas, A., Kloskowski, R., Walker, A., Travis, K., Smeets, L., Jones, R., Vanderbroeck, V., Van Der Linden, A., Broerse, S., Klein, M., Layton, J., Jacobsen, O. S. & Yon, D. Soil persistence models and EU registration. European Commision. Walker, A., Welch, S., & Turner, I. J. (1995) Studies of time-dependent sorption processes in soil. BCPC Monograph , 62, 13-18. Wauchope, R. & Meyers, R. (1985) Adsorption-desorption kinetics of atrazine and linuron in fresh-water sediment aqueous slurries. Journal of Environmental Quality, 14, 132-136. Weber, W. & Young, T. (1997) A distributed reactivity model for sorption by soils and sediments. 6. Mechanistic implications of desorption under supercritical fluid conditions. Environmental Science and Technology, 31, 1686-1691. Weissmahr, K., Haderlein, S., Schwarzenbach, R., Hany, R. & Nuesch, R. (1997) In-situ spectroscopic investigations of adsorption mechanisms of nitroaromatic compounds at clay minerals. Environmental Science and Technology, 31, 240-247. Wheeler, W., Straton, G., Twilley, R., Ou, L.-T., Carlson, D. & Davidson, J. (1979) Trifluralin degradation and binding in soil. Journal of Agricultural and Food Chemistry, 27, 702-705. White, J., Hunter, M., Nam, K., Pignatello, J. & Alexander, M. (1999) Correlation between biological and physical availabilities of phenanthrene in soils and soil humin in aging experiments. Environmental toxicology and Chemistry 18 1720-1727. Zablotowicz, R., Locke, M., Gaston, L. & Bryson, C. (2000) Interactions of tillage and soil depth on fluometuron degradation in a Dundee silt loam soil. Soil and Tillage Research, 57, 61-68.
|
||||||||||||||||||

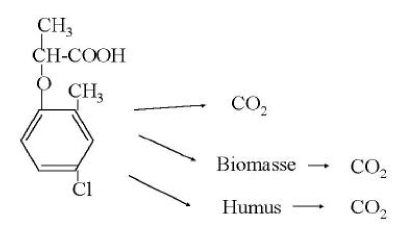
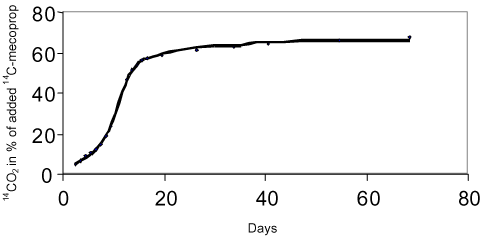

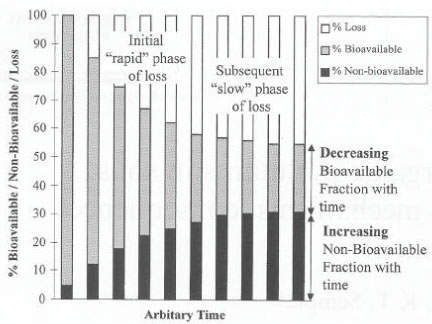
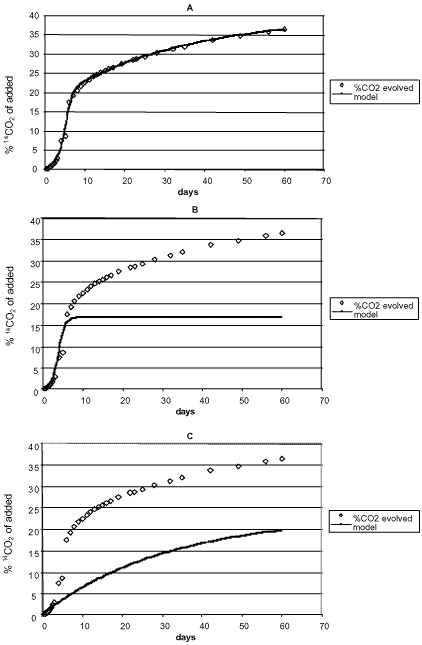
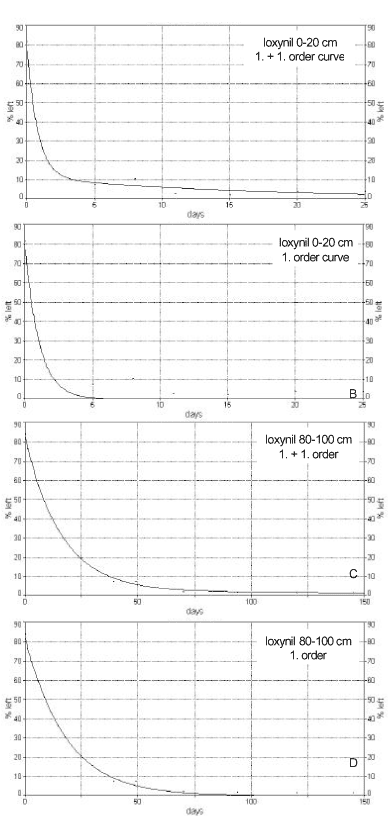
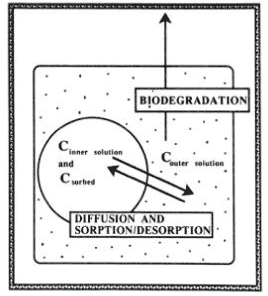
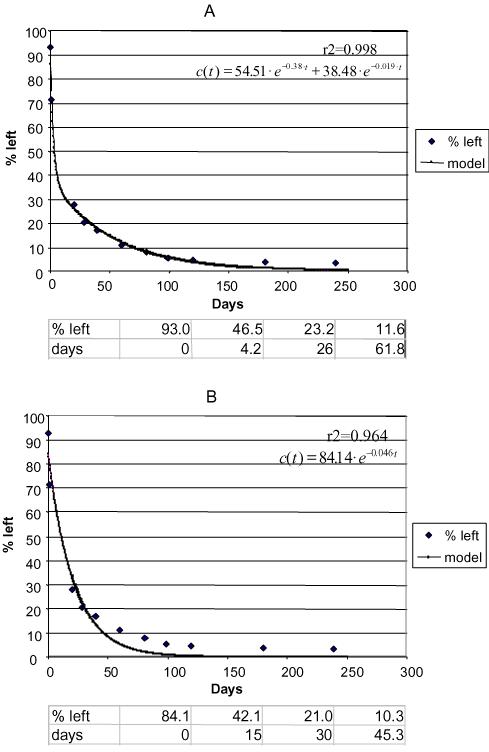
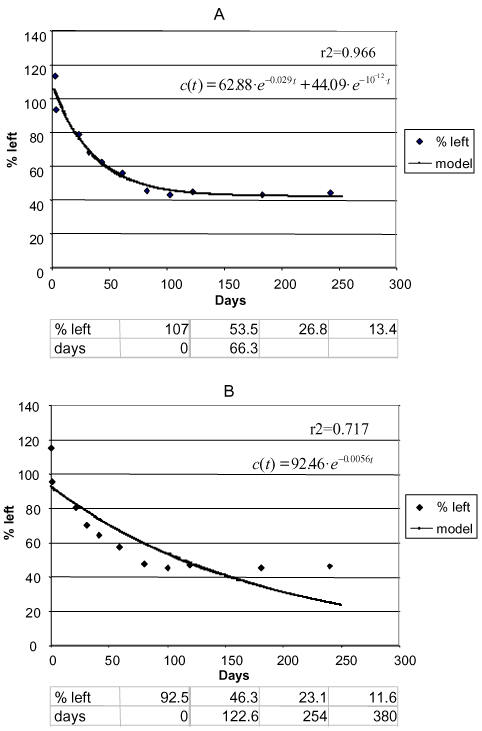
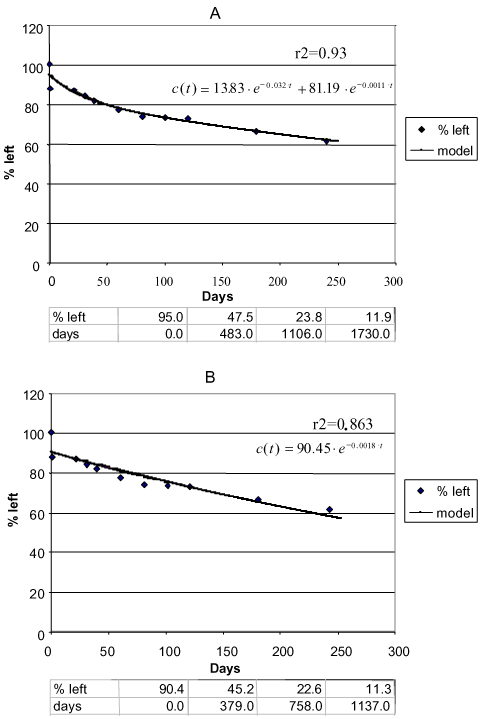
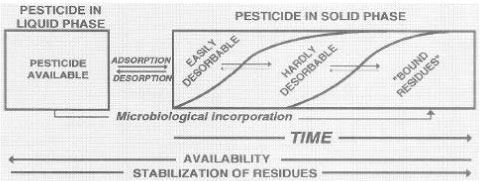
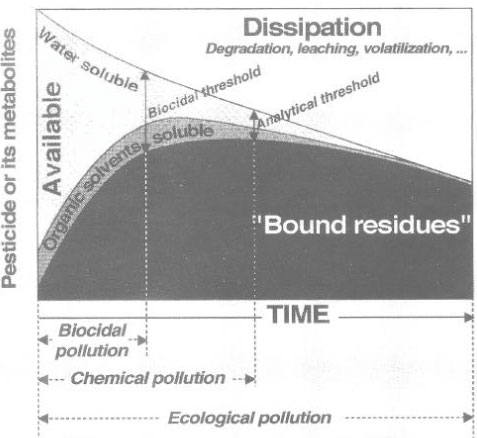
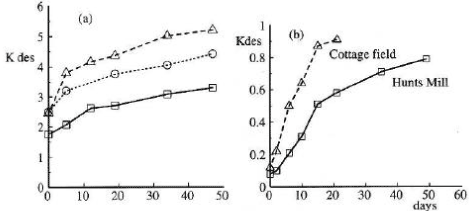
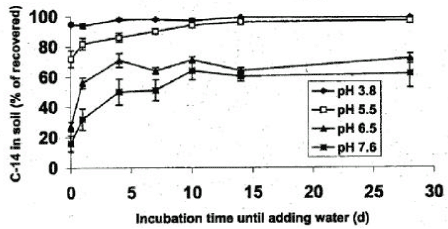
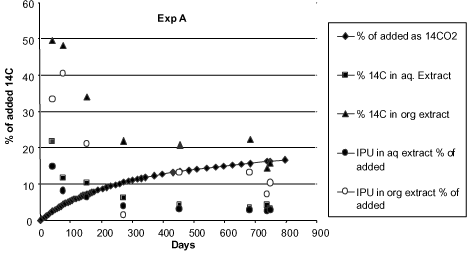
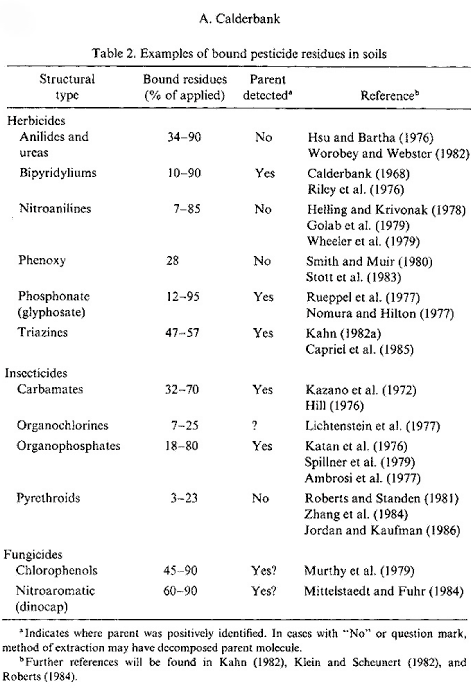
 /p>
/p>
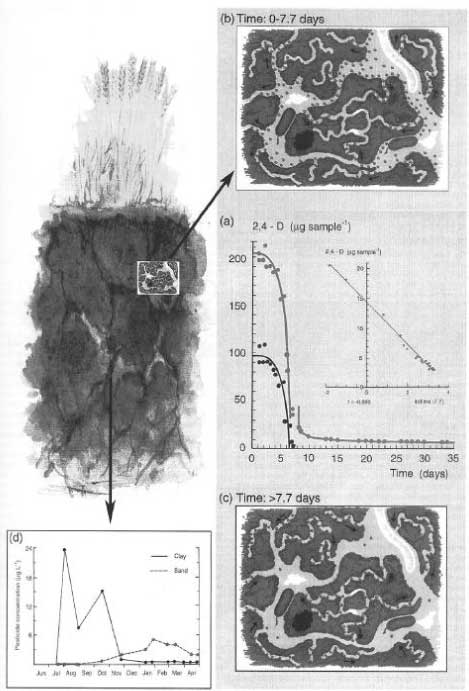 /p>
/p>