|
Miljøprojekt nr. 905, 2004 Teknologiudviklingsprogrammet for jord- og grundvandsforurening Vurdering af metoder til analyse af olie i jordIndholdsfortegnelse
5 Optimering af ekstraktionstid
10 Verifikation af ekstraktions-metoder
12 Naturligt forekommende kulbrinter
13 Tilsætning af olie til kompost
14 Svensk standard til beregning af totalkulbrinter ved GC-MS
Appendiks A : Analyseforskrift BILAG:
ForordNærværende rapport omhandler et projektarbejde vedrørende videreudvikling af metoder til analyse af olie i jord. Projektarbejdet er iværksat af Miljøstyrelsen under Miljøstyrelsens teknologiprogram for jord- og grundvandsforurening. Formålet med projektet har været:
Projektarbejdet er udført ved A/S AnalyCen fra primo 2002 til medio 2003. Projektmedarbejderne har været Jørgen Andersen, Lise-Lotte Hessellund Eriksen, Karen Halling, Hanne Sofie Klausen, Kristina Knudsen, Johnny Madsen, Kristina Bolbro Nielsen, Niels Weibel og Tina Wendelbo. Projektledelsen har været udført af Hanne Jensen, A/S AnalyCen. Til følgende personer rettes en stor tak til i forbindelse med gennemførelse af projektet: Per Wrang, AAUE – for introduktion til olieoprindelse og biomarkører. Kathrine Møhl, Dansk Jordrens og Søren Jensen, RGS 90 – for levering af forurenede jordtyper. Per Kjemtrup, Bascon – for kyndig vejledning i forbindelse med udtagning af prøver. Lars Porskjær Christensen, Danmarks JordbrugsForskning og Helle Weber Ravn, DMU – for hjælp vedrørende de naturlige kulbrinter. Kim Haagensen, Rambøll – for hjælp vedrørende opklaring af kulbrinter i prøver fra udgravninger i Københavns havnebassin. Per Ivarsson, A/S AnalyCen, Sverige – for opfindsomhed i forbindelse med GC-MS analysen. Jacqueline Anne Falkenberg, NIRAS – for kritisk gennemgang af den endelige projektrapport mm. Projektet har været fulgt af en følgegruppe med følgende medlemmer: Irene Edelgaard, Miljøstyrelsen Sammenfatning og konklusionerDenne projektrapport omfatter afprøvning og optimering af en analysemetode til vurdering af indholdet af kulbrinter i jordprøver, og den skal fungere som vejledning i forbindelse med evaluering af kulbrinteindhold i jordprøver. Rapporten er opdelt i to hoveddele; en del vedrørende afprøvning, optimering og validering af ISO/DIS 16703 metoden "Soil Quality – Determination of Mineral Oil Content by Gas Chromatography", Draft International Standard; en del vedrørende metode til at skelne mellem kulbrinter af petrogen oprindelse og naturlige kulbrinter. Del 1: Metoden, der er afprøvet i forbindelse med projektet (efterfølgende kaldet ISO/DIS 16703), er testet overfor de mest almindeligt anvendte ekstraktionsmetoder til analyse af olie i jord, herunder ekstraktion med pentan og 0,05M natriumpyrophosphat, som der tidligere er anvendt som standardmetode af de danske analyselaboratorier /2/. I forbindelse med afprøvning af ISO/DIS 16703 er der foretaget en del optimeringer af metoden, således at "den nye metode", der valideres i denne rapport, er en modificeret og forbedret udgave af ISO/DIS 16703. I ISO/DIS 16703 er der stillet skærpede krav til laboratoriernes betingelser på gaskromatograferne, som anvendes ved kvantificering af indholdet af olie i jord i forhold til de tidligere anvendte standardmetodes performancekrav. I forbindelse med nærværende redegørelse er der foretaget en optimering af analysemetoden på gaskromatografen, og der er anført et eksempel på indstilling af de forskellige variable på en gaskromatograf med splitless injektion. ISO/DIS 16703 anvender en blanding af acetone og heptan til ekstraktion. Det er konkluderet i denne rapport, at ISO/DIS 16703 metoden er den mest effektive analysemetode til ekstraktion af kulbrinter fra jord. I denne redegørelse er vist, at den ekstraktionstid, som der er anført i den oprindelige ISO/DIS 16703 metode på ½ time slet ikke er tilstrækkelig og der anbefales i denne rapport en ekstraktionstid på 12 – 16 timer. Heptan er erstattet af n-pentan, hvilket giver mulighed for kvantificering af benzen, toluen og xylener mm. ved samme ekstraktion. Kvantificering af kulbrinter i ISO/DIS 16703 foretages udfra en blanding af to olieprodukter. Denne kvantificeringsmetode har vist sig at give stor interlaboratorieafvigelse, hvorimod kvantificering af kulbrinter i jord overfor kalibrering med specifikke n-alkaner har vist sig at give større præcision ved interlaboratoritests /3/. Det er vist i denne rapport, at genfinding af olie med kogepunktsinterval fra og med benzen op til og med n-alkan C40 kan foretages udfra specifikke n-alkaner. Det har været projektets ønske, at kunne kvantificere totalkulbrinter ved GC-MS, men det har i forbindelse med dette projektarbejde vist sig ikke umiddelbart at kunne lade sig gøre. I ISO/DIS 16703 metoden indgår der, dels et vasketrin af ekstraktet, og dels et oprensningstrin med florisil. Det er i rapporten vist, at der mistes kulbrinter af både petrogen oprindelse og naturlige kulbrinter ved at udføre disse trin. Det anbefales derfor, at der som standard ikke foretages nogen oprensning af ekstraktet, der skal anvendes til injektion på gaskromatografen. I specielle tilfælde kan oprensning foretages, og dette er beskrevet i rapporten. Endelig er der foretaget en validering af metoden med henblik på kvantificering af følgende kulbrinter; Benzen – C10, >C10 – C25, >C25 – C35, >C35 – C40 som fremover omtales "totalkulbrinter", BTEX'er og PAH-er (16 EPA-PAH-er), og i afsnit 10 er der en henvisning til appendiks A hvor den endelige analyseforskrift for metoden ligger. Del 2: Denne del af rapporten omfatter specifikke petrogene kulbrinter til at skelne olie og naturlige kulbrinter. Der er udvalgt en række kulbrinteforbindelser, der forkommer i olier, og som er lette at identificere. Disse er beskrevet, og indholdet i rene olier beregnet. En række indexes, der kan anvendes i forbindelse med vurdering af kulbrinteindholdets oprindelse, er nævnt i denne rapport. CPI-indeks, isoprenoidforhold og "Unresolved complex matter" er beregnet for en hel række prøver, og der er udarbejdet en standardiseret kvantificering og vejledning i tolkning af opnåede værdier. Specielle PAH-forbindelser, herunder naphthalener og phenanthrener er undersøgt i denne redegørelse, og der er valgt en række af disse, der umiddelbart kan anvendes som indikator parametre i forbindelse med vurdering af, om der er tale om kulbrinter af petrogen oprindelse eller naturlige kulbrinter. Særlige forbindelser som findes i højtkogende olieprodukter som hopaner og stearaner er omtalt for sig. Der er i rapporten vist eksempler på genkendelighed af mønster af hopaner ved både GC-FID og GC-MS. Disse forbindelser er meget anvendelige som indikator for indhold af kulbrinter af petrogen oprindelse/naturlige kulbrinter specielt i fraktionen n-alkan (C25 til C40). For at kortlægge indholdet af kulbrinter, som der eventuelt kan findes i "rene" prøver, dvs. naturlige kulbrinter og nedbrydningspotentialet af disse, er der udtaget prøver fra udvalgte rene lokaliteter, hvor der forventes at være et indhold af naturlige kulbrinter, f. eks. overfladejord fra en bøgeskov og eng. Der er dels udført profilprøver – prøver udtaget i forskellige dybder – for at kortlægge nedbrydningspotentialet af de enkelte komponenter, og dels er der udført "skrabeprøver" af topjorden i ca. 2 cms dybder. Disse prøver er analyseret ved GC-MS i scan-mode, og de enkelte toppe er forsøgt identificeret ved hjælp af deres massespektrum og kogepunkt. Der er for nogle udvalgte komponenter foretaget analyse af rene referencestoffer. Der er en række gennemgående træk i alle de rene lokaliteter. Der er et forholdsmæssigt stort indhold af phytosteroler, n-alkaner og fedtsyrer. Phytosterolerne er tildels plantearts specifikke, der er dog enkelte phytosteroler, der forekommer i stort set alle de analyserede prøver. Dette er for eksempel sitosterol (beta- el. gamma-). Denne komponent er derfor anvendt som indikator for kulbrinter af biogen (naturlig) oprindelse. Der er ud fra gennemgangen af resultaterne opbygget en GC-MS metode til hurtig kvantificering af de ulige n-alkaner og phytosterolerne, som umiddelbart kan fratrækkes det indhold af kulbrinter, som der er opnået ved GC-FID analysen. Som et selvstændigt kapitel er den svenske standard til analyse af kulbrinter i jord beskrevet. Dette er dels fordi der anvendes GC-MS som kvantificeringsmetode af kulbrinter. Denne metode adskiller sig væsentligt fra den danske metode til at kvantificere kulbrinter på, og myndighedernes krav er derfor også anderledes end de danske krav. Den svenske metode har en patenteret metodik til at fratrække phytosteroler med ion 93 m/z. Der er på baggrund af de opnåede erfaringer i forbindelse med dette projektarbejdet udarbejdet en vejledning, der kan anvendes til evaluering af kulbrinteindholdet i jordprøver. Projektarbejdet er afsluttet med analyse af 49 prøver (dobbeltbestemmelser), hvor den udarbejdede vejledning er anvendt i forbindelse med evaluering af kulbrinteindholdet i disse jordprøver. Summary and conclusionsThis report presents the testing and optimisation of a new method for determination of the content of mineral oil in soil, and a method to calculate the content of hydrocarbons in soil samples. The report is intended as a guideline for evaluating the extent of mineral oil contamination in soil samples. The report is divided into two sections; one concerning the testing, optimisation and validation of the ISO/DIS 16703 method "Soil Quality – Determination of Mineral Oil Content by Gas Chromatography", Draft International Standard, and the second section concerning a method to determine whether the content of hydrocarbons originates from mineral oil or from natural organic matter. Part 1: The method ISO/DIS 16703 is tested together with the most commonly used methods to determine the oil content in soil, for example extraction with pentane and 0.05M Sodium pyrophosphate /2/, which is at present the Danish reference method. The ISO/DIS 16703 has been optimised during the development and testing, so that the method is improved with respect to Danish soil quality requirements. This new method requires a greatly enhanced performance for the gas chromatographs, compared to the previous reference method. Therefore, it has been necessary to optimise the performance and analytical method of the gas chromatograph. This report shows an example of how to set up a method for the gas chromatograph for split-less injection. ISO/DIS 16703 uses a mixture of acetone and n-heptane for extraction. In this report it is concluded that the ISO/DIS 16703 method is the most efficient method for extraction of hydrocarbons from soil. It is shown that the ½ hour extraction time, which is used in ISO/DIS 16703 is not sufficient for complete extraction. Equilibrium of the extraction is not reached until 12 - 16 hours, and in this report, this is recommended as the minimum time of extraction. Heptane is replaced by n-pentane, which gives the opportunity to quantify benzene, toluene, and the xylenes from the same extract as used for determination of mineral oil in soil. The quantification method in ISO/DIS 16703 is performed with a calibration mixture of two distinct mineral oil products. This method of quantification has been shown to give relatively large interlaboratory deviations /3/, but when using specific n-alkane mixtures, the precision is increased. It is shown that n-alkanes can be used for calibration and calculation of the content of oil in soil samples. It was the project's intention to quantify the mineral oil by GC-MS, but this procedure was not successful. In the ISO/DIS 16703, an analytical step with washing of the extract and purification of the extract with florisil is recommended. It is shown in this report that part of both the mineral oil and the natural hydrocarbons is lost during these analytical steps. It is therefore recommended not to perform any cleaning of the extract before analysis by gas chromatograph. In special cases with problematic samples the cleaning can be performed, and this is described in the report. Furthermore, the method is validated;:Total Hydrocarbons (Benzen – C10, >C10 – C25, >C25 – C35, >C35 – C40), BTEX and PAH (16 EPA-PAH), and the analytical procedure is described in section 10 and Appendix A. Part 2: This part of the report concerns biomarkers in mineral oil and in natural organic matter (hydrocarbons). A selected group of hydrocarbons, which are easy to identify, and which are present in mineral oil, were chosen, and the content is quantified in some mineral oils. Some indexes, which can be used in evaluating the origin of the oil are mentioned. CPI-index, isoprenoid content and unresolved complex matter are calculated for a number of samples, and a method for evaluating the results is included. Special PAH's: naphthalenes and phenanthrenes, are investigated, and some of these can be used as biomarkers in the evaluation of the origin of the hydrocarbons. Certain compounds, which are found in the higher boiling range of mineral oil products, the hopanes and stearanes, are mentioned in this report. These are true indicators for hydrocarbons of petrogen origin. Examples of the pattern of hopanes by GC-FID and GC-MS techniques are shown. These compounds are very useful as indicators to determine if the content of hydrocarbons is derived from mineral oil or is of natural origin, especially for the fraction of n-alkanes C25-C40. To map the content of natural hydrocarbons that may be present in "clean" soil samples, and to determine the extent to which these are degraded, samples were taken from selected clean areas, where the content of hydrocarbons from natural sources is expected to be high, for example in woods etc. The samples were collected in a depth profile to map the changes due to degradation of the natural hydrocarbons. Samples were also taken in a smaller fraction (scrape) of about 2 cm. These samples were all analysed by GC-MS in scan-mode, and every peak was, if possible, identified by the mass spectrum and boiling point range. A few selected reference compounds were also analysed. Some general features were seen at the "clean" locations. A relatively large amount of phytosterols, n-alkanes and fatty acids were detected. Phytosterols are mainly plant specific compounds, but some phytosterols are seen in all the analysed samples, for example, Sitosterol (beta- or gamma-). This compound can be used as a biogenic (natural hydrocarbon) biomarker in soil samples. Based on experience with these samples a GC-MS method was developed for fast quantification of the odd n-alkanes and phytosterols, and the calculated amount can be deducted from the total hydrocarbons found by the GC-FID method. As a separate chapter, the Swedish reference method for analysis of hydrocarbons in soil is described. This standard method uses GC-MS for quantification of hydrocarbons. It is very different from the Danish method of quantification of hydrocarbons, and the authorities also have different requirements. The Swedish method has a patented method of deduction of the phytosterols from the total hydrocarbon content in the soil samples. The experience achieved during this project has lead to the development of a guideline by which the content of hydrocarbons can be evaluated in soil samples. Finally, 49 samples were analysed by the new method, the content of hydrocarbons was calculated and evaluated according to the guideline. 1 Indledning1.1 BaggrundI forbindelse med Jordforureningsloven /4/ og de amtslige vejledninger som omhandler håndtering af forurenet jord i Danmark, bl.a. amterne på Sjælland /5/ er der i større grad kommet fokus på indholdet og arten af kulbrinter i jord. Interferenser kan give forhøjede resultater ved analyse eller medføre tvivl om det faktiske indhold af oliekulbrinter. Bidrag fra naturlige kulbrinter til det totale kulbrinteindhold er uhensigtsmæssigt, når jord skal klassificeres og administreres med store udgifter til følge. Det kan være svært at give en præcis vurdering af, hvorvidt de fundne indhold af kulbrinter helt eller delvis består af petrogene kulbrinter, som oprinder fra olie. Der har derfor været et stort ønske om, at finde egnede metoder til at undgå interferenser fra naturligt forekommende kulbrinter (ikke-petrogene) på analyseresultaterne. Analyseresultater for jordanalyser danner baggrund for, hvorledes jord skal håndteres. Der er i "Vejledning i prøvetagning og analyse af jord" /27/ beskrevet en række værdier, der fastlægger jordkvalitetskriterier for olie/benzin produkter i jord. Jordkvalitetskriterierne angiver den koncentration i jord, som skal sikre, at anvendelse af jord er sundhedsmæssigt forsvarligt også ved de mest følsomme arealanvendelse (private haver, børnehaver og legepladser). Jordkvalitetskriteriet for indholdet af kulbrinter er for benzin fastlagt til 25 mg/kg TS målt som C5-C10 kulbrinter og for Diesel-, fyrings- og gasolie fastlagt til 100 mg/kg målt som C5-C35 kulbrinter. Derudover har mange amter udarbejdet supplerende retningslinier for, hvorledes jord med højere værdier end jordkvalitetskriterierne skal håndteres. I tabel 1.1 er angivet de kvalitetsklasser for håndtering af jord, som amterne på Sjælland sammen har opstillet i den såkaldte Sjællandsvejledning /5/. I denne vejledning er der opstillet yderligere 3 kvalitetsklasser. Kvalitetsklasserne er udformet således, at kravene for olie i jord er opstillet efter olietype. Det er afgørende for administration af jorden, hvilken kvalitetsklasse resultaterne falder inden for.
Tabel 1.1: Inddeling i kvalitetsklasser 1-4 /5/. Jord i kvalitetsklasse 1 betragtes som "ren" jord, og kan i princippet anvendes frit i henhold til miljølovgivningen. Kvalitetsklasse 2 defineres som letttere forurenet jord, og skal så vidt muligt genanvendes i for eksempel bygge- og anlægsarbejder. Klasse 3 defineres som forurenet jord, der skal renses og/eller deponeres og klasse 4 er kraftigt forurenet jord, som formentlige vil blive anvist til rensning, med eventuelt efterfølgende deponering. Denne inddeling i de 4 kvalitetsklasser anvendes i vid udstrækning til håndtering af forurenet jord i Danmark. Kravene til "olie, total" anvendes uanset, om der er interferens fra f. eks. naturlige kulbrinter. Ud over krav til olie, stilles krav til en hel række andre stofgrupper, bl. a. polycykliske aromatiske hydrocarboner (PAH'er) og metaller. Analysen for olie i jord udføres typisk ved ekstraktion af en given jordmængde med en blanding af f. eks. pentan og 0,05M natriumpyrophosphat, og efterfølgende kvantificeres indholdet af de ekstraherbare komponenter i forhold til en række udvalgte n-alkaner /2/. Denne analysemetode har vist sig ved gaskromatografiskanalyse med flammeionisationsdetektor (GC-FID) primært at være anvendelig til "let kogende" olieprodukter, dvs. olieprodukter med et kogepunktsinterval fra ca. 80°C til ca. 500°C. Der er i øjeblikket en officiel ISO standard til høring (25-10-2001 til 25-03-2002: "Soil quality – Determination of Mineral Oil Content by Gas Chromatography", Draft International Standard ISO/DIS 16703/1/. Efterfølgende benævnt med ISO/DIS 16703. Denne standard omhandler kvantificering af total kulbrinter fra olieprodukter i intervallet fra n-alkan C10 til n-alkan C40, hvor det i dag er praksis kun at kvantificere til og med n-alkan C35. Kvantificering af BTEX'er og PAH'er er ikke omfattet af metoden. ISO/DIS 16703 er baseret på ekstraktion af jordprøver med acetone/n-heptan. Polære komponenter fjernes ved adsorption til florisil, og det rensede ekstrakt analyseres ved GC-FID. Total toparealet i det kromatografiske område, der er defineret som retentionstiden fra n-alkanen n-decan (C10) til n-alkanen n-tetracontan (C40) måles. Indholdet af mineral-olie i prøven kvantificeres mod en ekstern standard indeholdende to forskellige typer af mineralolie. Det har vist sig, at kvantificeringen overfor n-alkaner giver lavere total analyseusikkerhed mellem laboratorierne (RSD), og det er at foretrække fremfor kvantificering overfor blandingsprodukter. Ekstraktion med pentan/0,05M pyrophosphat giver dog et for lavt ekstraktionsudbytte af specielt højtkogende olieprodukter /3/. Der er således et behov for at afprøve ISO/DIS 16703 i forhold til metoder, som benyttes i dag. Samt videreudvikle ISO/DIS 16703 således at der kan kvantificeres overfor specifikke n-alkaner. Desuden er der et behov for at udvikle en metode til at skelne mellem petrogene oprindelse og naturligt forekommende kulbrinter set i relation til kvalitetskravene til jord, så der kan opstilles nogle kvalitetkrav, der er uafhængige af, om der er bidrag fra naturlige kulbrinter i en jordprøve. 1.2 FormålDet er formålet med projektet, at finde en egnet analysemetode til bestemmelse af totalkulbrinter op til n-alkan-C40. En analysemetode består af forskellige trin, hvorunder indholdet af totalkulbrinter skal ekstraheres over i et organisk ekstrakt, som eventuelt behøver oprensning, således at der kan foretages en analyse af indholdet af totalkulbrinter. Det er en del af projektets formål at udarbejde en standardiseret kvantificering af markører for petrogen oprindelse/naturlige kulbrinter, herunder af kendte naturlige kulbrinter ved GC-FID og GC-MS-scan analyse. 1.3 ProjektomfangProjektet omfatter udvikling og validering af en analysemetode til olie i jord med udgangspunkt i ISO/DIS 16703. Projektet omfatter desuden en kortlægning af hvilke petrogene kulbrinter, der er relevante i forbindelse med jordforurening, og en kortlægning af de mest almindeligt forekommende naturlige kulbrinter. 1.4 Rapportens indholdRapporten er opdelt i to dele: Del 1 omhandler metodeudvikling og validering af en analysemetode til bestemmelse af olie i jord og del 2 omhandler metoder til at skelne petrogene og naturlige kulbrinter. Del 1: Rapportens første del består af de forskellige trin, det er fundet nødvendige at undersøge i forbindelse med videreudvikling og optimering af analysemetoden ISO/DIS 16703, og starter ud med en beskrivelse og en optimering af GC-betingelserne i kapitel 2. Kapitel 3 beskriver et større forsøg, der finder frem til den bedst egnede ekstraktionsmetode ved at sammenligne 5 forskellige allerede eksisterende ekstraktionsmetoder herunder metoden, der er optimeret: ISO/DIS 16703. Herefter bliver der foretaget en optimering af ISO/DIS 16703 metoden, hvor der i kapitel 4 ses på rækkefølgen af tilsætning af ekstraktionsmidler samt prøvemængde og pentanmængde. Kapitel 5 omhandler flere forsøg i forbindelse med optimering af ekstraktionstiden. Der ses på vask og oprensning af ekstraktet i kapitel 6. I kapitel 7 bliver der beskrevet forsøg i forbindelse med bestemmelsen af metodens kvantificeringsmodel. Genfinding af olier, BTEX, PAH forbindelser og referencematerialer bliver beskrevet i kapitel 8. I kapitel 9 er den nye optimerede metode ISO/DIS 16703-Mod. valideret. Analyseforskriften for metoden ses i appendiks A. Som afslutning på første del af rapporten, er den nye metodes effektivitet eftervist i kapitel 10, ved en metodeafprøvning. ISO/DIS 16703-Mod. er sammenlignet med de analysemetoder, der anvendes i dag, og den originale ISO/DIS 16703. Et flowdiagram over metodeudvælgelse og optimeringsforløb af metoden, som den foregår i rapportens del 1, ses i figur 1.1. Beskrivelse af de enkelte metoder ses i bilag 1.
Figur 1.1- Flowdiagram af metodeudvælgelse og ekstraktionsmetodens optimeringsforløb. Del 2: Kapitel 11 omhandler valg af de petrogene kulbrinter. Kapitlet omhandler en række udvalgte kulbrinter, som er de mest anvendte i litteraturen til beskrivelse af olie. Der er vist eksempler på indhold i referencemateriale og en række prøver, med kendt indhold af olie. Der er omtalt hvilke indeks, der kan være relevante i forbindelse med vurdering af, om der er tale om olie i jorden eller ej. Kapitel 12 omhandler naturlige kulbrinter. Laboratoriet har lavet en screening af 9 forskellige lokaliteter med henblik på identifikation af indholdet af naturligt forekommende kulbrinter. Der er i kapitlet foretaget en vurdering af nedbrydningspotentialet af disse. Kapitel 13 beskriver tilsætningsforsøg med 3 typer af olie til kompost, opbygning af en GC-MS metode og beregning af de forskellige indeks. Kapitel 14 giver en kort introduktion til, hvilken metode der er anvendt i Sverige til beregning af kulbrinter i jord. Denne metode er ved GC-MS, og den benytter en i dag patenteret metode til at fratrække det bidrag som naturligt forekommende phytosteroler bidrager med til total kulbrinter. Kapitel 15 beskriver en trinvis metode til evaluering af kulbrinteindholdet i en given prøve udfra dels GC-FID screening og dels GC-MS-Scan af prøven. Kapitel 16 omhandler en omfattende metodeafprøvning på kendte jorde. Kapitel 17 beskriver en analyse af 49 prøver hvor den udarbejdede vejledning er anvendt i forbindelse med evaluering af kulbrinteindhold. En samlet konklusion for rapportens 1. og 2. del er samlet i kapitel 18, og i kapitel 19 er hele rapportens referenceliste samlet. Appendiks A indeholder analyseforskriften for den nye metode, ISO/DIS 16703-Mod. 2 GC-betingelser2.1 FormålTidligere præstationsprøvninger mellem danske laboratorier har vist, at der i mange tilfælde er problemer med at få gaskromatograferne til at kromatografere tungtkogende alkaner, så de giver et højt ensartet respons. Formålet med dette afsnit er at beskrive et forsøg, der dokumenterer, at det er muligt at optimere en gaskromatograf (GC) til at leve op til de krav, der stilles til kromatografering af tungtkogende forbindelser. 2.2 Krav til GC-betingelserFor at kunne analysere totalkulbrinter præcist ved GC-FID er det nødvendigt at have et GC-system, der lever op til bestemte krav. I forskellige standarder er der opstillet specifikke krav for hvad GC'ens performance skal leve op til. Kravene specificerer nøjagtigt hvilke krav, der stilles til responset af de tungest kogende alkaner i forhold til lettere. Eksempler på disse krav fra forskellige standarder er listet op i tabel 2-1.
Tabel 2-1. Oversigt over krav til gc-performance Kravene i DHI-metoden /2/ er som følger: Fraktion 2 (C10-C25) kvantificeres overfor summen af C12, C16, C20 og C24. Fraktion 3 (C25-C35) kvantificeres overfor C28, C30, C32 og C34. Det stillede krav er, at det laveste respons inden for et interval skal udgøre mindst 2/3 af det største. I DHI-metoden står der desuden, at der ikke må være uhensigtsmæssig diskriminering af tungere forbindelser. Under projektarbejdet er det erfaret, at det mest kritiske punkt i enhver form for gaskromatografisk analyse er injektionen. For at sikre, at det er muligt at leve op til kravene i ISO/DIS 16703 udføres derfor et faktorforsøg, hvor der optimeres på injektionsbetingelserne. 2.3 Optimering af injektionsbetingelserVed forsøget er der 2 parametre, der optimeres på. Den ene er injektionstemperaturen og den anden er purgetiden (splitlesstiden). Begge disse faktorer undersøges på 3 niveauer som det fremgår af tabel 2-2.
Tabel 2-2. Oversigt over forsøgsparametre til splitless injektion. De valgte faktorer giver mulighed for at lave 9 forskellige kombinationer, og da forsøgene udføres som dobbeltbestemmelse, skal der i alt udføres 18 bestemmelser. Der analyseres på en n-alkan standard fortyndet i pentan. n-alkan standarden er; Florida TRPH Standard fra Restec med Katalog nr. 31266 og Lot nr. A024463. Floridastandarden indeholder alle lige n-alkaner fra C8 og til C40 og er opløst i hexan. 2.4 ForsøgsresultaterAnalyseresultaterne er behandlet statistisk. Forudsætningerne, normalfordeling, statistisk uafhængighed og varianshomogenitet er tilfredsstillende opfyldt. De statistiske analyser viser, at både injektionstemperaturen og purgetiden har signifikant indflydelse på resultaterne, se figur 2-1. Jo højere temperatur og jo længere purgetid, jo tættere kommer forholdet C40/C20 på 1, samtidig med at der opnås det største totalareal for komponenterne.
Figur 2-1. C40/C20-forhold ved forskellige indstillinger. Figur 2-1 viser udviklingen af C40/C20-forholdet ved forskellige indstillingsmuligheder. I figur 2-2 og 2-3 ses, hvor galt det kan gå, hvis der vælges forkerte injektionsbetingelser.
Figur 2-2. C40/C20-forhold ved 250°C. Figur 2-2 viser, at ligegyldigt hvor lang purgetid, der vælges, kan der ikke opnås brugbare resultater, hvis injektionstemperaturen er 250°C. I kromatogrammet figur 2-3 kan det ses, hvordan kromatogrammet kan se ud når performance af GC'en langt fra er tilfredsstillende. C40 er næsten væk, og forholdet mellem C40/C20 er = 8,9/158 = 0,056. Figur 2-3. Eksempel på kromatogram, hvor GC'en ikke yder en tilfredsstillende performance. Ved en god daglig vedligeholdelse af GC'en og optimering af GC-programmet, især injektionstemperatur og purgetid, kan det lade sig gøre at få en tilfredsstillende kromatografisk performance. Figur 2-4. Eksempel på kromatogram med tilfredsstillende performance. Til kromatogrammet i figur 2-4 er benyttet den optimerede GC-metode "350_10", hvor injektionstemperaturen er 350°C og purgetiden 1,0 min. Her er C40/C20 = 144/167 = 0,86. Dette overholder kravet om, at GCén skal have et responsforhold for C40/C20 der er over 80 %. Kromatogrammet i figur 2-4 viser også overensstemmelse med kravene i DHI-metoden /2/, hvor forholdet mellem det laveste respons inden for et interval skal udgøre mindst 2/3 af det største respons. Responsforholdene for de to fraktioner ses i tabel 2-3.
Tabel 2-3. Responsforhold efter DHI-metoden for kromatogram i figur 2-4. Ved anvendelse af metoden "350_10" samt god vedligeholdelse af GC kan kravene i afsnit 2.2, jf. tabel 2.1 således overholdes. 2.5 KonklusionDet er vist, at det er muligt at lave et GC-program, der lever op til kravene i standarderne. Dette er gjort ved at optimere på injektionsbetingelserne. Der benyttes splitless injektion med en injektionstemperatur på 350 °C og en purgetid på 1 min. Generelt er det projektets erfaring, at det kræver en høj grad af daglig vedligeholdelse af kolonne og injektionsport for at kunne opretholde en tilstrækkelig god kromatografisk standard. I tabel 2-4 ses betingelserne for den GC-metode, der er anvendt i projektet.
Tabel 2-4. Gc-metoden "350_10" som anvendes i projektet. I analyseforskriften i Apendiks A, afsnit 9.1.1 ses nærmere beskrivelse af vedligehold af GC-apparat. 3 Valg af ekstraktionsmetode3.1 FormålFormålet med denne del af rapporten er at sammenligne metoden ISO/DIS 16703 /1/ med andre eksisterende analysemetoder med henblik på at udvælge den mest egnede metode til videre afprøvning. Ekstraktionseffektiviteten vurderes for kulbrinter i området fra n-alkan C10 til og med n-alkan C40 samt for de 7 MST PAH'er fluoranthen, benzo(bjk)fluoranthener, benzo(a)pyren, indeno(1,2,3-cd)pyren og dibenz(a,h)anthracen. Der udføres en række sammenlignende ekstraktioner på 4 forskellige jordtyper, hvoraf de tre stammer fra reelle forureningslokaliteter i Danmark. Metoderne sammenlignes og vurderes ud fra statistiske analyser både med hensyn til ekstraktionseffektivitet og metodernes reproducerbarhed. I vurderingen inddrages desuden overordnede betragtninger i forbindelse med anvendelsen af de forskellige analysemetoder. 3.2 Forsøgsplanlægning og indledende forsøgI kapitel 2, GC-betingelser, er der udviklet en GC-metode "350_10" som, når apparatet er vedligeholdt, yder en tilfredsstillende performance i forhold til en n-alkan standard "Floridastandard" [2] indeholdende lige n-alkaner fra C8 til C40. Denne GC-metode, "350_10" er anvendt i resten af metodeudviklingen, der er beskrevet i denne rapport, med mindre andet er angivet i teksten. 3.2.1 Afprøvede ekstraktionsmetoderDet er valgt at finde det bedst egnede ekstraktionsmiddel/ekstraktionsmetode ved sammenligning af 5 forskellige, allerede eksisterende ekstraktionsmetoder:
Ved anvendelsen af ISO/DIS 16703 /1/ og PAH-metoden /28/ er der taget udgangspunkt i de oprindelige standardinstrukser. Som intern standard anvendes dog brombenzen og o-terphenyl ligesom for de resterende ekstraktionsmetoder. Ved anvendelsen af ISO/DIS 16703 foretages ikke vask og oprensning af ekstraktet. Behovet for dette er undersøget i afsnit 6, Vask og oprensning. Den modificerede PAH-metode bygger på PAH-metoden fra VKI, men anvender ikke lufttørrings- og inddampningstrin. Pentan- og dichlormethanekstraktion er standardmetoder, som anvendes internt i laboratoriet. De 5 ekstraktionsmetoder ses sammenlignet i tabel 3-1, hvor forskellen på prøvemængder, rystetider, ekstraktionsmidler med mere fremgår. Det skal bemærkes, at der til forsøgene er anvendt sammenlignelige rystetider på både ½, 4, 8 og 16 timer.
Tabel 3-1. Sammenligning af 5 ekstraktionsmetoder. 3.2.2 Anvendte jordtyperFor at få et så bredt vurderingsgrundlag som muligt, er de sammenlignende ekstraktioner udført på fire forskellige jordtyper med forskellig kulbrintesammensætning. Indholdet af kulbrinter i de tre første jordtyper stammer fra aktuelle forureningslokaliteter, og de er erhvervet igennem jordrensningsfirmaet Dansk Jordrens i Vemmelev. Den sidste jordtype, en havekompost, er rekvireret hos Jydske Haveselskab, og kulbrinteindholdet er primært af naturlig oprindelse. Der er udført en tekstur- analyse samt pH-bestemmelse på de fire jordtyper, som ses i tabel 3-2.
Tabel 3-2. Teksturanalyse af de fire jordtyper. Med det tilhørende JB-nr /30/ kan jordtyperne karakteriseres som i tabel 3-3:
Tabel 3-3. Karakterisering af de fire anvendte jordtyper i henhold til JB-nr. Ved at inddrage forskelligartede jordtyper kan det endelige valg af ekstraktionsmetode foretages mere generelt, idet også vanskelige jordmatricer indgår i undersøgelsen. Det er valgt ikke at anvende certificeret referencemateriale til de sammenlignende ekstraktioner. Referencemateriale udmærker sig ved en høj dokumenteret prøvehomogenitet, men anses i denne sammenhæng ikke som optimal, idet ekstraktionseffektiviteten ønskes undersøgt for jordtyper med forskellige fysiske karakteristika. Forudgående for de sammenlignende ekstraktioner er der udført en jordhomogenisering i laboratoriet for at sikre en acceptabel homogenitet af de fire jordtyper. Fremgangsmetoden beskrives i afsnit 3.2.3. For at vurdere indholdet af kulbrinter for de fire jordtyper både kvalitativt og kvantitativt er der udført en indledende ekstraktion med dichlormethan i 4 timer. Ekstrakterne er derefter analyseret kvantitativt på GC-FID, hvor fraktionen n-alkan C10-C25 (2.fraktion) er kvantificeret overfor C12, C16,C20 og C24 i en alkanstandard. Fraktionerne n-alkan C25-C35 (3. fraktion) samt n-alkan C35-C40 (4.fraktion) er kvantificeret overfor n-alkan C26, C28, C30 og C32 i standarden, hvilket er normal praksis på laboratoriet i dag. Prøverne er analyseret med det optimerede GC-program, jvf. afsnit 2. Resultaterne fra kvantificeringen kan ses i tabel 3-4.
Tabel 3-4. Indhold af totalkulbrinter for de 4 jordtyper bestemt efter ekstraktion med dichlormethan i 4 timer. Som det ses af tabel 3-4, er der et forholdsvist højt kulbrinteindhold i samtlige jordtyper. Indholdet af kulbrinter i den sidste fraktion, især for den gasolieforurenede jordtype, er dog forholdsvist lavt. Indholdet af kulbrinter i fraktionen C35-C40 i de tre resterende jordtyper vurderes dog at være rimelig til at vurdere metodernes ekstraktionseffektivitet i denne fraktion. I figur 3-1 ses en alkanstandard med fordelingen af n-alkaner fra C8 til C40. Figur 3-1. Alkanstandard. Da alle analyser gennem hele projektforløbet, er foretaget under de samme betingelser, kan retentionstiderne tilnærmelsesvis anvendes til vurdering af analyseresultaterne af de 4 jordtyper. I GC-FID kromatogrammerne i figur 3-2 til 3-5 ses fordelingen af kulbrinter for de 4 jordtyper som omtalt ovenfor. Figur 3-2. GC-FID-kromatogram af den tjæreforurenede jord. Som det ses af figur 3-2, viser kogepunktsfordelingen og det høje indhold af PAH'er, en typisk tjæreforurenet jord. Figur 3-3. GC-FID-kromatogram af den gasolieforurenede jord. Kulbrintefordelingen i figur 3-3 svarer til en relativt frisk gasolie, som ved nærmere eftersyn, kan ses af forholdene mellem phytan og C18 samt pristan og C17. De tydelige høje toppe for den ligelige fordeling af n-alkaner er karakteristiske for en frisk gasolie. Figur 3-4. GC-FID-kromatogram af den motorolieforurenede jord. Indholdet af kulbrinter i den tredje jordtype, som ses på figur 3-4, er overvejende højtkogende forbindelser i ét stort kontinuum, UCM (unresolved complex matter), og ud fra kogepunktsintervallet kan indholdet i overvejende grad karakteriseres som motor/smøreolie eller lignende. Der ses dog også indhold af de højtkogende PAH'er, fluoranthen, pyren m.fl., der ofte ses i forbindelse med forurening i byområder. Det vælges i denne sammenhæng at benævne indholdet som motor/smøreolie, da dette vurderes at være mest sigende for det aktuelle kogepunktsinterval. Figur 3-5. GC-FID-kromatogram af kompostjorden. De ekstraherede forbindelser fra kompostjorden på figur 3-5 dækker et forholdsvist bredt kogepunktsinterval. Typisk ses et højt indhold af mange usystematiske enkeltkomponenter, hvilket sjældent ses i forbindelse med petrogene forureningskilder. Størstedelen af de ekstraherede kulbrinter befinder sig i fraktionen C25-C40. Fra Dansk Jordrens i Vemmelev er der indhentet supplerende oplysninger om opgravningslokationen for de tre petrogent forurenede jordtyper, jf. tabel 3-5.
Tabel 3-5. Opgravningslokationer for de tre jordtyper fra Dansk Jordrens, Vemmelev. En samlet vurdering af de 4 jordtyper viser, at de repræsenterer et typisk udsnit af de kulbrintefordelinger, som ses i den daglige laboratoriepraksis. Jordtyperne indeholder desuden kulbrinter, som dækker hele kogepunktsintervallet fra C10-C40. Dette ses som en fordel i forbindelse med den endelige vurdering. 3.2.3 Homogenisering af de 4 jordtyperFor at sikre mindst mulig variation, som følge af inhomogen jordprøve, er der foretaget en mekanisk homogenisering i laboratoriet før anvendelse. De fire jordtyper er blevet sigtet over et fintmasket soldevæv (maskestørrelse ca. 2 mm) for at fjerne større sten, grene m.v. Jorden har herefter gennemgået en omhyggelig sammenblanding, dette er forgået ved piskning og omrøring med piskeris på en boremaskine. Denne homogeniseringsmetode er anvendt på samtlige jordtyper anvendt under projektet. Ud fra en visuel vurdering medfører dette en ensartet homogenitet for de tre første jordtyper med petrogen forurening. Kompostjorden er pga. sin oprindelse mere inhomogen og ud fra en visuel vurdering vanskeligere at homogenisere pga. dens høje indhold af ufuldstændigt nedbrudt, organisk materiale. Før homogeniseringen er der udført en 5-fold dichlormethanekstraktion af 60 g jord for de tre første jordtyper med efterfølgende GC-FID-analyse i henhold til standardmetoden /2/, med en injektionsmængde på 1 L. Forud for analysen er der analyseret en alkan-standard på 5 mg/L for at sikre performance af gaskromatografen. Mængden af ekstraherede kulbrinter er angivet som arealrespons i fraktionerne n-alkan C10-C25 (2. fraktion), C25-C35 (3.fraktion) samt C35-C40 (4.fraktion). Som et udtryk for homogeniteten er den relative standardafvigelse beregnet for de tre fraktioner samt for totalfraktionen C10-C40. Alle arealer er korrigeret med den interne standard o-terphenyl, og resultaterne fremgår af tabel 3-6.
Tabel 3-6. Relativ standardafvigelse beregnet for de enkelte kulbrintefraktioner ved ekstraktion af ikke homogeniserede jordtyper med dichlormethan. Til sammenligning er samme ekstraktion foretaget efter gennemført homogenisering. Ekstrakterne er ligeledes analyseret på GC-FID dog med et injektionsvolumen på 2 L. Resultaterne fremgår af tabel 3-7.
Tabel 3-7. Relativ standardafvigelse beregnet for de enkelte kulbrintefraktioner ved ekstraktion af homogeniserede jordtyper med dichlormethan. Som det ses af resultaterne, er den relative standardafvigelse reduceret væsentligt for fraktionerne for samtlige jordtyper. Det højere injektionsvolumen vurderes til at have en ubetydelig indflydelse på reproducerbarheden under normale laboratorieforhold. Der er ligeledes udført en 5-fold ekstraktion på kompostjorden efter gennemførelse af homogeniseringen. Resultaterne fremgår af tabel 3-8.
Tabel 3-8. Relativ standardafvigelse beregnet for de enkelte kulbrintefraktioner ved ekstraktion af homogeniseret kompostjord med dichlormethan. Som det ses af tabellen, er den relative standardafvigelse på niveau med de resterende jordtyper. For at kunne vurdere om den opnåede homogenitet er acceptabel, er det nødvendigt at sammenholde resultaterne med den variation, som ses i forbindelse med de senere sammenlignende ekstraktioner. Såfremt inhomogenitetsvariansen er større, eller har samme størrelse, vil det være vanskeligt at drage anvendelige konklusioner ud fra datamaterialet. Af hensyn til læseren foretages denne vurdering først senere, afsnit 3.3.1, når fremgangsmetoden for de sammenlignende ekstraktioner er beskrevet, og der foreligger data herfor. 3.2.4 GenfindingsforsøgDet er valgt at anvende brombenzen og o-terphenyl som interne standarder (IS)for samtlige ekstraktionsmetoder. Forud for de sammenlignende ekstraktioner er der udført et genfindingsforsøg for at vurdere, om responset på de interne standarder er afhængigt af den valgte metode og det valgte opløsningsmiddel. Til genfindingsforsøget er der fremstillet en motoroliestandard på 10,04 g/L i dichlormethan, som spikes til rent sand. Forsøget er udført som 6-fold bestemmelse for hver ekstraktionsmetode. Forsøget er udført ved standardbetingelser, men ekstraktionstiden er valgt til 2 timer for alle metoder, da matricen bestod af ren sand. Der er tilsat en mængde motorolie svarende til en koncentration på 250 mg/kg TS. I tabel 3-9 ses resultaterne af genfindingsforsøget beregnet ud fra de IS-korrigerede data.
Tabel 3-9. Beregnet genfinding og relativ standardafvigelse. Af resultaterne ses, at genfindingen ligger i intervallet 82-115 % for samtlige metoder. Det har været nødvendigt at udelukke enkelte bestemmelser fra PAH-modificeret, PAH samt ISO/DIS-metoden pga. dårlig kromatografiering. Det er forsøgt at beregne indholdet med og uden korrektion med intern standard. Ved anvendelse af intern standard ses en mere præcis genfinding, hvilket viser, at det er en fordel at anvende intern standardkorrektion. Forsøgene tyder ikke på, at responset fra den interne standard er forskellig ved anvendelse af henholdsvis pentan, heptan og dichlormethan som opløsningsmiddel. 3.2.5 ForsøgsplanlægningFormålet med de sammenlignende ekstraktioner er at undersøge, om en eller flere af de udvalgte ekstraktionsmetoder er specielt anvendelig med henblik på at kvantificere indholdet af kulbrinter i fraktionen n-alkan C10-C40. Derudover vil metodernes reproducerbarhed, kromatografieringsegenskaber samt praktiske forhold i forbindelse med udførelsen blive vurderet. Forud for forsøgene er der opstillet en forsøgsplan, hvor det vælges at variere den anvendte ekstraktionsmetode og ekstraktionstid. De resterende analysefaktorer holdes konstante under hele forsøget. Ud fra erfaring og praktiske forhold er det valgt at anvende ekstraktionstider fra ½ time til 16 timer med to mellemliggende tider på 4 og 8 timer. Det er forud for forsøget vurderet, at en tilstrækkelig ekstraktionseffektivitet vil kunne opnås inden for dette tidsrum. Det er valgt at fastholde en konstant rystehastighed på 200 ryst/min ved alle ekstraktioner. I forsøgsdesignet indgår således 2 variable faktorer med niveauer som følgende:
Et egnet forsøg er et fuldfaktor forsøg, hvor de to forsøgsparametre sammensættes i alle mulige kombinationer. For én jordtype skal der udføres (5 ekstraktionsmetoder x 4 ekstraktionstider) = 20 delforsøg. Det vil sige i alt 80 delforsøg for de 4 jordtyper. For at øge det statistiske grundlag udføres dobbeltbestemmelse for hver behandling. I forbindelse med opstilling af forsøgsdesignet er der mange faktorer, som skal indgå i overvejelserne, og som i denne sammenhæng umuliggør en korrekt udførelse af fuldfaktorforsøget. Først og fremmest bør hele forsøgsudførelsen være randomiseret for at undgå periodiske variationsbidrag i datamaterialet. I praksis betyder dette, at hele prøveforberedelsen (ekstraktion af de 160 delprøver) skal udføres i tilfældig rækkefølge. Dette vil være en meget vanskelig opgave at udføre i praksis, uden at der opstår fejl. I så fald ville det betyde, at alle jordtyper, alle ekstraktionstider og alle metoder skulle tages i arbejde på kryds og tværs af hinanden på samme tid. Dette faktum har medført en alternativ plan for udførelsen af ekstraktionerne:
Denne alternative fremgangsmetode øger risikoen for systematiske fejl i datamaterialet som følge af fejl under ekstraktionen. Under vurderingen af dataene er det derfor vigtigt at være opmærksom på, at denne risiko eksisterer. I forbindelse med selve analysen er der ligeledes en række faktorer, som skal vurderes. Optimalt set burde alle ekstrakter randomiseres og analyseres på GC-FID i én lang kø på 160 prøver. I praksis er dette dog ikke muligt pga. en systematisk faldende apparatperformance, som nødvendiggør løbende standard- og blindkontrol samt vedligehold af liner, kolonne m.v. I praksis er det derfor også her nødvendigt at anvende en alternativ fremgangsmetode:
Alle prøver hørende til samme jordtype er analyseret på samme apparatsignal for at undgå unødig variation i datamaterialet. Ved at anvende ovenstående metode sikres apparatperformance i videst mulig omfang. Ulempen ved at anvende denne fremgangsmetode er dag til dag variationen på gaskromatografen, bl.a. fordi signalrespons og diskriminering ikke er konstante. Ideelt set burde analysedagen indgå som en blokfaktor i den endelige statistiske analyse, men med den valgte fremgangsmetode er dette ikke muligt, fordi der i praksis analyseres prøver for én ekstraktionsmetode om dagen [4]. Dette problem kan løses ved at randomisere analysen af alle 160 ekstrakter, således at der ikke tages hensyn til, hvilket opløsningsmiddel, der er brugt. Dette vil dog øge antallet af nødvendige standarder og blindprøver (forskellige opløsningsmidler) i både start og slutning af analysekøen og er derfor af praktiske årsager ikke gjort. I stedet er det valgt at korrigere de enkelte fraktioner med middelsummen af de to analyserede alkanstandarder i henholdsvis starten og slutningen af den opsatte analysekø. Herved sikres samme basis for de analyserede ekstrakter. Forsøgsplanen for de 4 jordtyper kan ses i bilag 2 . Under forsøgsudførelsen er der lagt vægt på at sikre en tilfredsstillende apparatperformance i henhold til krav om, at forholdet mellem arealet for C40 og C20 mindst skal være 80 % for alkanstandarden, jf. tabel 2-1. Der er under forsøgsudførelsen brugt meget tid på at sikre overholdelsen af dette krav, da det i perioder har været vanskeligt at overholde dette trods effektiv service og vedligehold af det anvendte apparatur. I forbindelse med de sammenlignende ekstraktioner har det derfor i få tilfælde været nødvendigt at acceptere en performance, som er mindre end 80 % . Betydningen for analyseresultaterne er beskrevet i afsnit 7.6.2. Alle analyserede blindprøver er udrystet i ca. ½ time med de aktuelle opløsningsmidler for at inddrage eventuelle ligevægtsbidrag mellem de anvendte opløsningsmidler. Blindprøverne for PAH-metoden har dog ikke gennemgået en inddampningsproces. Der er under dette afsnits forsøg, samt alle andre forsøg i projektperioden, udtaget ekstrakt til 4 vials, som opbevares i fryser, for evt. senere at kunne verificere de fremkomne data. Forud for ekstraktionsforsøgene er der foretaget GC-FID-analyser af de rene opløsningsmidler til kontrol. Alle standarder og IS-opløsninger er fremstillet på samme tid ud fra certificerede standardampuller og er efterfølgende analyseret på GC-FID. Da alle analyser foretages med GC-metoden "350_10", er det valgt kun at fremstille to sæt standarder svarende til Standard 1 og Standard 2 i n-pentan og n-heptan. For alle heptan-ekstrakter er starttemperaturen i GC-ovnen hævet til 90 °C for at undgå dårlig kromatografiering. 3.3 Resultater og databehandlingI bilag 3 er beregnede værdier angivet for de sammenlignende ekstraktioner med korrektion med intern standard. For hver jordtype er der udarbejdet tre særskilte tabeller for kulbrinteanalyserne, som anvendes til beregningerne. Forklaringer hertil er ligeledes angivet på bilag 3. For PAH-analyserne er der udarbejdet 2 særskilte tabeller for hver jordtype. Da der kun ses indhold af PAH'er i den tjæreforurenede jordtype samt den motorolieforurenede jordtype, indgår kun disse i forsøget. Resultater fremgår af bilag 3. Alle analyseresultater er korrigeret med en blindværdi fra den tilhørende analysekø. I det ideelle tilfælde bør der analyseres en blindprøve på hver side af alle prøverne for at kunne korrigere for variationer i blindværdien eksempelvis som følge af slæb. Udføres dette i praksis, kan der opstå problemer med apparatperformance pga. en for lang analysekø, som igen kræver analyse af flere standardrækker. Ud fra denne betragtning er det valgt kun at analysere en blindprøve i starten og i slutningen af hver analysekø. Til korrektion er anvendt den blindprøve, som repræsenterer blindniveauet bedst i de enkelte prøver. 3.3.1 Vurdering af jordhomogenitetDen relative standardafvigelse (RSD) er beregnet for de 8 behandlinger for hver ekstraktionsmetode og for hver jordtype. En metode til at vurdere om den opnåede jordhomogenitet er acceptabel, er at sammenligne RSD fra de indledende homogeniseringsforsøg, afsnit 3.2.3 med RSD fra ekstraktion med dichlormethan i de sammenlignende ekstraktioner. De samhørende RSD-værdier er angivet i tabel 3-10.
Tabel 3-10. Sammenligning af RSD for de 4 jordtype fra de indledende homogeniseringsforsøg med RSD fra ekstraktion med dichlormethan fra de sammenlignende ekstraktioner. Som det ses af tabellen, er den relative standardafvigelse generelt større for dataene fra de sammenlignende ekstraktioner. Der ses dog en forholdsvis lille forskel mellem resultaterne for kompostjorden i 2. og 3. fraktion. Den større variation tyder på at variationen, som følge af forskellige rystetider, generelt er større end variationen som følge af eventuel inhomogenitet. Det vælges at udføre en række variansanalyser for at klarlægge om det er muligt at se nogle systematiske trends i datamaterialet. 3.3.2 TotalkulbrinterFor at kunne sammenligne ekstraktionseffektiviteterne for de 5 ekstraktions-metoder er der udført en en-sidet variansanalyse. I variansanalysen inddrages alle behandlinger, udført for de fire jordtyper, på én gang. Herved anvendes 7 frihedsgrader til at vurdere de enkelte metoders evne til at ekstrahere kulbrinter fra de 4 jordtyper. Såfremt indholdet af kulbrinter for én jordtype ligger højt i forhold til de resterende jordtyper, kan det være nødvendigt med en forudgående databehandling (normalisering). Hvis en ekstraktionsmetode udviser høj ekstraktionseffektivitet overfor netop denne jordtype, kan dette medføre en urealistisk høj samlet ekstraktionseffektivitet idet gennemsnitsværdien i variansanalysen er midlet over alle 4 jordtyper. Dette til trods for at metodens ekstraktionseffektivitet overfor de andre jordtyper ikke er optimal. Der er ikke taget hensyn til dette i de kommende variansanalyser, da undersøgelser heraf ved sammenligning af normaliserede og ikke-normaliserede data viser samme resultat. Alle statistiske analyser i rapporten er udført i programmet "Statgraphics Plus, version 5.0" udgivet af Manugistics Inc. /29/. 3.3.2.1 Ekstraktionseffektivitet Undersøgelse af varianshomogenitet er udført ved Bartlets test. Testværdierne er angivet i tabel 3-11.
Tabel 3-11. P-værdier fra Bartlets test for fraktionen C10-C40. Som det ses af tabel 3-11 er p-værdien < 5 %, hvilket indikerer at der ikke umiddelbart kan antages varianshomogenitet i dette datamateriale (5 % signifikansniveau). Dette kan hænge sammen med udførelsen af forsøgene, da det ikke har været muligt at anvende en fuld randomiseret forsøgsplan. Da variansanalysen er forholdsvis robust overfor afvigelse fra varianshomogenitet, og da p-værdien ikke ligger meget under de 5 %, vælges det alligevel at udføre variansanalysen. Med hensyn til normalfordelte residualer har dette i begge tilfælde været tilfredsstillende opfyldt. Hvad angår statistisk uafhængighed, kan dette også antages at være nogenlunde opfyldt. Variansanalysen er udført på et 5%-signifikansniveau, og den poolede standardafvigelse er anvendt til at beregne de angivne konfidensintervaller. De anførte forkortelser henviser til de anvendte ekstraktionsmetoder som følgende: D = Dichlormethan-ekstraktion (dichlormethan/vand). Ud af x-aksen i figur 3-5 er angivet behandlingsnummeret, som svarer til de behandlinger, som er angivet i bilag 2. Behandling 1-5 repræsenterer ekstraktionsudbyttet (y-akse) ved en ekstraktionstid på ½ time, behandling 6-10 ved en ekstraktionstid på 4 timer, behandling 11-15 ved en ekstraktionstid på 8 timer, mens behandling 16-20 svarer til en ekstraktionstid på 16 timer. En enkelt afviger er udeladt fra dataanalysen (F3-A3 [6]).
Figur 3-6. Variansanalyse for summen af ekstraherede kulbrinter i fraktionen C10-C40. Betragtes variansanalysen på figur 3-6, ses generelt en stigende ekstraktionseffektivitet med stigende ekstraktionstid, som forventet. Tydeligst er denne tendens for PM- og P-metoden. Analysen indikerer endvidere at en høj ekstraktionseffektivitet opnås hurtigst ved anvendelse af D-, I- og PA-metoden. Desuden ses, at ekstraktionseffektiviteten ved en ekstraktionstid på 16 timer er nogenlunde ens for D-, I-, PM- og PA-metoden. Dog ser det ud til at der opnås den højeste ekstraktionseffektivitet ved anvendelse af I-metoden ved 16 timers ekstraktion. Af analysen ses en generelt dårligere ekstraktionseffektivitet ved anvendelse af P-metoden i forhold til de resterende metoder. Under ekstraktionen af kompostjorden er der bemærket en meget dårlig befugtningsevne ved anvendelse af P-metoden. Dette viser sig i form af en meget dårlig ekstraktionseffektivitet overfor denne jordtype sammenlignet med de resterende ekstraktionsmetoder. De enkelte metoders ekstraktionseffektivitet kan også vurderes ved at betragte den samlede ekstraktionseffektivitet for hver metode bestemt ved et gennemsnit, som er en midling over alle ekstraktionstider og alle jordtyper. Dette er illustreret i figur 3-7.
Figur 3-7. Middelværdi for mængden af ekstraherede kulbrinter bestemt ud fra inddragelse af alle ekstraktionstider og alle jordtyper for hver ekstraktionsmetode. Som det ses af figuren, opnås den størst samlede ekstraktionseffektivitet ved anvendelse af I-metoden eller D-metoden, som begge er signifikant forskellige fra de resterende metoder. Analysen viser dog ikke, hvorvidt den samme ekstraktionseffektivitet kan opnås ved en bestemt ekstraktionstid for forskellige metoder, men viser derimod en mere generel tendens. Der er ligeledes udført en variansanalyse for de 5 ekstraktionsmetoder i forhold til kulbrinteindholdet i den tunge fraktion C25-C40 (3.+4.), hvilket ses på figur 3-8. Test af varianshomogenitet er anført i tabel 3-12. Den gasolieforurenede jordtype indgår ikke i datamaterialet, pga. det lave indhold af tunge kulbrinter i fraktionen C35-C40.
Tabel 3-12. P-værdier fra Bartlets test for fraktionen C25-C40. Betragtes variansanalysen på figur 3-8, ses de samme tendenser som fra variansanalyserne på fraktionen C10-C40 (figur 3-5).
Figur 3-8. Variansanalyse for summen af ekstraherede kulbrinter i fraktionen C25-C40. Udføres en variansanalyse, hvor middelværdien er bestemt ud fra alle ekstraktionstider og alle jordtyper, så ses samme tendens som for fraktionen C10-C40 vist i figur 3-7. Som tillæg til genfindingsforsøget er der udført en variansanalyse på arealerne af den interne standard o-terphenyl i forhold til anvendt ekstraktionsmetode. Resultatet af analysen kan ses på figur 3-9.
Figur 3-9. Variansanalyse udført på arealet af o-terphenyl fra samtlige behandlinger fra de sammenlignende ekstraktioner. Af analysen ses generelt høje arealværdier for D-, I-, PM- og P-metoden, mens inddampningstrinnet for PA-metoden medfører en noget lavere respons, som forventet. Testes middelværdierne i forhold til hinanden, er der ikke signifikant forskel på responsen for I- , P- og D-metoden, hvorfor responsen på de interne standarder ikke kan siges at være forskellige. Statistisk set er der ingen signifikant forskel mellem I- og PM-metoden, men responsen på den interne standard for PM-metoden kan siges at være mindre end for D- og P-metoden på et 5%-signifikansniveau. Dette kan betyde, at ekstraktions-effektiviteten kan være estimeret en smule for højt for PM-metoden. Den meget lave respons for PA-metoden viser, at det er nødvendigt at anvende IS-korrektion for at kunne tage højde for tab af ekstraherede kulbrinter under inddampningsprocessen. Det anbefales dog at anvende flere interne standarder i en endelig metode med forskellige kogepunkter, for at kunne korrigere mere optimalt for de forskellige kulbrintefraktioner. Da der kun er anvendt én intern standard i forbindelse med de sammenlignende ekstraktioner, kan ekstraktionseffektiviteten for nogle af jordtyperne generelt være estimeret for højt. Ud fra de sammenlignende ekstraktionsforsøg for totalkulbrinter i området C10-C40 kan følgende tendenser fremhæves ved de undersøgte ekstraktionstider:
3.3.2.2 Reproducerbarhed
Tabel 3-13. Relativ standardafvigelse for ekstraherede kulbrinter for de anvendte ekstraktionsmetoder ved forskellige ekstraktionstider. De relative standardafvigelser er beregnet ud fra ekstraktion af alle jordtyper. De grå-farvede felter i tabel 3-13 angiver den laveste RSD for hver ekstraktionstid og for hver beregningssituation. Betragtes resultaterne for de enkelte ekstraktionstider for fraktionen C10-C40 ser det ud til, at den bedste reproducerbarhed opnås ved anvendelse af PM-metoden. Generelt ses de bedste reproducerbarheder ved anvendelse af PM-, D- og PA-metoderne ud fra betragtning af sum-værdien samt ekstraktionstiderne på 8 og 16 timer. Reproducerbarheden for P- og I-metoden ser ud til at være dårligere end for de resterende. For fraktionen C25-C40 ses den bedste reproducerbarhed for I-metoden ved en ekstraktionstid på 0,5 og 4 timer. Totalt set opnås den bedste reproducerbarhed også her ved anvendelse af PM-metoden, hvilket ligeledes er gældende ved 8 og 16 timers ekstraktion. Ved 8 og 16 timers ekstraktion ses sammenlignelige ekstraktionseffektiviteter for D- ,I- , og PA-metoden. Ud fra en samlet vurdering indikerer variansanalyserne at:
Der er en vis usikkerhed behæftet ved ovenstående beregninger, hvorfor værdierne kun må betragtes som vejledende. Det er nødvendigt at undersøge reproducerbarheden mere dybdegående for den endelige udvalgte ekstraktionsmetode, for at kunne vurdere dens anvendelighed. Dette gøres senere i form af en metodevalidering, kapitel 9. 3.3.3 7 MST-PAHDe tjære- og motorolieforurenede jordtyper er anvendt til at vurdere de fem metoders ekstraktionseffektivitet overfor de 7 MST PAH'er fluoranthen, benzo(bjk)fluoranthener, benzo(a)pyren, indeno(1,2,3-cd)pyren samt dibenz(a,h)anthracen. Råarealerne er korrigeret på samme måde som for totalkulbrinterne, dog er der anvendt en PAH-standard til at korrigere for dag til dag variationen på GC-FID'en. Som intern standard er også her anvendt o-terphenyl. 3.3.3.1 Ekstraktionseffektivitet
Tabel 3-14. P-værdier fra Bartlets test for summen af de 7 MST PAH'er. Som det ses af tabellen, kan der antages varianshomogenitet i datamaterialet. Behandling F3-A3 er ligesom for analysen af totalkulbrinterne udeladt fra variansanalysen pga. fejl i den interne standard. Kravet om normalfordelte og statistisk uafhængige residualer er ligeledes undersøgt og er tilfredsstillende opfyldt i datasættet. Ud af x-aksen er angivet behandlingsnummeret, som svarer til de behandlinger, som er angivet i bilag 2. Behandling 1-5 repræsenterer ekstraktionsudbyttet (y-akse) ved en ekstraktionstid på ½ time, behandling 6-10 ved en ekstraktionstid på 4 timer, behandling 11-15 ved en ekstraktionstid på 8 timer, mens behandling 16-20 svarer til en ekstraktionstid på 16 timer.
Figur 3-10. Variansanalyse for summen af de 7 MST PAH'er ved anvendelse af forskellig ekstraktionsmetode og ekstraktionstid. Betragtes variansanalysen på figur 3-10, er det med den gældende usikkerhed ikke muligt at se nogen forskelle i ekstraktionseffektivitet for de 5 metoder. Behandling nr. 3 og 7 er de to eneste, som viser signifikant afvigelse fra nogen af de resterende behandlinger. De høje værdier for behandling 7 skyldes et afvigende datapunkt, som det ikke har været muligt at fjerne med dokumenteret grund fra datamaterialet. Dette er ligeledes gældende for behandling nr. 3, hvor variansen mellem bestemmelserne er forholdsvis stor. Ses alene på tendenserne i variansanalyserne, ser det ud til, at ekstraktionseffektiviteten for P-metoden ved 0,5 og 4 timer er generelt lavere end for de resterende metoder. Ved en ekstraktionstid på 16 timer ser det ud som om, at forskellen er udlignet. Udføres en 3-sidet variansanalyse med valgt ekstraktionsmetode og jordtype som kvalitative faktorer og ekstraktionstiden som kvantitativ faktor, viser det sig, at ekstraktionstiden ikke har signifikant betydning på mængden af ekstraherede PAH'er. Dette resultat svarer ikke umiddelbart til forventningerne og overlappet mellem konfidensintervallerne, kan derfor skyldes en for stor prøveinhomogenitet. Dette er dog ikke undersøgt nærmere i denne rapport. Ud fra de sammenlignende ekstraktioner for de 7 MST PAH'er kan følgende fremhæves:
3.3.3.2 Reproducerbarhed
Tabel 3-15. Relativ standardafvigelse for ekstraherede PAH'er for de anvendte ekstraktionsmetoder ved forskellige ekstraktionstider. De relative standardafvigelser er beregnet ud fra ekstraktion af den tjære- og motorolieforurenede jord. Betragtes resultaterne for de normaliserede data, opnås den bedst samlede reproducerbarhed ved anvendelse af I-metoden efterfulgt af D-metoden. Dette er ligeledes gældende ved en ekstraktionstid på 8 og 16 timer. Den dårligste reproducerbarhed ser ud til at opnås ved anvendelse af PA- eller PM-metoden. Vurderingen af disse tal er naturligvis behæftet med en vis usikkerhed, og det er nødvendigt at undersøge reproducerbarheden ved en validering af den endeligt udvalgte metode. Ud fra en overordnet vurdering kan følgende fremhæves:
3.4 Overordnede betragtningerI forbindelse med udvælgelse af en anvendelig ekstraktionsmetode til det videre projektarbejde bør en række andre aspekter ligeledes inddrages i den samlede vurdering. Disse behandles i det følgende. 3.4.1 Praktisk udførelseDen praktiske udførelse af ekstraktioner i laboratoriet har en stor betydning for valg af ekstraktionsmetode. D-, P- og PM-metoderne er stort set ens med hensyn til den praktiske udførelse. I-metoden kan på dette analyseniveau sammenlignes med de nævnte metoder, dog følger i referencemetoden en række oprensningstrin, som er mere tidskrævende. Udelades disse, vil den praktiske udførelse af D-, I-, PM og P-metoden være på niveau med det, som allerede i dag udføres i laboratorierne. PA-metoden er uhensigtsmæssig på grund af de nødvendige faseadskillelses-, tørrings- og inddampningstrin. 3.4.2 Miljømæssige betragtningerAf miljømæssige hensyn, er dichlormethan uønsket som ekstraktionsmiddel og ønskes generelt udsluset fra brug i laboratoriet. Samtidig er det ikke hensigtsmæssigt med en stor mængde opløsningsmiddel, som f.eks. anvendes i PA-metoden. P-, I- og PM-metoden er ud fra denne betragtning de mest optimale at anvende. 3.4.3 Kromatografiske betragtningerDet er af stor betydning, at det er muligt at evaluere prøvernes indhold af kulbrinter ud fra kromatogrammerne fra GC-FID. Dette er i PA-metoden vanskeligt pga. høj baggrundsstøj fra kulbrinter i begyndelsen af kromatogrammerne. Dette skyldes ikke de anvendte opløsningsmidler, da disse i forvejen er renhedstjekket. Desuden er der forøget risiko for forurening under inddampningsprocessen. 3.4.4 BTEXMulighed for kvantificering af BTEX'er er afhængig af valg af opløsningsmiddel. I-metoden egner sig ikke til BTEX'er i den anvendte form med heptan som opløsningsmiddel. D- og P-metoden er i dag de metoder, som anvendes til kvantificering af BTEX'er. PA-metoden kan ikke anvendes pga. inddampningstrinnet. PM-metoden kan i princippet anvendes, men der kræves dog en kromatografisk optimering, for at muliggøre kvantificering af specielt benzen. 3.4.5 Flygtige halogenerede kulbrinterMulighed for kvantificering af flygtige halogenerede kulbrinter (trichlormethan m.m.) er ikke muligt for D-metoden. Laboratoriet har ikke undersøgt muligheden for kvantificering af de halogenerede kulbrinter med I-metoden ved ECD, hvor kun de halogenerede kulbrinter detekteres. I dag anvendes primært P-metoden til kvantificering af de halogenerede kulbrinter, og PM-metoden adskiller sig ikke væsentligt i ekstraktionsprincippet. Denne metode kan i princippet anvendes. Dog er det endnu ikke afprøvet i laboratoriet. 3.4.6 Anvendelse af interne standarderI forbindelse med evaluering af resultaterne, har laboratoriet overvejet at indføre en højere kogende intern standard eksempelvis squalan. Årsagen er, at o-terphenyl har retentionstidssammenfald med en hel række andre kulbrinter – især i gasolieforureninger, og kan blive fejlintegreret pga. interferens. En højere kogende intern standard kan i nogle tilfælde afhjælpe dette problem. Laboratoriet har desuden overvejet, om det i forbindelse med PA-metoden vil være en fordel, at anvende 3 interne standarder, således at alle fraktioner korrigeres overfor hver sin interne standard. Dette vil delvis korrigere for tab, som følge af inddampningstrinet, som ikke er ligeligt fordelt over hele det kromatografiske område. 3.5 KonklusionUd fra de sammenlignende ekstraktioner for kulbrintefraktionerne C10-C40 samt C25-C40 er det med de gældende usikkerheder vanskeligt at udvælge én metode, som medfører den mest optimale ekstraktionseffektivitet. Valg af ekstraktionsmetode afhænger af, hvilken ekstraktionstid der anvendes. Anvendes en ekstraktionstid i størrelsesordenen 8-16 timer, som vurderes at være realistisk, ses en mindre forskel i ekstraktionseffektivitet for I-, D-, PA-, og PM-metoden. Ved en ekstraktionstid på 16 timer ses dog en tendens til at der opnås en højere ekstraktionseffektivitet ved anvendelse af I-metoden. Resultaterne viser dog tydeligt en generel dårligere ekstraktionseffektivitet for P-metoden ved samtlige ekstraktionstider. Valg af metode ud fra de sammenlignende ekstraktioner kan kun foretages ud fra ekstraktion af kulbrinter i området C10-C40 og C25-C40. Dette skyldes, at der ikke kan drages nogen anvendelige konklusioner ud fra ekstraktion af de 7 MST PAH'er med de gældende usikkerheder. Ud fra betragtning af resultaterne fra de sammenlignende ekstraktioner vurderes, at D-, I-, PA-, og PM-metoden er de mest velegnede at arbejde videre med, fordi der opnås sammenlignelige ekstraktionseffektiviteter ved 8 og 16 timers ekstraktionstid. En yderligere afgrænsning af metoder kan foretages ud fra praktiske og miljømæssige vurderinger. D-metoden ønskes udsluset pga. de miljømæssige konsekvenser ved anvendelse af dichlormethan. Projektgruppen har været bevidst om dette faktum forud for projektet, og D-metoden er primært inddraget for at kunne vurdere ekstraktionseffektiviteten af de andre metoder i forhold hertil. D-metoden anses derfor ikke som en anvendelig metode. I PA-metoden indgår et faseadskillelses- og inddampningstrin, som er to tidskrævende processer. Den ekstra håndtering ved omhældning til skilletragt og rotationsfordamper øger desuden risikoen for kontaminering af prøven. De kromatografiske forhold for metoden er ligledes ikke optimale, idet der ses uhensigtsmæssigt meget baggrundsstøj fra kulbrinter i starten af kromatogrammet. Dette vil have indflydelse på muligheden for kvantificering af BTEX'er og flygtige halogenerede forbindelser. På grund af inddampningstrinet kan PA-metoden dog ikke anvendes til kvantificering af BTEX'er og flygtige halogenerede forbindelser. Metoden anvender en forholdsvis stor mængde af opløsningsmidler, som af både økonomiske- og miljømæssige årsager ses som en ulempe. En prøvestørrelse på 10 g, i modsætning til 20-60 g for de andre metoder, vurderes ikke at være hensigtsmæssig. Med den stigende fokus på udtagning af repræsentative delprøver i laboratoriet bør dette også indgå i overvejelserne. Det vurderes således, at anvendelsen af PA-metoden ikke er hensigtsmæssig. PM-metoden kan i princippet anvendes både til kvantificering af BTEX'er og halogenerede flygtige forbindelser. Der kræves dog en optimering af de kromatografiske forhold for at muliggøre kvantificering af specielt benzen. I-metoden har i forhold til PM-metoden den ulempe, at der anvendes n-heptan som opløsningsmiddel. Dette umuliggør kvantificering af BTEX'er og flygtige halogenerede forbindelser på GC-FID pga. sammenfald. I ISO/DIS 16703 standarden gives der dog mulighed for anvendelse af pentan som opløsningsmiddel. Herved bør det være muligt at kvantificere både BTEX'er og de flygtige halogenerede forbindelser. Ud fra sammenligning af data for metodernes reproducerbarhed ses den bedste reproducerbarhed for kulbrintefraktionerne generelt for PM-metoden. Dataene er dog behæftet med en vis usikkerhed, og reproducerbarheden bør derfor undersøges nærmere. PM-metoden anvender en prøvemængde på 50 g mens I-metoden kun tager 20 g i arbejde. For at opnå en god repræsentativ prøveudtagning i laboratoriet må PM-metoden således foretrækkes. I-metoden forventes inden for den nærmeste fremtid at blive godkendt som europæisk standard. Såfremt dette bliver en realitet vil det af praktiske årsager være en fordel at arbejde videre med I-metoden. Herved undgås implementering af endnu en ny metode i laboratorierne. Der vil blive arbejdet videre med metoderne ISO/DIS 17603 samt PAH modificeret for at kunne foretage en yderligere afgrænsning. De to metoder ligner hinanden en del, og i kapitel 4 vil de to metoder blive afprøvet overfor hinanden. 4 Ekstraktionsmidler4.1 FormålI dette afsnit beskrives nogle indledende optimeringer af ISO/DIS 16703 (I-metoden). Her sammenlignes ISO/DIS 16703 med metoden PAH modificeret (PM-metoden). Det afprøves at ændre ekstraktionsmidlet heptan til pentan, samt at opskalere prøvemængden og mængden af ekstraktionsopløsninger, jf konklusionen i afsnit 3.5. 4.2 Sammenligning ISO/DIS 16703 med PAH modificeret4.2.1 Indledning/formålEkstraktionsmetoden fra ISO/DIS 16703 er sammen med PAH modificeret, udvalgt til at være de to metoder, der arbejdes videre med udfra betragtninger om ekstraktionseffektivitet og hensyn til miljøet. I metoden fra ISO/DIS 16703 anvendes acetone/heptan til ekstraktion, samt efterfølgende destilleret vand til at adskille faserne. Men i standarden for metoden påpeges det, at man i stedet for heptan kan anvende andre ikke-polære opløsningsmidler f.eks. pentan. I det efterfølgende har vi valgt, at afprøve pentan i stedet for heptan, da pentan har et lavere kogepunkt end heptan. Derved bliver det muligt at identificere enkeltkompontenter, der har lavere kogepunkt. Det vil således give mulighed for at beregne BTEX'er, modsat den nuværende metode, der anvender heptan. Det kan være af betydning i hvilken rækkefølge der tilsættes acetone og vand. Denne betragtning er undersøgt i forbindelse med sammenligning af ISO/DIS 16703-metoden og PAH modificeret metoden. I ISO/DIS 16703 metoden tilsættes acetone før udrystning og vandet tilsættes efter udrystning. Til sammenligning tilsættes der i ekstraktionsmetoden PAH-modificeret, opløst pyrophosfat først og acetone umiddelbart derefter. Der er således foretaget en analyse, der sammenligner de to metoder, for at se om disse forhold har nogen indflydelse på ekstraktionen af kulbrinter. Resultatet skal anvendes til, at fastlægge hvilke ekstraktionsmidler, der skal benyttes i den endelige metode, samt hvilken rækkefølge de tilsættes. Dette vurderes ud fra det højeste udbytte af ekstraktionen af totalkulbrinter og den mindste analyseusikkerhed for de to ekstraktionsmetoder. I denne afprøvning af ekstraktionsmetoden ISO/DIS 16703 og i metoden PAH-modificeret bliver der anvendt pentan som ekstraktionsmiddel. Derudover er der tilsat acetone for at befugte prøven og effektivisere ekstraktionen.Vand er tilsat for at fjerne acetonen igen, altså adskille de to faser, således at acetonen er i vandfasen og analytterne i pentanfasen. Forskellen på de to metoder er, at der i ekstraktionsmetoden fra ISO/DIS 16703 først tilsættes vand efter ekstraktionen er foretaget, hvorimod der ved den modificerede PAH-metode tilsættes vand (i form af 0,05 M pyrophosfatopløsning) samtidig med pentan og acetone. I tabel 4-1 er de to metoder sat op mod hinanden. For at kunne sammenligne de to metoder er der for begge metoder valgt at lade ekstraktet ryste i 12 timer, da den optimale ekstraktionstid endnu ikke er fundet, se kapitel 5. Desuden er der valg at anvende 20 g jordprøve til begge ekstraktionsmetoder.
Tabel 4-1. Sammenligning af ISO/DIS 16703 og PAH modificeret. 4.2.2 ForsøgsplanlægningDa usikkerheden på bestemmelser i jordprøver er relativ stor, er der foretaget 10-dobbeltbestemmelse af hver metode. Dette giver 20 behandlinger. Disse behandlinger er fordelt på GC-apparatet, således at 10 behandlinger er foretaget på forreste injektionsport og 10 behandlinger er foretaget på bagerste injektionsport. Der er således to faktorer, der analyseres for; ekstraktionsmetoden og injektionsporten. Hver af disse faktorer har to niveauer, som det ses i tabel 4-2.
tabel 4-2. Faktorer og faktorniveauer. 4.2.3 Udførelse af forsøgsplanenDen anvendte jord til alle prøverne i denne analyse er en homogen blanding bestående af kompostjord og motor-smøreolieforurenet jord, som er beskrevet i afsnit 3.2.2. Hver af de 20 behandlinger er baseret på 20 g af denne blandejord, hvor der er tilsat de respektive ekstraktionsmidler, som beskrevet i tabel 4-1, for hver af de to ekstraktionsmetoder. Ekstraktionstiden er foreløbigt fastsat til 12 timer, da indledende forsøg pegede på, at optimum er nået ved denne tid. Efter ekstraktionen er der udtaget ekstrakt, der er overført til GC-vials. De 20 behandlinger er herefter analyseret på GC-FID i randomiseret rækkefølge for at eliminere ændringer i GC-performance. 4.2.4 ResultatbehandlingResultaterne af forsøgsrækken er omregnet fra de totale toparealer i GC-kromatogrammet til indholdet af totalkulbrinter i mg/kg TS pr. prøve. En jord med motor- og smøreolieforurening blandet med kompostjord ligger i kogepunktsintervallet mellem C14 og C36. Arealet herunder er integreret som en totalsum og det totale kulbrinteindhold er beregnet overfor n-alkanerne C10 til C36. Beregningerne er udført med o-terphenyl som den interne standard. Resultaterne ses i tabel 4-3.
Tabel 4-3. Resultater for de to metoder. Datamængden er herefter undersøgt for normalfordeling og varianshomogenitet. Da dette er tilfældet, er der foretaget en variansanalyse over resultaterne, som viser, at der ikke er statistisk bevis for, at hverken ekstraktionsmetoden eller placeringen i GC-en har nogen indflydelse på ekstraktionsudbyttet af totalkulbrinter i prøverne. Ved at opstille et konfidens interval plot, for de to ekstraktionsmetoder, fremkommer resultatet i et grafisk udtryk. Figur 4-1.
Figur 4-1. Konfidensintervalplot for de to metoder. Illustrationen viser, at det kunne se ud som om, at udbyttet er en anelse større, når der ekstraheres efter ISO/DIS 16703 metoden. 4.2.5 DelkonklusionResultatet af analyserne viser, at de to metoder statistisk set ikke er forskellige. Men ved at se lidt nærmere på ekstraktionsudbyttet er der dog en lille tendens til at ISO/DIS 16703 har et lidt større udbytte end PAH-metoden. For at kunne vurdere de to metoder optimalt, bør man også sammenligne ydre faktorer så som arbejdsmiljøet, priser, arbejdsbyrde mm. Nogle af disse ting er sammenholdt i tabel 4-4.
Tabel 4-4. Sammenligningsskema. Den samlede konklusion af ovenstående forsøg, er således at det foretrækkes at anvende ekstraktionsmidler og tilsætningsrækkefølge som i ekstraktionsmetoden fra ISO/DIS 16703, men med den modifikation at der anvendes pentan i stedet for heptan. I tabel 4-5 ses i korte træk den valgte ekstraktionsmetode, som i den nuværende modificerede udgave, kaldes for "ISO/DIS 16703-a.
Tabel 4-5. Den foreløbige metode. 4.3 Prøvemængde og pentanmængdeDet er nu valgt at anvende ISO/DIS 16703 metoden som udgangspunkt for den nye metode. 4.3.1 FormålFormålet med forsøget er at bestemme om prøvemængden på ca. 20 g jord, der anvendes i den oprindelige ekstraktionsmetode fra ISO/DIS 16703, kan skaleres op til en prøvemængde på ca. 60 g jord. En prøvemængde på 60 g i 100 mL Duranglas er det, der anvendes på de fleste danske laboratorier jf. standard metoden, i dag. Ved at få en større jordmængde i arbejde, 60 g i stedet for 20 g, opnås et mere repræsentativt billede af den faktiske forureningsgrad, og derved også et mere præcis resultat af indholdet i prøven. Desuden er det en fordel for laboratorierne ikke at skulle udtage delprøver. En prøvemængde på ca. 60 g, svarer til at prøvetageren skal fylde et 100 mL Duranglas omkring halvt op. Tørstofindholdet vil således komme til at svare til omkring 50 g jord. 4.3.2 ForsøgsplanlægningDer er foretaget sammenligningsforsøg, hvor der ekstraheres jordprøver på 20 g og prøver på 60 g. I prøverne med 60 g jord er det nødvendigt at tilsætte større mængder opløsningsmidler. Selve ekstraktionsmidlet, pentan, blev opskaleret således, at der er tilsat dobbelt så meget pentan med intern standard til prøvemængden på 60 g jord som i den oprindelige metode. Mængden af acetone og vand er ikke fordoblet, da den i forskriften anviste mængde viste sig at være passende. Derfor er der ikke udført yderligere forsøg med andre mængder opløsningsmidler. Rent praktisk er disse mængder passende i forhold til de anvendte prøveglas på 100 mL, således at der stadig er volumen nok over prøven og ekstraktionsmidlerne, til at der kan foretages en effektiv rystning. Den nye metode, en modifikation af ISO/DIS 16703-a, med 60 gram jordprøve og 20 mL pentan (IS), kaldes i det følgende for ISO/DIS 16703-b. I tabel 4.6 ses jordprøvemængden og de forskellige opløsningsmidler i de to testede metoder.
Tabel 4-6. Prøvemængde og mængde af ekstraktionsstoffer. Forsøget kommer således til at bestå af én faktor med to faktorniveauer. Der er foretaget 5-dobbelbestemmelse af hvert faktorniveau, således at der er 10 behandlinger i alt, som er analyseret randomiseret. 4.3.3 Udførelse af forsøgsplanenDer er anvendt en homogen jordblanding bestående af lige dele kompostjord (naturlige kulbrinter) og motorolieforurenet jord, se afsnit 3.2.2 for beskrivelse af jorden. Der er afvejet henholdsvis omkring 20 g og 60 g af jordblandingen til hver af de 5 prøveglas og opløsningsmidler er tilsat efter tabel 4-6. 4.3.4 ResultatbehandlingResultatet af forsøgsrækken foreligger som det totale topareal af hver behandling. Indholdet i prøverne er beregnet udfra en n-alkanstandard analyseret på samme sekvens som prøverne. Der er anvendt de n-alkaner som ligger i samme kogepunktsområde som prøven. Der er anvendt o-terphenyl som intern standard. Resultaterne ses i tabel 4-7.
Tabel 4-7. Resultater for forsøgsrækken. De sædvanlige forudsætninger for en variansanalyse kontrolleres og er accepteret. Der kan nu foretages statistisk resultatbehandling. Resultatgennemsnit samt konfidensintervaller er plottet. Ses i figur 4-2.
Figur 4-2. plot af middelværdier og konfidensinterval. Trods det, at der grafisk ses en lille tendens til, at der er større udbytte ved ekstraktion af 20 g jordprøve, er der intet statistisk bevis i variansanalysen for, at der er en forskel mellem udbyttet på ekstraktioner foretaget med de to ekstraktionsmetoder. 4.3.5 DelkonklusionAnalysen viser, at der ikke er statistisk signifikans for, at en prøvemængde på 20 g jord og en prøvemængde på 60 g jord udviser forskelligt ekstraktionsudbytte. Men analysen viser, at der er en mindre spredning mellem resultaterne for prøverne med 60 g jord se tabel 4-7. Derfor vælges at gå videre med 60 g jord pga. de under afsnit 4.3.1 angivne fordele. Ovenstående analyse viser således, at ekstraktionsmetoden godt kan skaleres op fra en prøvemængde på 20 g til den ønskede prøvemængde på 60 g. På baggrund af valget af 60 g jord vil der således blive anvendt 20 mL pentan til ekstraktionen. 4.4 KonklusionMed udgangspunkt i ISO/DIS 16703 er det nu afprøvet at benytte 60 g prøve, at tilsætte 20 mL acetone og 20 mL pentan som ekstraktionsmiddel og derefter 30 mL vand. Denne metode er let anvendelig, og giver et højt udbytte med lav usikkerhed. Metoden ser nu som i tabel 4-8, og den omtales efterfølgende som den modificerede ISO/DIS 16703-b:
Tabel 4-8. Modificeret ISO/DIS 16703-b. I analyseforskriften for den nye metode vil der blive åbnet mulighed for anvendelse af andre ikke-polære opløsningsmidler f.eks. cyclohexan, n-heptan eller n-hexan. Dette kræver dog en selvstændig afprøvning og validering. 5 Optimering af ekstraktionstid5.1 FormålI afsnit 3.5 er det konkluderet, at der kun er mindre forskel i ekstraktionseffektivitet for de enkelte metoder, men at udrystningstiden er betydningsfuld. Som et led i optimeringen af ekstraktionsbetingelserne er formålet her at bestemme den ekstraktionstid, der giver det højeste udbytte og den mindste analyseusikkerhed. Indledningsvis afprøves ekstraktionstider for PAH-modificeret (PM-metoden). Ekstraktionstiden er defineret som den tid, prøven skal rystes med ekstraktionsmidlet, for at det totale kulbrinteindhold i prøverne er ekstraheret over i pentanfasen. 5.2 Indledende rystetidsforsøg5.2.1 FormålDette indledende forsøg er udført sideløbende med forsøgene i afsnit 4. På dette tidspunkt i metodeudviklingen er der anvendt en ekstraktion med pyrophosphat/pentan/acetone. 50 g jordprøve udrystes med 20 mL pyrophosphat + 20 mL pentan + 20 mL acetone. Formålet med forsøget er at få en ide om, i hvilket tidsområde den optimale rystetid for ekstraktion findes. 5.2.2 AfgrænsningEkstraktionstiden er defineret som den tid, prøven skal rystes med ekstraktionsmidlerne, for at det totale kulbrinteindhold i prøverne er ekstraheret over i pentanfasen. Den optimale rystetid kan teoretisk ligge fra ganske kort tid fx. 15 min og helt op til flere døgn. Erfaring har vist at:
Ud fra disse betragtninger samt en praktisk synsvinkel er der valgt følgende 5 niveauer:
Da niveauerne er kvantitative, vil det være muligt ved regressionsanalyse at bestemme en optimal rystetid, som godt kan ligge mellem de enkelte niveauer. Den optimale rystetid vil være afhængig af jord- og forureningstype. For at dække forskellige typer vælges følgende:
De fire jordtyper er de samme som er anvendt i forsøgene afsnit 3.2.2. Der er lavet tilsætning med gasolie til jordtype D, således at metodens genfinding kan kontrolleres. Erfaring viser, at usikkerheden på bestemmelser af jordprøver er relativt stor. Forsøget kræver flerdobbeltbestemmelse på hvert niveau for at få et bedre estimat for spredningen samt mulighed for udelukkelse at evt. outliers. Det er valgt at lave 4-dobbeltbestemmelse på hvert niveau. Ovenstående valg giver følgende dimensionering af forsøget: 5 rystetider * 4 jordtyper * 4 bestemmelser = 80 behandlinger 5.2.3 ForsøgsplanlægningDe 80 behandlinger skal analyseres på gaskromatografen i randomiseret rækkefølge for at kunne eliminere ændringer i GC-performance. Erfaring viser, at det rent praktisk er muligt efter kørsel af standarder at analysere op til 15 på hinanden følgende prøver på gaskromatografen med tilfredstillende resultat. Derefter skal gaskromatografen vedligeholdes før yderligere analyse. Det er valgt at analysere standard, kontrol samt blind i starten og slutningen af en sekvens. Således kan det kontrolleres, at apparatets performance er tilfredsstillende over hele sekvensen. Rent praktisk giver dette, at der kan analyseres 12 behandlinger på en sekvens/kø. Der kan vælges at analysere alle prøverne på samme GC-signal over 12 dage, eller der kan analyseres på flere forskellige signaler. Idet apparatets performance ændres fra dag til dag pga. vedligehold (linerskift mm.), er det nødvendigt at indføre sekvensen som en blokfaktor under databehandlingen. Det er valgt, at analysere alle prøverne på samme GC, dog fordelt på både A og B signal (front/back). De 80 behandlinger analyseres på 7 sekvenser. 5.2.4 UdførelseDer er udført prøveforberedelse på alle prøver sideløbende. Det vil sige, alle prøver er vejet af og har fået tilsat ekstraktionsmiddel samtidigt. Derefter er alle prøverne sat op til rystning og løbende taget af efter deres respektive rystetid. Hver prøve er aftappet i 4 vials. Vials er opbevaret i fryser indtil analyseringstidspunktet. Prøverne er analyseret i randomiseret rækkefølge. 5.2.5 ResultatbehandlingTotalkulbrinteindholdet i prøverne er beregnet ud fra totalarealet og overfor de lige n-alkaner, der ligger i tilsvarende kogepunktsintervaller. Indholdet i de enkelte prøver er beregnet i forhold til et gennemsnit af de to standarder analyseret i start og slut af pågældende sekvens. Beregningen er udført overfor intern standard (o-terphenyl). Resultaterne for de fire forskellige jordtyper ses i tabel 5.1.
Tabel 5-1. Beregnet indhold i prøverne. Resultaterne mærket * er på grund af for høje residualer betragtet som outliers og er udelukket af analysen. Der laves variansanalyse på de resterende resultater. Sekvensnummeret indføres i første omgang som en blokfaktor, som dog viser sig ikke at være signifikant. Blokfaktoren fjernes fra variansanalysen. De sædvanlige forudsætninger for en variansanalyse kontrolleres. Datamaterialet viser varianshomogenitet, normalfordeling samt statistisk uafhængighed, og vi kan derfor gå videre med den statistiske resultatbehandling. Der er lavet variansanalyser for samtlige jordtyper samt plot, der viser middelværdi og konfidensintervaller, se figur 5-1 til 5-4.
Figur 5-1. Middelværdier og konfidensintervaller for den tjæreforurenede jord. Ud fra figur 5-1 konkluderes det for den tjæreforurenede jord:
Figur 5-2. Middelværdier og konfidensintervaller for gasolieforurenet jord. Ud fra figur 5-2 konkluderes for den gasolieforurenede jord:
Figur 5-3. Middelværdier og konfidensintervaller for motorolieforurenet jord. Ud fra figur 5-3 konkluderes for den motorolie forurenede jord:
Figur 5-4. Middelværdier og konfidensintervaller for spiket sandjord . Ud fra figur 5-4 konkluderes det for jorden med tilsætning af gasolie:
Gasolieprøverne er spiket med en kendt mængde gasolie. Der er afvejet 50 g sand (med tørstofprocent på 100%). Denne er spiket med 1 mL af 40,2 g/L stamopl. = 804 mg/kg TS. Der er genfundet følgende middelværdier, se tabel 5-2:
Tabel 5-2. Genfinding. Genfindingen er tilfredsstillende for alle rystetider, men standard afvigelsen er klart størst ved 2 timer. Der er tendens til at 12 timer giver den mindste afvigelse. De samme resultater er anvendt i regressionsanalyser, da rystetiden er en kvantitativ faktor. Der benyttes en formel for kurve, der nærmer sig et maksimum, da dette forventes at være tilfældet her.
Resultaterne fra regressionsanalysen ses i figur 5-5 som viser et eksempel på et plot ud fra formlen, der nærmer sig et maksimum. Plottene for de forskellige jordtyper leder til samme konklusioner som figurene 5-1 til 5-4 ovenfor.
Figur 5-5. Plot af modellen fra regressionsanalysen. 5.2.6 DelkonklusionDet kan konkluderes, at rystetiden skal være længere end 4 timer. Desuden viser forsøget med den motorolieforurenede jord, at rystetiden bør være længere end 8 timer. Der kan ikke vises nogen signifikant forskel på rystetider fra 12 – 16 timer. Det kan konkluderes, at den halve times rystetid, som er anbefalet i ISO/DIS 16703, ikke er tilstrækkelig. I næste afsnit skal der findes en optimal rystetid indenfor de 12 – 16 timer. Desuden skal det undersøges, om en rystetid længere end 16 timer evt. giver bedre resultater. 5.3 Ekstraktionstidsforsøg5.3.1 FormålDer skal i dette afsnit findes den bedste udrystningstid for optimal ekstraktion af kulbrinterne fra forskellige jordtyper med den modificerede ISO/DIS-b metode. 5.3.2 Afgrænsning/baggrundTidligere i projektprocessen er der udført nogle indledende rystetidsforsøg foretaget med ekstraktion ud fra den modificerede PAH metoden, afsnit 5.2. Resultaterne af de indledende forsøg viser, at rystetiden skal være større end 4 timer. Den viser også, at for nogle jord- og forureningstyper skal man op på rystetider på mellem 12 – 16 timer, og at en rystetid på ½ time, som angivet i ISO/DIS 16703 metoden /1/, er utilstrækkelig. Ud fra disse indledende resultater blev et rystetidsforsøg for den nye modificerede ekstraktionsmetode planlagt. Oprindeligt anbefales der en rystetid på kun 30 minutter. Men erfaringen fra de indledende undersøgelser viser som nævnt en tendens til, at længere rystetider er nødvendige for nogle jordtyper. Dette sammenholdt med erfaringer fra eget analyselaboratorium gør, at der blev valgt at undersøge, om der er en markant forbedring af udbyttet ved at lade rystetiden bliver længere. 5.3.3 ForsøgsplanlægningUdgangspunktet for den modificerede ekstraktionsmetode (ISO/DIS-b) er således en rystetid på omkring 12 timer, jf. afsnit 5.2. Desuden er det undersøgt, om prøver, der udsættes for specielt korte ryste/ekstraktionstider (0,5 time og 6 timer), opnår signifikant lavere ekstraktionsudbytter. Det er selvfølgelig en fordel med så korte rystetider som muligt, da miljøanalyser ofte skal afvikles inden for en stram tidsplan. Herudover er der medtaget prøver med længere rystetider, for at se om optimum er nået ved en rystetid på omkring de 12 timer, eller om en længere ekstraktionstid også giver højere udbytte (24-48 timer). Da jordprøver er svære at få helt homogene, kan usikkerheden på bestemmelser af prøverne til tider være relativ stor. Det vil således være nødvendigt med flerdobbeltbestemmelse på hvert prøve for at få et bedre estimat for spredningen, samt at eventuelle outliers kan udelukkes, uden at det går ud over mængden af forsøgsdata. Her er valgt at lave 4-dobbeltbestemmelse på hvert niveau. 8 rystetider * 4 bestemmelser = 32 behandlinger I tabel 5.3 er faktorniveauerne angivet.
Tabel 5-3. Faktorniveauer. 5.3.4 Udførelse af forsøgsplanenDer er anvendt en homogen jordblanding bestående af lige dele kompostjord (naturlige kulbrinter) og motorolieforurenet jord. Se afsnit 3.2.2 for beskrivelse af jorden. Der er afvejet ca. 60 gram jord til hver delprøve til analysering. Hver prøve er derefter tilsat ekstraktionsmidlerne efter ISO/DIS 16703-b, jf. afsnit 4.4. Herefter er de sat på rystebord i henhold til de respektive timer. Efterfølgende er der tilsat vand, centrifugeret og udtaget til GC-vials. De 32 behandlinger er analyseret på gaskromatografen i randomiseret rækkefølge for at kunne eliminere ændringer i GC-performance. Alle prøverne er analyseret på samme GC, dog fordelt på både A og B signal (front/back). De 32 behandlinger er analyseret på 4 sekvenser samt på en omkørsel. 5.3.5 ResultatbehandlingResultaterne af forsøget foreligger som det totale topareal af hver behandling. Arealet er beregnet som det totale areal under toppene fra C12 til C38 og er integreret som en totalsum. Overfor n-alkaner og brug af intern standard, o-terphenyl, er prøvernes indhold af totalkulbrinter beregnet, således at de fremstår som mg totalkulbrinter pr. kg TS. Se tabel 5-4.
Tabel 5-4. Resultater for forsøgsrækken. Indhold af totalkulbrinter. For at der kan foretages statistiske analysemetoder, som variansanalyser og regressionsanalyser, forudsættes det, at datamængden er statistisk uafhængig, normalfordelt samt udviser varianshomogenitet. Dette er undersøgt og tilfredsstillende opfyldt. Der er herefter foretaget en regressionsanalyse, der tester faktorene, se tabel 5-5. Ud over rystetiden, er der medtaget faktorerne dag og inlet.
Tabel 5-5. Regressionsanalyse foretaget i StatGraphics. Af tabel 5-5 ses at dag og inlet ikke er signifikante for modellen da P-value er større end 0,05. Dag og inlet fjernes fra regressionsanalysen og den nye regressionsanalyse (ikke vist) viser med statistisk signifikans, at antallet af rystetimer har en indflydelse på indholdet af totalkulbrinter i ekstraktet. Grafisk ser forsøget ud som i figur 5-6, hvor der er vist et middelværdiplot af modellen.
Figur 5-6. Middelværdiplot af model for effektiviteten af rystetider på udbyttet. Figur 5-6 viser at rystetider på 0,5 og 2 timer giver lavere udbytte end en rystetid, der er længere end 4 timer. Der er ikke signifikant forskel for rystetider på 4 –12 timer. På baggrund af usikkerhed af resultaterne for 48-timer, tillægges det højere resultat for denne behandling ikke større betydning. På baggrund af figur 5-6, tabel 5-4 og 5-5 konkluderes det, at:
5.3.6 DelkonklusionSammen med den indledende rystetidsanalyse i afsnit 5.2 viser dette rystetidsforsøg også, at det bedste resultat opnås hvis der anvendes en ekstraktionstid på minimum 10 timer. Så resultatet er, at ved rystetider fra 10-24 timer er der et højt udbytte, samt en analyseusikkerhed, der kan anses for at være rimelig. 5.4 Ekstra forsøg5.4.1 FormålDet er besluttet, at det ville være fordelagtigt at underbygge ekstraktionsforsøgene med et ekstra forsøg, der er baseret på en mere tung og lerholdig jord. Desuden er der kun fokus på ekstraktion med rystetider omkring 8-16 timer, således at der er tale om mere samlede datasæt. 5.4.2 Afgrænsning/baggrund - ForsøgsplanlægningDer er udvalgt en meget tung og lerholdig jord med en gammel forurening fra tjære og motorolie som prøvejord. Jorden er beskrevet som sandblandet lerjord med JB nr. 5, jf. yderligere oplysninger og kromatogram af jorden i afsnit 9.2.3 under valideringen. De rystetider, der er undersøgt er 8, 12 og 16 timer. 3 rystetider * 10 bestemmelser = 30 behandlinger Dimensioneringen af forsøget bliver således 30 behandlinger, som kan behandles statistisk som et balanceret forsøg. Derudover er der også to andre faktorer med, da analysen er foretaget over 2 dage og prøverne har været placeret ligeligt fordelt både på front- og back-inlet i GC-apparatet. 5.4.3 Udførelse af forsøgsplanenDer er afvejet ca. 60 gram jord til hver prøve. Hver prøve er derefter tilsat ekstraktionsmidlerne efter ISO/DIS 16703-b. Herefter er de sat på rystebord i de respektive antal timer. Efterfølgende er der tilsat vand, centrifugeret og tappet i vials. De 30 behandlinger er analyseret på gaskromatografen i randomiseret rækkefølge for at kunne eliminere ændringer i GC-performance. 5.4.4 ResultatbehandlingResultaterne af forsøget foreligger som det totale topareal af hver behandling. Arealet er beregnet som det totale areal under toppene fra C12 til C38 og er integreret som en totalsum. Indholdet er beregnet overfor n-alkanerne i samme kogepunktsinterval. Resultaterne ses i tabel 5-6.
Tabel 5-6. Resultater for forsøgsrækken. Indhold af totalkulbrinter i tung lerjord. For at der kan foretages statistiske analyser, som variansanalyser og regressionsanalyser, forudsættes det, at datamængden er statistisk uafhængig, normalfordelt samt har samme varians. Dette er undersøgt og fundet tilfredsstillende. Der er foretaget en regressionsanalyse, der tester faktorene. Ud over rystetiden er der medtaget de to faktorer for dag og inlet. Som i tidligere analyser viser regressionsanalysen, at dag og inlet ikke er signifikante for modellens forklaring og er derfor fjernet fra modellen. Den ny regressionsanalyse uden dag og inlet viser, at der er statistisk signifikans for, at antallet af rystetimer har en indflydelse på indholdet af total kulbrinter i ekstraktet. Grafisk ser forsøget ud som i figur 5-7, hvor der er foretaget et middel plot af modellen.
Figur 5-7. Middelværdiplot af model for effekten af rystetiden på udbyttet i tung lerjord. Heraf ses, at rystetider ved 12 og 16 timer ikke er signifikant forskellige. Ved at se på en multipel range test, tabel 5-7, ses det netop, at der er statistisk signifikans for, at ekstraktionsudbyttet ved en rystetid på 8 timer er lavere end rystetiderne på 12-og 16-timer.
Tabel 5-7. Multiple range test, der viser, at en rystetid på 8 timer er signifikant forskellig fra de øvrige. Ved at se på analyseresultaternes middelværdier og spredninger i tabel 5-8, konstateres det, at middelværdierne for 12- og 16-timers rystetid også er højere end for 8 timers rystetid, således at højeste udbytte opnås ved rystetider mellem 12 og 16 timer. Spredning og de relative standardafvigelser ligger omtrent på samme niveau for alle tiderne, og inden for et rimeligt niveau, således at analyseusikkerheden anses for at være acceptabel.
Tabel 5-8. Middelværdi, standardafvigelse og den relative standardafvigelse. 5.4.5 Delkonklusion
5.4.6 Sammenligning af rystetider på 12 og 16 timerDet har ikke i nogen af de udførte rystetidsforsøg (afsnit 5.2 - 5.4) været muligt at vise en forskel på resultaterne for en rystetid på henholdsvis 12 eller 16 timer. Der er foretaget en sammenligning af alle forsøg med en rystetid på 12 timer og en rystetid på 16 timer for at dokumentere, at der ikke er forskel på resultaterne for de to rystetider, jf. tabel 5-9. Disse forsøg bygger på forskellige jordtyper.
Tabel 5-9. Middelværdier og forholdet mellem rystetider på 12- og 16-timer. I tabel 5-9 er det procentvise forhold mellem 12- og 16- timers udrystning vist. Forholdet af 12 timer ligger på mellem 94 – 105 % i forhold til 16 timers rystetid. Der er således belæg for, at det er lige så godt at anvende en rystetid på 12 timer som en på 16 timer. Dette betyder, at de enkelte laboratorier kan tilpasse rystetiden frit imellem 12 – 16 timer afhængigt af, hvordan det passer ind i produktionen. 5.5 KonklusionJordprøverne skal ekstraheres med acetone/pentan i mindst 12 timer. Det har vist sig, at en ekstraktionstid på 16 timer giver det samme resultat som 12 timers ekstraktionstid, og et analyselaboratorium kan derfor vælge den tid, der er mest praktisk for deres arbejdstilrettelæggelse inden for 12 – 16 timer. I standarden for den nye metode vil der komme til at stå, at man kan vælge at ekstrahere i 12 til 16 timer, da der ikke er signifikante afvigelser i dette tidsrum. Den nye ekstraktionsmetode med de nye tiltag, der er foretaget i dette kapitel, omtales nu som ISO/DIS 16703-c og metoden ses i tabel 5-10.
Tabel 5-10. ISO/DIS 16703-c. 6 Vask og oprensning6.1 FormålI standarden for ISO/DIS 16703 metoden beskrives to trin til oprensning af ekstraktet før analyse. De to trin er tidskrævende og medfører omhældning af ekstraktet, hvilket umuliggør analyse af flygtige komponenter. Det bliver i dette kapitel afprøvet om de to oprensningstrin er relevante for en dansk metode til bestemmelse af totalkulbrinter. 6.2 Vaskeforsøg6.2.1 IndledningI ISO/DIS 16703 er det angivet, at ekstraktet skal vaskes med vand efter ekstraktionen. Vask af ekstraktet har til formål at fjerne evt. polære forbindelser stammende fra indhold af naturlige kulbrinter i jordprøverne. Det er interessant, om vasken har en rensende effekt på ekstraktet. Desuden skal det undersøges, om vasken evt. fjerner noget af olieforureningen. 6.2.2 AfgrænsningForsøget er afgrænset til tilsætning af gasolie og motorolie til en jord med indhold af naturlige kulbrinter (kompost). Der anvendes en homogeniseret kompostjord, jf. afsnit 3.2.2, med et indhold af naturlige kulbrinter på ca. 500 mg/kg TS ved dichlormethanekstraktion i 4 timer i fraktionen C10-C40. Der analyseres på kompostjord, kompostjorden spiket med gasolie samt kompostjorden spiket med motorolie. På ekstrakterne udføres henholdsvis ingen vask, vask med vand eller vask med svag basisk vand (0,01 M NaOH).
Tabel 6-1. Forsøgsplanen. Der er foretaget dobbeltbestemmelse. 3 vask * 3 olietilsætning * 2 bestemmelser = 18 behandlinger Analysen af alle ekstrakterne er foretaget over to dage på apparatet, denne faktor skal der tages højde for i resultatbehandligen. Alle faktorer er kvalitative. 6.2.3 Udførelse af forsøgsplanenDer er afvejet 18 prøver af kompostjorden, ca. 50 g prøve i hvert glas. 6 prøver er spiket med ca. 1500 mg gasolie/kg TS og 6 prøver er spiket med ca. 1500 mg motorolie/kg TS. Prøverne er ekstraheret med acetone/pentan og rystet i 12 timer. Efter udrystning er der tilsat 30 mL vand til prøverne, og der er rystet 1 min som foreskrevet i ISO/DIS 16703. Selve vasketrinet følger derefter og udføres ved at aftappe hele det organiske ekstrakt til et centrifugerør, og dertil endnu engang tilsætte vand. Derefter rystes i yderligere 1 min. Efter aftapning af ekstrakterne er disse behandlet efter forsøgsplanen. Ekstrakterner analyseret i randomiseret rækkefølge for at opnå statistisk uafhængighed. 6.2.4 ResultatbehandlingKromatogrammerne er integreret og totalkulbrinte indholdet er beregnet overfor n-alkanstandard med korrektion for intern standard (o-terphenyl). Resultaterne ses i tabel 6-2.
Tabel 6-2. Resultater for forsøgsrækken. Totalkulbrinteindhold. Det er fundet, at datamaterialet er varianshomogent, normalfordelt og statistisk uafhængigt. Det vurderes, at der kan udføres statistiske test på datamaterialet. 6.2.5 ResultaterFor bedre at kunne vurdere resultaterne er de opstillet i følgende tabel 6-3.
Tabel 6-3. Totalkulbrinte indhold i mg/kg TS samt genfindingsprocent. Resultaterne, for prøverne uden tilsætning, er middelværdier af værdierne fra tabel 6-2. Resultaterne, for de spikede prøver, er middelværdien af de fundne indhold af totalkulbinter fra tabel 6-2, men fratrukket det oprindelige indhold af naturlige kulbrinter. Resultaterne skal således kunne sammenlignes direkte med spikeningsniveauet. Der laves en variansanalyse. Middelværdier med konfidensintervaller plottes for hver jordtype/koncentration. Variansanalysen viser, at der ikke er signifikans for, at vaskemetoden har indflydelse på resultatet for de tre jordtyper. Dog er det muligt at se og vurdere lidt på de tendenser, der kan ses af figur 6-1, 6-2 og 6-3.
Figur 6-1. Naturlige kulbrinter ca. indhold 500 mg/kg TS. Når man ser på indholdet af naturlige kulbrinter i de rene kompostprøver, figur 6-1, fås gennemsnitligt et lidt lavere resultat efter vask med vand. Dette er ikke signifikant.
Figur 6-2. Prøven spiket med motorolie 1481 mg/kg TS. Når prøven er spiket med motorolie, jf. figur 6-2, ses samme tendens. Resultatet for ekstrakterne, der er vasket med vand, er lidt lavere, dog ikke statistisk signifikant. Indholdet fra de naturlige kulbrinter er fratrukket i grafen, dvs. genfindingen af motorolie ligger på ca. 75 %.
Figur 6-3. Prøven spiket med gasolie 1468 mg/kg TS. Når prøven er spiket med gasolie ses af figur 6-3 samme tendens i forhold til vask med vand. Det ses også, at der mistes yderligere indhold ved at vaske med svag base. 6.2.6 DelkonklusionUd fra dette mindre forsøg, er der ikke statistisk belæg for at konkludere, at vask af ekstraktet har nogen betydning for indholdet af totalkulbrinter. Tendensen er dog, at der kan fjernes en mindre del af de naturlige kulbrinter i et kompostjords ekstrakt ved at vaske ekstraktet med vand før analyse. Der er dog også tendens til, at der mistes en del af en olieforureningen. 6.3 Oprensningsforsøg6.3.1 FormålI ekstraktionsmetoden fra ISO/DIS 16703 bliver ekstraktet som sidste led tørret med vandfrit natriumsulfat og oprenset med florisil med henblik på fjernelse af polære komponenter. Formålet med dette forsøg er at teste, om det er hensigtsmæssigt og nødvendigt at foretage denne oprensning. 6.3.2 Afgrænsning/baggrundFor at finde ud af, om oprensningsproceduren fra ISO/DIS 16703 er nødvendig, tilsættes florisil direkte til ekstraktet som foreskrevet. Da der i standarden er anmærket, at det også er muligt at anvende en fastfase kolonne med florisil, og at en sådan vil være mere effektiv, er der også afprøvet en fastfasekolonne med florisil, IST. Desuden er der foretaget forsøg med oprensning på en anden type fastfase kolonne, der også kan anvendes til at tilbageholde polære komponenter, en PPL kolonne. Oprensning jf. ISO/DIS 16703: 0,25 - 0,50 g vandfrit natriumsulfat + 3,0 g Florisil. Ryst i 10 min. IST kolonne/søjle: Fastfase kolonne med 200 mg Florisil. PPL søjle: Fastfase kolonne med 200 mg cross-linked copolymerer. 6.3.3 ForsøgsplanlægningDer er oprenset på et ekstrakt af en homogeniseret kompostjord, hvor der med sikkerhed ses et indhold af kulbrinter af biogen oprindelse ved GC-FID jf. afsnit 3.2.2. Der er ligeledes oprenset på ekstrakt af samme jord spiket med hhv. motorolie og gasolie. Der er udført dobbeltbestemmelser. Ud fra totalkulbrinteindholdet ved GC-FID vurderes de tre oprensningsmetoder mod ingen oprensning. 4 oprensningsmetoder * 3 prøvetyper * 2 bestemmelser = 24 behandlinger De fire oprensningmetoder er de tre ovennævnte samt ingen oprensning. Prøvetyperne er kompostjord samt spiket kompostjord med henholdsvis gasolie/motorolie.
Tabel 6-4. Faktorniveauer. 6.3.4 Udførelse af forsøgsplanenDer er afvejet 24 prøver af den homogeniserede kompostjord ca. 50 g prøve i hvert glas. 8 prøver er spiket med ca. 1500 mg gasolie/kg TS og 8 prøver er spiket med ca. 1500 mg motorolie/kg TS. Prøverne er ekstraheret med acetone/pentan og rystet i 12 timer, tilsat 30 mL vand, rystet i 1 min og tappet over i centrifugeglas. Derefter er ekstrakterne vasket med vand efter ISO/DIS 16703. Herefter er ekstrakterne enten oprenset efter en af de tre metoder eller ikke oprenset. De 24 behandlinger er analyseret på gaskromatografen i randomiseret rækkefølge. Alle prøverne er analyseret på samme GC apparat, dog fordelt over to dage og to signaler. 6.3.5 ResultatbehandlingKromatogrammerne er integreret og totalkulbrinteindholdet er beregnet overfor n-alkan standard. Resultaterne ses i tabel 6-5.
Tabel 6-5. Resultater for forsøgsrækken. Indhold [mg/kg TS]. 6.3.5.1 Indhold af naturlige kulbrinter
Figur 6-4. Middelværdiplot for prøver med indhold af naturlige kulbrinter. 6.3.5.2 Kompostjord spiket med motorolie
Figur 6-5. Middelværdiplot for prøver spiket med motorolie. 6.3.5.3 Kompostjord spiket med gasolie
Figur 6-6. Middelværdiplot for prøver spiket med gasolie. Af figur 6-6 ses, at man ved oprensning med metode 2. ISO/DIS eller ved 3. PPL mister en del af totalkulbrinteindholdet i prøven. Eftersom vi ikke fandt nogen tendens til at miste naturlige kulbrinter, se figur 6-4, må det være gasolie, der mistes, ved disse to oprensningsmetoder. Tabet af kulbrinter kunne skyldes omhældning og håndtering af ekstrakt, men idet vi ikke ser en ligeså kraftig tendens ved 4. IST må der formodes at være et reelt tab under oprensningen. 6.3.6 DelkonklusionUdfra dette mindre forsøg med oprensning efter ISO/DIS 16703 syntes der ikke at være meget at vinde ved at oprense ekstraktet. Der findes intet statistisk belæg for, at der kan fjernes nogle at de naturlige kulbrinter ved en af disse oprensningstyper. Til gengæld risikeres at fjerne en del af gasolien. Dette forsøg er udført på kompostjord, og det kunne være interessant også at afprøve oprensningen på en anden type naturlige kulbrinter. Dette vil blive afprøvet i et yderligere forsøg med endnu en jordtype. Denne jord stammer fra Chritianshavn, og heri findes et højt indhold af kulbrinter af uvis oprindelse, sandsynligvis stammende fra havbund/gytje eller lignende. 6.4 Vaske og oprensningsforsøg 26.4.1 FormålI de foregående forsøg har vask og oprensning været afprøvet på kompostjord samt spikening med olie af denne. På kompostjorden har det ikke været mulig at vise, at der kan fjernes naturlige kulbrinter ved vask og oprensning. Det skal i dette forsøg afprøves, hvilken indflydelse vask og oprensning har på en anden jordtype indeholdende naturlige kulbrinter. 6.4.2 AfgrænsningTil forsøget er anvendt en homogeniseret jord fra Christianshavn i København. Jorden har et indhold af kulbrinter på ca. 300 mg/kg TS. Kulbrinterne er i overvejende grad af naturlig oprindelse. Jorden er fremskaffet gennem jordrensefirmaet RGS 90. Jorden stammer fra Christianshavn, som er begyndt opfyldt fra år 1733. På grunden har der tidligere været pakhuse og havneanlæg. Teksturanalyse af jorden er anført i tabel 6-6.
Tabel 6-6. Teksturanalyse af jorden fra Christianshavn. Med det tilhørende JB-nr kan jorden karakteriseres som angivet i tabel 6-7:
Tabel 6-7. Karakterisering af jorden i henhold til JB-nr. I figur 6-7 ses et GC-FID kromatogram af jorden fra Christianshavn. Figur 6-7. Kromatogram af Christianshavnjord med indhold af naturlige kulbrinter. 6.4.3 ForsøgsplanlægningDer skal laves en sammenligning af totalkulbrinteindholdet i et ekstrakt: før og efter vask, og efter de tre følgende oprensningsmetoder:
6.4.4 Udførelse af forsøgetDer er afvejet 15 prøver af den homogeniserede jord af 60 g i Duranglas. Prøverne er ekstraheret efter den endelige metode og udrystet i 12 timer. Efter endt rystetid er der tappet 5 prøver, kaldet: før vask Herefter er alle 15 ekstrakter vasket med vand. Hele ekstraktet er tappet fra prøverne og overført til centrifugerør. Der er tilsat 10 mL demineraliseret vand til centrifugerøret og røret er rystet i 1 min. Fra centrifugerørene er der tappet 5 prøver, kaldet: efter vask De 15 vaskede ekstrakter opdeles i 3 grupper af 5 prøver, der hver skal oprenses på forskellig måde. Først er ekstraktet tappet fra vandet i centrifugeglassene. Fastfase kolonnerne (5 stk PPL og 5 stk IST) er gjort klar og konditioneret med methanol. Et ekstrakt er påsat hver søjle, som så er elueret ved et flow ca. 10 mL/min. Eluatet er opsamlet i vials klar til GC-kørsel. De sidste 5 prøver tilsættes natriumsulfat og florisil, som beskrevet ISO/DIS 16703. Prøverne rystes 10 min på rystebord, centrifugeres og tappes derefter til vials. Prøverne kaldes henholdvis: efter PPl, efter IST og efter florisil. De 5-dobbeltbestemmelser gange 5 forskellige behandlinger, i alt 25 GC-analyser analyseres på to signaler på GC'en (front og back). Alle vials randomiseres før analyse. 6.4.5 ResultatbehandlingAlle kromatogrammer er integreret som et totalareal samt opdelt i fraktioner. Totalkulbrinter er beregnet overfor et gennemsnit af n-alkanstandarder analyseret først og sidst på sekvensen, hvor pågældende prøve er analyseret. Der er regnet i forhold til intern standard. Resultaterne ses i tabel 6-8.
Tabel 6-8. Resultater for total kulbrinter fra forsøget. Resultaterne er behandlet statistisk. Forudsætningerne for en variansanalyse er testet og i orden. Variansanalysen giver, at der er statistisk belæg for, at de fem metoder er forskellige, jf. figur 6-8. Metodernes usikkerhed ses i figur 6-9.
Figur 6-8. plot af middelværdi [mg/kg TS]. Hvor 1 = Før vask, 2 = Efter vask, 3 = Efter PPL søjle, 4 = Efter IST søjle, 5 = Efter florisil.
Figur 6-9. Fordelingen af residualer for de 5 behandlinger. Hvor 1 = Før vask, 2 = Efter vask, 3 = Efter PPL søjle, 4 = Efter IST søjle, 5 = Efter florisil. Det ses tydeligt, at resultaterne for ekstraktet før vask ligger pænt samlet, mens resultaterne allerede efter vask af ekstraktet bliver mere spredte. For de tre oprensningsmetoder er det tydeligt, at oprensning på IST søjle er den mest reproducerbare. På figur 6-10 ses et eksempel på et kromatogram efter oprensning over florisilsøjle. Ved sammenligning af figur 6-7 med figur 6-10 ses ikke umiddelbart en ændring i fordelingen af enkeltkomponenter. Figur 6-10. Kromatogram efter oprensning på IST-søjle. Af ovenstående resultater kan det konkluderes, at der kan fjernes en del af totalkulbrinteindholdet i ekstraktet på jord fra Christianshavn ved vask og oprensning. Ved nærstudier af datamaterialet kan det dog ses, at dette er en sandhed med begrænsninger. Det konstateredes nemlig allerede ved integration af de interne standarder i prøverne, at de i visse tilfælde svinder meget. I eksemplet i figur 6-11 er arealet af o-terphenyl faldet til under det halve i forhold til den rene pentan med interne standarder, uden at de andre to interne standardarealer har ændret sig væsentligt. Figur 6-11. Kromatogram af ekstraktet efter oprensning med florisil. I tabel 6-9 arbejdes med middelværdier for hvert behandlingsniveau. I tabellen er indsat nominerede værdier for totalareal samt arealet af den interne standard o-terphenyl. Med nomineret menes, at arealerne er divideret med den interne standard i en blindprøve (den pentan IS der er anvendt til ekstraktion) fra den pågældende sekvense, hvor prøven er analyseret. Dette er nødvendigt for at tage højde for forskellen i apparat performance specielt i forhold til front og back signal.
Tabel 6-9. Middelværdier for de enkelte behandlinger, IS = o-terphenyl. Der laves en variansanalyse på de nominerede totalarealer for hvert behandlingsniveau. Se figur 6-12.
Figur 6-12. plot af middelværdier. Hvor 1 = Før vask, 2 = Efter vask, 3 = Efter PPL søjle, 4 = Efter IST søjle, 5 = Efter florisil. Tilsvarende plot for arealerne for den internestandard ses i figur 6-13.
Figur 6-13. plot af middelværdier. Hvor 1 = Før vask, 2 = Efter vask, 3 = Efter PPL søjle, 4 = Efter IST søjle, 5 = Efter florisil. Det ses af tabel 6-9 samt figur 6-12 og 6-13, at det nominerede total areal samt areal for den interne standard efter vask stort set ikke er ændret. Til gengæld falder begge dele kraftigt efter oprensning med PPL eller florisil. 6.4.6 DelkonklusionResultatet tyder på at oprensning med PPL og Florisil ukritisk fjerner kulbrinter uden hensyntagen til polaritet. Altså mistes en del af den reelle forurening, ligeså vel som der fjernes en del af de naturlige kulbrinter. Til gengæld ser det ud til, at oprensning over IST florisilkolonne kan fjerne en lille del af de naturlige kulbrinter uden indflydelse på den interne standard, og dermed mistes sandsynligvis heller ikke den egentlige petrogene forurening. Forsøget baseret på 5 gentagelser viser at vask af ekstraktet med vand ikke har nogen betydning. Af de tre afprøvede oprensningsmetoder er oprensning med IST florisil søjle klart at foretrække frem for de andre, hvor der generelt mistes kulbrinter uanset oprindelse. Oprensning med IST florisil søjle kan sandsynligvis være relevant i særlig problematiske tilfælde, hvor jorden i høj grad er forurenet med polære forbindelser eksempelvis vegetabilsk og eller animalsk olie stammende fra et køkken eller lign. Dette findes dog sjældent i forbindelse med sager om olieforurening i jord, og er ikke afprøvet i projektperioden. En sådan forurening af polær karakter vil dog være let genkendelig i kromatogrammet med karakteristiske brede toppe. Eksempler ses i figur 6-14 og 6-15. Figur 6-14. GC-FID Kromatogram af sojaolie. Figur 6-15. GC-FID kromatogram af olivenolie. 6.5 Konklusion på vask og oprensningDer er ikke fundet signifikante resultater på de jordtyper, der er afprøvet i projektperioden, der viser, at vask og/eller oprensning kan fjerne en del af indholdet af naturlige kulbrinter, uden at der også mistes en del af den reelle petrogene forurening. Dette er selvfølgelig meget afhængig af jordtypen. Det vurderes i forbindelse med bestemmelse af jordforurening, at oprensning med florisil yderst sjældent er relevant, idet polære komponenter som fedtstoffer ikke er et problem i forbindelse med jordforurening. Vask og oprensningsdelen, som der anbefales i ISO/DIS 16703 metoden, stammer fra en tidligere spildevandsmetode; DS/EN ISO 9377-2 som omhandler bestemmelsen af olie i vand. I vand er fjernelsen af fedtstoffer langt mere relevant. Set i relation til dette, er det en forklaring på, hvorfor der er medtaget ekstra vaske og oprensningstrin i ISO/DIS 16703, som ikke har så stor relevans i forbindelse med jordforurening. Det findes således, at disse trin ikke er relevante i forbindelse med bestemmelsen af totalkulbrinter og dermed heller ikke i den nye metode, ISO/DIS 16703-Mod. i forhold til adskillelse af forurening af biogen- og petrogen oprindelse. En anden vigtig betragtning er, at den nye metode, ISO/DIS 16703-Mod. kan anvendes til analyse for flygtige kulbrinter, så her er vask og oprensning udelukket, idet der i forbindelse med disse trin vil ske afdampning på grund af omhældninger med mere i forbindelse med oprensningsprocedurene. Det skal bemærkes, at der i selve metoden indgår et naturligt vaskeled i forbindelse med ekstraktionen, hvor der indledningsvis bliver fjernet polære forbindelser herunder naturlige kulbrinter. Efter udrystningen og dermed ekstraktionen tilsættes 30 ml vand, hvorefter der sker faseseparation, hvor ekstraktet findes i pentanfasen, og de uønskede og eventuelt polære stoffer ligger i vandfasen. I den ny udviklede metode, ISO/DIS 16703-Mod., anbefales det ikke at medtage yderligere vask og oprensning som en del af standardproceduren for analyse af kulbrinter i forurenet jord. Disse trin vil således ikke blive medtaget i afprøvningen og valideringen af den nye metode i kapitel 8 og 9. Den endelige metode, ISO/DIS 16703-Mod. ses i tabel 6-10.
Tabel 6-10. ISO/DIS 16703-Mod. 7 Kvantificeringsmodel7.1 FormålI ISO/DIS 16703 kvantificeres indholdet af kulbrinter i jordprøver overfor en blanding af to rene mineralolier. I "Bestemmelse af olie i jord" /2/ benyttes n-alkaner til kvantificeringen i området benzen til n-alkan C35. Da beregning med n-alkaner ved tidligere præstationsafprøvninger har vist sig at mindske usikkerheden imellem laboratorier, er målet med dette kapitel at vise, at n-alkaner kan anvendes til kvantificering op til og med n-alkan C40 ved både GC-FID og GC-MS. 7.2 FremgangsmådeFor at undersøge de to kvantificeringsmodeller er der udført lineære regressionsanalyser af henholdsvis ISO/DIS 16703 blandingsstandarden og n-alkan blandingen. Til sammenligning er der kvantificeret i området n-alkan C10 til n-alkan C40, da ISO/DIS 16703 kun omfatter dette område. Desuden er der lavet fortyndingsrækker af en række rene højtkogende olieprodukter, som kvantificeres med de to ovennævnte metoder. De to kvantificeringsmodeller er undersøgt på både GC-FID og GC-MS. GC-MS kørslerne er udført ved at opsamle data i SCAN-mode fra m/z 35 og op til m/z 500. 7.3 LinearitetDer er lavet fortyndingsrækker af en n-alkan blandingen samt ISO/DIS 16703 blandingsstandarden. ISO/DIS 16703 blandingsstandarden fremstilles som beskrevet i ISO/DIS 16703. Der afvejes lige mængder af to mineralolier, en gasolie og en tungerekogende olie. Den benyttede tungerekogende olie er Motorolie Shell X-100, bilag 4. Denne blandingsstandard fortyndes til koncentrationsområdet 40 – 8000 mg/L med 7 punkter i området. n-Alkan fortyndingsrækken, er fremstillet udfra en ampul indeholdende lige n-alkaner fra C10 til C40 (beskrevet i afsnit 2.3). Koncentrationsområdet er 0,25 –50 mg/L pr. enkeltkomponent (totalkoncentrationsområdet for alle 16 n-alkaner er 4 – 800 μg/L) med 8 punkter i området. 7.3.1 Lineær regression udført på GC-FID kørslerDen lineære regression udført på fortyndingsrækkerne viser, at de begge kan beskrives ved simple lineær regression (se figur 7-1). For n-alkan standardrækken (beregnet udfra summen af alle n-alkaner i blandingen) er r2 = 99,72 % og for ISO/DIS standardrækken er r2 = 99,99 %. Dette betyder, at modellen beskriver hhv. 99,72 % og 99,99 % af variationen i arealer for de to fortyndingsrækker. Fortyndingsrækkerne lavet udfra n-alkan standarden er desuden undersøgt for de tungtkogende alkaner, de letterekogende n-alkaner, og enkeltkomponenter C10 og C40. Alle de udførte lineære regressioner udviser linearitet.
Figur 7-1. Graferne viser den udførte lineære regression for n-alkan standard (øverst) og ISO/DIS standard (nederst) ved GC-FID. Standardrækkerne udført for ISO/DIS standarden og n-alkan standarden er begge lineære. 7.3.2 Lineær regression udført på GC-MS kørslerDen lineære regression udført på fortyndingsrækkerne (GC-MS) viser, at de begge kan beskrives ved simpel lineær regression, se figur 7-2. For n-alkan standardrækken er r2 =99,54% og for ISO/DIS standardrækken er r2 =99,51%.
Figur 7-2. Graferne viser den udførte lineære regression for n-alkan standard (øverst) og ISO/DIS standard (nederst) ved GC-MS. De tungeste n-alkaner (C36, C38 og C40) viser tegn på, at disse komponenter ikke er lineære, endvidere er responset for disse komponenter ustabilt hen igennem en kørsel på GC-MS. Endeligt har det vist sig umuligt at opnå et tilfredsstillende C40/C20 forhold på de analyserede standarder. Dette skyldes, at det er nødvendigt at sænke ovnprogrammets sluttemperatur til ca. 280°C, for at undgå at kontaminere MS-detektoren med kolonnemateriale. På baggrund af disse erfaringer vurderes det, at brugen af n-alkan standard ikke er velegnet til kvantificering af olieprodukter ved GC-MS. Hvad angår lineariteten af ISO/DIS standarden er resultaterne mere lovende; men her besværliggøres muligheden for at opgive resultaterne i kogepunktsintervaller. Endvidere gør problemet med ustabilitet igennem en sekvens analyseret på GC-MS sig også gældende her. 7.4 Kvantificering af rene olieprodukterFor at verificering af at n-alkaner kan benyttes til kvantificering, fremstilles en række fortyndingsrækker af rene højtkogende olieprodukter, som kvantificeres overfor n-alkan blandingen samt ISO/DIS standarden. De benyttede olieprodukter til denne verificering er listet i bilag 4. Til kvantificeringen af olieprodukter med n-alkan standarden er benyttet alle lige n-alkaner fra C10 til og med C40. Kvantificeringen er endvidere foretaget i fraktionerne C10-C20, C20-C30 og C30-C40. Ved ingen af beregningerne er der korrigeret med intern standard. Resultaterne er præsenteret i figur 7-3. Kvalitetskriteriet for kulbrinter i jord med kogepunktsinterval svarende til motorolie med kulbrinter op til n-alkan C35 er i dag 100 mg/kg TS, hvilket svarer til en koncentration i injektionsopløsning til gaschromatografen på ca. 250 mg/L. Typisk er detektionsgrænsen for dette interval 25 mg/kg TS. Denne koncentration svarer i opløsning til 55 mg/L. Detektionsgrænsen for totalkulbrinter er for det samlede område 32,5 mg/kg TS, hvilket svarer til en koncentration i injektionsopløsningen til gaschromatografen på 78 mg/L. Figur 7-3. Søjlediagrammer, der viser genfindingen af forskellige olier med hhv. n-alkan standard og ISO/DIS standard. I figur 7-3 ses resultaterne fra kvantificeringen fra forskellige fortyndinger af olieprodukter ved GC-FID.
Efter beregningen af disse resultater er det eftervist, at resultatet ikke ændres signifikant ved at udføre beregningen med fraktionerne opdelt på følgende måde; fraktion C10 – C25 (beregnet med C12 + C16 + C20 + C24), fraktion C25 – C35 (beregnet med C28 + C30 + C32 + C34), og fraktion C 35 – C40 (beregnet med C36 + C38). Under kørslen på GC-FID er der så vidt muligt benyttet standarder, hvor responset for C40 er mindst 80 % af responset for C20, således som det er krævet i ISO/DIS 16703 standarden. Beregningen med ISO/DIS standarden er foretaget, som beskevet i ISO/DIS 16703. Hele arealet fra C10 – C40 er integreret og kvantificeret over for den fremstillede ISO/DIS standard. Kvantificering af tungtkogende kulbrinteprodukter som f.eks. stenkulstjære, bitumen og tung fuelolie er efterfølgende blevet undersøgt. Kvantificeringen af denne type produkter blev besværliggjort af produkternes meget lille opløselighed i pentan. Dette bevirkede, at de koncentrationer, der kunne opløses, er tæt på metodens detektionsgrænse. Endvidere har denne type produkt en tendens til at udfælde inden analysen på GC-FID. De bestemmelser der er udført resulterede i en genfinding på 57 – 66 % for henholdsvis tjære og fuelolie. 7.4.1 DelkonklusionResultaterne, der er opnået med de to beregningsmetoder, viser, at begge metoder kan anvendes til kvantificering af højtkogende olieprodukter. For meget højtkogende olieprodukter, der strækker sig udover kogepunktsområdet, kan det ikke forventes at opnå tilstrækkelig god genfinding. Meget tungtkogende olietyper kromatografieres ikke fuldstændigt. Som det fremgår af kapitel 2, GC-betingelser, er der flere parametre ved gaskromatografen, der bevirker, at de tungtkogende kulbrinter ikke kromatografieres, selvom de er i olien. Disse komponenter sidder tilbage i kolonnen, og frigives langsomt som "slæb" eller kolonneblødning, ved de efterfølgende injektioner. Det er svært at undgå med almindelige gaskromatografiske betingelser, som samtidig skal kunne kromatografiere f. eks. let- og mellemkogende olieprodukter som gasolie etc. Det er heller ikke altid muligt at afgøre, om der rent faktisk er tale om "slæb", eller om der er tale om et reelt kulbrinteindhold. Den meget dårlige genfinding ved indhold nær detektionsgrænsen i disse forsøg kan, trods god kromatografi og vedligehold, alligevel skyldes basislinieforskydning pga. slæb. Da der opnås større interlaboratoriepræcision ved anvendelsen af n-alkaner til kvantificering, og der herved er mulighed for at opdele de beregnede resultater i fraktioner, anbefales denne beregningsmetode. 7.5 Gentagne kørsler af rene olieprodukterPå baggrund af at visse af resultaterne i figur 7-3 muligvis udviser systematik i form af, at den ene beregningsmodel genfinder mere end den anden, og der kunne opstå mistanke om, hvorvidt der er fejl i kalibreringen, er der udvalgt to olietyper som er analyseret over 6-9 forskellige dage. Herved skal det undersøges, hvor stor betydning kalibreringen har på de opnåede resultater. De to valgte olietyper er Shell Donax TA og Shell X-100 Motor oil. Disse to olier er hver analyseret i to koncentrationer, og kvantificeret overfor hhv. ISO/DIS standarden og n-alkanstandarden.
Tabel 7-1. Resultater opnået med kvantificering med ISO/DIS standard hhv. n-alkanstandard. Som det fremgår af tabel 7-1, ses der ikke signifikant forskel på, hvorvidt der anvendes ISO/DIS standard eller n-alkaner til beregning af indholdet i de to olietyper. Dette fremgår af de beregnede genfindingsprocenter ved hhv. den ene metode sammenlignet med den anden, sammenholdt med spredningen, der er på resultaterne, her udtrykt ved RSD%. Den forholdsmæssig lave genfindingsprocent af motorolien må tilskrives andre faktorer end kalibreringen. 7.5.1 Verificering af ISO/DIS standardenSom verifikation af den af laboratoriet fremstillede ISO/DIS standard er der fremskaffet en ISO/DIS standard (BAM KS 5004) bestilt ved Fachgruppe I.2 i Tyskland. Denne standard er nævnt i ISO/DIS 16703, og er en færdigblandet standard indeholdende lige dele diesel fuel (DK 1037, Deutsche Shell AG)og smøreolie (HVI 50, Deutsche Shell AG). Der er foretaget en sammenligning af beregning af to forskellige typer motoroliers indhold ved hhv. kalibrering af ISO/DIS standard fremstillet på laboratoriet, ISO/DIS standard fra BAM og n-alkan standard, som tidligere beskrevet. Resultaterne er vist i tabel 7-2.
Tabel 7-6. Resultater af beregning med n-alkan std, ISO/DIS færdigblandet standard (BAM KS 5004) og ISO/DIS standard blandet på laboratoriet. Resultaterne i tabel 7-2 viser, at der ikke er signifikant forskel på, hvorvidt der anvendes laboratoriets fremstillede ISO/DIS standard, ISO/DIS standard fra BAM og n-alkan standard. Den lave genfinding af motorolien må tilskrives andre faktorer en kalibreringsstandarden. 7.5.2 DelkonklusionDet konkluderes, at begge beregningsmodeller kan anvendes til kvantificering af kulbrinter i områder n-alkan C10 til n-alkan C40. 7.6 KonklusionKvantificering af totalkulbrinter bør udføres ved GC-FID, da denne type detektion har vist sig, at give det mest stabile respons, specielt hvad angår de tungerekogende komponenter. GC-MS-scan benyttes efterfølgende til kvantificering af enkeltkomponenter, karakterisering af specifikke petrogene kulbrinter osv. Beregning af totalkulbrinteindholdet kan med fordel foretages med n-alkaner som kalibrant og med opdeling i tre fraktioner. De tre fraktioner er opdelt på følgende måde;
Der er valgt at opdele beregningen i disse fraktioner, da responset fra n-alkanerne har en tendens til at give et noget lavere respons i fraktion C35-C40. Derfor vil beregningen i kun to fraktioner give en fejlestimering af indholdet i fraktion C35-C40. Ved rapportering af resultater er det dog valgt, at resultatet for C25- C40 opgives samlet, da ingen olieprodukter kromatografieres udelukkende i fraktionen C35 til C40. 8 Genfindingsforsøg8.1 FormålDen modificerede analysemetodes (ISO/DIS 16703-c) nøjagtighed, bliver her indledningsvis testet før den endelige validering, ved hjælp af en række tilsætningsforsøg samt analyse af certificeret referencemateriale. 8.2 Afgrænsning/baggrundPå GC-FID er metoden afprøvet i forhold til totalkulbrinter ved spikning af prøver af tre forskellige olier indeholdende et kendt indhold af totalkulbrinter og for BTEX med tilsætning af BTEX standard i kendt koncentration samt ved analyse af certificeret referencemateriale indeholdende indeholdende kulbrinter og BTEX. På GC-MS er metoden afprøvet i forhold til PAH'er og BTEX ved spikning af prøver med kendt indhold af PAH og BTEX i jord samt ved analyse af certificeret referencemateriale indeholdende indeholdende PAH og BTEX. 8.3 Anvendte jordtyperTil genfindingsforsøgene er der anvendt 2 forskellige "rene" jordtyper. Med "rene" menes jord uden olieforurening. Efter test med forskellig jord, blev der fundet frem til to forskellige jordtyper med et lavt indhold af totalkulbrinter. De to "rene" jordtyper; sand og lerjord er vist i figur 8-1 og 8-2. Figur 8-1. Kromatogram af "ren" sandjord. Figur 8-2. Kromatogram af lerjord. Jordene er homogeniseret og ekstraheret efter den nye metode (ISO/DIS 16703-c), hvorefter indholdet i jordene er beregnet. I tabel 8-1 ses resultater af homogenitetstest og undersøgelse af jordens baggrundsindhold af kulbrinter bestemt ved GC-FID.
Tabel 8-1. Totalkulbrinteindholdet i de to jordtyper. Disse to jordtyper er anvendt i de efterfølgende forsøg:
8.4 Genfinding af totalkulbrinter på GC-FID8.4.1 Forsøgsplanlægning/udførelseTil forsøgene er der anvendt tre typer af olie; gasolie, motor-smøreolie samt fuelolie. 8.4.1.1 Motor- og gasolie. Oliekoncentrationer:
Der er lavet 6-dobbeltbestemmelser. Behandlinger = 2 jordtyper * 2 olier *4 konc. * 6 gentagelser = 96 behandlinger, minus 12 (da disse er gentagelser uden tilsætning) dvs. 84 behandlinger.
Tabel 8-2. Oversigt over de 84 behandlinger. 84 sand- og jordprøver er spiket og afvejet med forskellig oliekoncentration efter tabel 8-2. Prøverne er ekstraheret efter den modificerede metode /ISO/DIS 16703-c), og der er foretaget kvantificering. Alle prøverne er analyseret over 3 dage på to signaler. Der er randomiseret over alle vials. 8.4.1.2 Fuelolie Der er afvejet fire sandprøver og fire lerjordprøver af ca. 60 g. Prøverne er spiket med fuelolie på 500 mg/kg TS. Prøverne ekstraheres efter den modificerede metode (ISO/DIS 16703-c) og rystes i 12 timer. Herefter er den organiske fase aftappet over i vials, og prøverne er analyseret i randomiseret rækkefølge på GC-FID. 8.4.2 ResultatbehandlingDer er beregnet totalkulbrinteindhold udfra n-alkan standard der er analyseret på pågældende sekvens. I beregningerne er der taget højde for intern standard. Kromatogrammerne er integreret som en totalaresalsum. Eksempler på kromatogrammer for sand eller jord med spikede indhold af de tre olier ses i figur 8-3 til 8-5. Figur 8-3. Kromatogram af sand spiket med gasolie 1000 mg/kg TS. Figur 8-4. Kromatogram af lerjord spiket med motorolie 500 mg/kg TS. Figur 8-5. Kromatogram af sand spiket med fuelolie 500 mg/kg TS. Middelværdierne samt genfindingen for de spikede prøver ses i tabel 8-3 og 8-4, hvor det er middelværdier af resultaterne fra både sand og lerjord, der er listet.
Tabel 8-3. Middelværdier og genfindingsprocenter bestemt i både sand og lerjord.
Tabel 8-4. Genfinding af fuelolie Genfindingen for motorolie tilsat til sand og lerjord ligger på mellem 99% – 126 %. For tilsætning af gasolie til sand og lerjord er genfindingen mellem 111-157 %. Der er beregnet en genfinding på 88 % for lerjord og 78 % for sand ved tilsætning af fuelolie udfra de fire bestemmelser. De relative standardafvigelser er mindre end 10%. Generelt anses genfindinger på mellem 80-120 % for tilfredsstillende. 8.4.3 DelkonklusionGenfindingen for motorolie er tilfredstillende. Genfindingen for gasolie ligger for højt specielt på den lave koncentration. Dette bliver yderligere undersøgt ved metodevalideringen. 8.5 Genfinding af PAH på GC-MS8.5.1 Forsøgsplanlægning/udførelseTil genfindingsforsøget med tilsætning af PAH er der anvendt de samme to jordtyper, homogeniseret lerjord samt sand. Indholdet af PAH'er i sandet før spikning ligger under detektionsgrænsen. Indholdet af PAH i lerjorden ligger på maksimum 0,047 mg/kg TS pr. enkeltkomponent, som der vil blive korrigeret for under beregningerne af det spikede indhold. Der er afvejet fire sand- og fire lerjordprøver af 60 g som er spiket med 16 EPA PAH'er til en koncentration på 3,33 mg/kg TS pr. komponent. Prøverne er ekstraheret efter den nye metode (ISO/DIS 16703-c) i 12 timer og er analyseret ved GC-MS i SIM-mode. 8.5.2 ResultatbehandlingPAH'erne er identificeret og integreret udfra targetion, kvalificeringsion samt retentionstid. Indholdet af PAH i prøven er beregnet overfor en PAH-standard analyseret på pågældende sekvens. Indholdet er beregnet i forhold til den intern standard o-terphenyl, som har været anvendt under kørsel på GC-FID. Indholdet i jordprøverne er korrigeret for baggrundsindhold. I figur 8-6 ses et eksempel på et kromatogram med PAH'er i sand. Figur 8-6. Eksempel på kromatogram af PAH'er i sand analyseret på GC- MS. Nedenfor i tabel 8-5 og 8-6 ses middelværdier og genfindingsprocent af de fire gentagelser for hver jordtype.
Tabel 8-5. Resultaterne fra genfinding af PAH i lerjord.
Tabel 8-6. Resultaterne fra genfinding af PAH i sand. Genfindingen for lerjord ligger på 76-109 % og for sand mellem 72-98 %. Genfindingen i lerjord er acceptabel, mens den for sand er lidt lav. 8.5.3 DelkonklusionGenfindingen af de fleste af PAH'erne er acceptabel, men de ligger dog generelt lidt for lavt. Specielt for de tunge PAH'er i sand er genfindingen ikke tilfredstillende. Det formodes, at genfindingen kan forbedres til et acceptabelt niveau ved at anvende isotopmærkedePAH'er som interne standarder. Dette vil blive afprøvet under valideringen af den nye metode. Den relative standardafvigelse ligger under 10 %, hvilket er tilfredsstillende. 8.6 Genfinding af BTEX på GC-FID og GC-MS8.6.1 Forsøgsplanlægning/udførelseTilsætningen af BTEX'er er foretaget sideløbende med tilsætningen af PAH'er, da BTEX'erne er tilsat til de samme prøver. Til forsøget er der anvendt to jordtyper, homogeniseret lerjord samt sand. Indholdet af BTEX'er før spikning ligger under detektionsgrænsen. Der er afvejet prøver af 60 g, som er spiket med BTEX til en teoretisk koncentration på 3,33 mg/kg TS pr. komponent. Prøverne er ekstraheret efter den nye metode i 12 timer. Ekstraktet er tappet i dobbelt antal vials og benyttes til både GC-FID og GC-MS. Prøverne er analyseret ved GC-MS i SIM-mode samt ved GC-FID. 8.6.2 Resultatbehandling8.6.2.1 GC-FID Figur 8-7. Kromatogram af BTEX'er i sand på GC-FID. Middelværdierne og genfindingsprocenter af resultaterne for alle bestemmelser er opgivet i tabel 8-7 samt er illustreret i figur 8-8.
Tabel 8-7. Resultater for genfinding af BTEX på GC-FID. Figur 8-8. Søjlediagram, der viser genfindingen af BTEX på GC-FID. Det ses af resultaterne, at genfindingen for TEX er tilfredsstillende, mens genfindingen for benzen ligger for lavt. Det vurderes, at den lave genfinding for benzen tildels skyldes, at den hurtigt fordamper, når stamopløsning tilsættes ned i prøven. Dette vil bliver undersøgt yderligere under valideringen, hvor der vil blive fokuseret mere på at undgå fordampning ved spikning. Den relative standardafvigelse ligger under 6 %, hvilket er flot for BTEX bestemt ved GC-FID. 8.6.2.2 GC-MS Figur 8-9. Eksempel på kromatogram af BTEX i lerjord ved GC-MS Middelværdierne af resultaterne for alle bestemmelser er opgivet i tabel 8-8.
Tabel 8-8. Resultater for genfinding af BTEX på GC-MS. Resultaterne er også illustreret i figur 8-10.
Figur 8-10. Søjlediagram, der viser genfindingen af BTEX på GC-MS. Det ses af resultaterne at genfindingen for TEX er tilfredsstillende. Genfindingen for benzen ligger igen lidt lavt, specielt for sand, som dog er bedre end ved GC-FID. Den relative standardafvigelse ligger under 6%, bortset fra toluen i sand (12%). 8.6.3 DelkonklusionGenfindinger er tilfredsstillende for TEX, mens det er nødvendigt at se yderligere på benzen under valideringen. Standardafvigelserne for BTEX ser ud fra dette forsøg både ved GC-FID og GC-MS tilfredsstillende ud. 8.7 Genfinding af totalkulbrinteindhold i referencemateriale8.7.1 Forsøgsplanlægning/udførelseEt certificeret referencemateriale (T.P.H [9]; mineraloil in soil) er ekstraheret og analyseret ved GC-FID. Referencematerialet er fra Resource Technology Corporation, Katalog nr.: PR 9583. Ekstraktet af referencemateriale analyseres over seks forskellige dage og analysesekvenser. Der er lavet en afvejning på ca. 20 g prøve. Prøven er ekstraheret efter den nye analysemetode og udrystet i 12 timer. Hele ekstraktet er tappet i vials med indsatser. Der blev 16 vials, som er analyseret på tilfældige sekvenser. 8.7.2 ResultatbehandlingPrøverne er beregnet i forhold til den standard, der er analyseret på den pågældende sekvens. Der er korrigeret i forhold til intern standard. I figur 8-11 ses et eksempel på et kromatogram af prøven. Figur 8-11. Kromatogram af referencematerialet T.P.H. Ved beregning af resultaterne over de seks dage er middelværdien for de 16 bestemmelser beregnet og ses i tabel 8-9, hvor også værdierne fra referencematerialets certifikat er opgivet til sammenligning. Der er oplyst på analysecertifikatet, at det certificerede materiale er analyseret på GC-FID med 42 gentagelser.
Tabel 8-9. Sammenligning af værdier for referencematerialets certifikat og forsøgets resultater. 8.7.3 DelkonklusionDen nye analysemetode giver tilfredsstillende resultater for total kulbrinter på referencematerialet T.P.H. (mineral oil in soil). 8.8 Genfinding af PAH i referencemateriale8.8.1 Forsøgsplanlægning/udførelseMetoden er afprøvet i forhold til bestemmelse af PAH i referencemateriale. Der er anvendt et referencemateriale fra Ressource Technology Corporation med Katalog nr.: CRM 104-100 Lot. nr.: CR104. Materialet er certificeret for en lang række af PAH'erne. Der er også opgivet vejledende værdier for andre PAH-forbindelser, som ikke er cerificeret. Værdier er opgivet i tabel 8-10, ikke certificerede med ( ) omkring.
Tabel 8-10. Værdier fra referencecertifikatet. * = sum af benzo(b)fluoranthen og benzo(k)fluorantehen. Der afvejes fire prøver á ca. 50 g referencemateriale. Prøverne er ekstaheret efter den nye analysemetode. Det var nødvendigt at tilsætte vand til prøverne inden rystning, da referencematerialet er et tørt pulver, der ikke opslemmes helt i de 50 mL (acetone/pentan). Prøverne er rystet i 12 timer og er tappet i GC-vials. 8.8.2 ResultatbehandlingPAH'erne er integreret ud fra targetion, kvalificeringsion samt retentionstid. Indholdet af PAH i prøven er beregnet overfor en PAH-standard analyseret på pågældende sekvens. Indholdet er beregnet i forhold til den interne standard o-terphenyl, som har været anvendt under kørsel på GC-FID. I tabel 8-11 ses middelværdi og genfindingsprocent, som er beregnet ud fra den fundne middelværdi og sammenholdt med referenceværdien.
Tabel 8-11. Middelværdier af de fire bestemmelser samt genfindingsprocent. 8.8.3 DelkonklusionGenfindingen ligger fra 57 – 120 %. Specielt for de tunge PAH'er er resultatet ikke tilfredsstillende. Det vurderes, at resultatet vil blive bedre ved brug af isotopmærkede PAH'er som interne standarder. Dette er afprøvet i et ekstra forsøg. 8.9 Ekstra forsøg PAH-referencemateriale8.9.1 FormålDer er for de tunge PAH'er ikke er fundet tilfredsstillenderesultater for referencematerialet, når o-terphenyl benyttes som intern standard. Der er derfor udført dette ekstra forsøg, hvor der anvendes isotopmærkede PAH'er som interne standarder. 8.9.2 Forsøgsplanlægning/udførelseDer analyseres med 3-fold bestemmelse på referencematerialet CRM 104-100 som er beskrevet i afsnit 8.9.1. Den eneste forskel, der er i dette forsøg, er at der tilsættes isotopmærkede interne standarder til pentan inden ekstraktion. De udvalgte interne standarder er phenanthren-d10, fluoranthen-d10 og benzo(a)pyren-d12. Prøverne er analyseret som forsøget i afsnit 8.9, dog indføres de isotopmærkede internestandarder i quantificeringen på GC-MS. I tabel 8-12 kan det ses hvilke PAH'er der er kvantificeret overfor hvilke interne standarder.
Tabel 8-12. interne standarder PAH'erne er beregnet overfor. Indholdet i prøverne er beregnet overfor en PAH-standard med koncentrationen 5 mg/L pr. komponent. 8.9.3 ResultaterReferenceværdier for det anvendte materiale er opgivet i afsnit 8.9.2, og i tabel 8-13 sammenholdt med de fundne middelværdier for de tre bestemmelser med disse.
Tabel 8-13. Middelværdier og genfindingsprocent i forhold til referenceværdi. Genfindingen for specielt de højerekogende PAH'er er blevet tilfredsstillende, 71 – 121%, ved brug af de isotopmærkede interne standarder. Genfindingen af naphtalen er ikke tilfredsstillende. Dette skyldes ikke de interne standarder, men nærmere at ekstrakterne ved et uheld kom til at stå i autosampleren på apparatet i et helt døgn før analyse pga. at apparatet var gået i stå (plugger error). Genfindingen af naphtalen lå i det tidligere forsøg jf. afsnit 8.9.3 på 86%, hvilket er helt tilfredsstillende, og det må formodes at dette er et bedre udtryk for metodens kunnen. 8.9.4 DelkonklusionGenerelt ligger genfindingerne for PAH'erne mellem 70,9 – 121,4% [10] med en middelværdi på 91,4%, hvilket må konkluderes at være aldeles tilfredsstillende. 8.10 KonklusionDet konkluderes, at den nye analysemetode giver tilfredsstillende genfinding på total kulbrinter til og med n-alkan C40. Indholdet i referencematerialet er genfundet tilfredsstillende, ligesom ved spikning med motorolie og fuelolie. Ved spikning med gasolie er genfindingen for høj, og dette undersøges nærmere under valideringen. Der skal selvfølgelig en fuld validering af analysemetoden til at afgøre, i hvilket måleområde metoden kan anvendes, samt hvilken usikkerhed der skal tillægges resultater i forskellige niveauer. Den nye analysemetode bliver i resten af rapporten således omtalt som ISO/DIS 16703-mod. Det vurderes ud fra spikningsforsøget med BTEX, at metoden kan anvendes til bestemmelse af BTEX ved GC-FID og GC-MS med tilfredsstillende resultat. Der er foretaget en fuld validering af bestemmelse af BTEX med den nye metode. Når der ses på begge de udførte forsøg med PAH referencemateriale i afsnit 8.9 og 8.10, producerer metoden tilfredsstillende resultater for alle 16 PAH'er ved at der anvendes de isotopmærkede PAH'er som interne standarder. Disse er også anvendt under valideringen jf. afsnit 9. 9 Validering af metoden9.1 FormålSom en del af projektet, med at videreudvikle en velegnet metode til analyse af jordprøver med indhold af højerekogende oliefraktioner, er der foretaget en validering af denne videreudviklede analysemetode. Denne valideringsrapport indeholder en validering af den videreudviklede analysemetode; Optimeret "ISO/DIS 16703 Soil Quality – Determination of Mineral Oil Content by Gas Chromatography – 2001 February," som er beskrevet i afsnit 5. Metodevalideringens formål er at dokumentere en afsluttet indkøring af den modificerede metode til analyse af totalkulbrinter i jordprøver. Metoden omfatter analyse på både GC-FID og GC-MS, således at der er analyseret for det totale kulbrinteindhold fra benzen til og med n-alkan C40, inklusiv indholdet af BTEX'er, samt analyse af indholdet af 16 relevante polycycliske aromatiske hydrocarboner (PAH-er). Der er således foretaget en validering af analysemetoden, herunder kvantificering af BTEX'er og PAH'er, i overensstemmelse med "Håndbog i metodevalidering"/6/. Valideringen er foretaget ved GC-FID for indholdet af total kulbrinter samt ved GC-MS for BTEX-indholdet og for indholdet af de 16 PAH-er. 9.1.1 DefinitionerTotalkulbrinter er defineret som summen af kulbrinter fra benzen til og med n-alkan C40, med opdeling i fraktionerne;
Hvor 3. og 4. fraktion beregnes hver for sig over for hver deres respektive n-alkaner for derefter at blive lagt sammen og angivet samlet. Grunden til denne sammenlægning er, at der ikke er olieprodukter, der ligger alene i 4. fraktion (C35-C40). BTEX er en samlet betegnelse for følgende stoffer; benzen, toluen, ethylbenzen, m- og p-xylen samt o-xylen. I Danmark er det i dag normalt at analysere for MST 6 PAH'er: Fluoranthen, benzo[b+j+k]fluoranthener, benzo[a]pyren, indeno[1,2,3-cd]pyren og dibenz[a,h]anthracen. Der er imidlertid også interesse i alle 16 EPA-PAH'er i visse miljøundersøgelser. Metoden valideres derfor med hensyn til alle disse PAH'er. De 16 PAH-er er følgende; naphthalen, acenaphtylen, acenaphten, fluoren, phenanthren, anthracen, fluoranthen, pyren, benz(a)anthrazen, chrysen (triphenylen), benzo(b)fluoranthen, benzo(j)fluoranthen, benzo(k)fluoranthen, herefter kaldt benzo[bjk]fluoranthener, benzo(a)pyren, indeno(1,2,3-cd)pyren, dibenz[ah]anthracen og benzo[ghi]perylen. 9.2 AnalysemetodeDen nyudviklede metode, som er valideret, har taget udgangspunkt i, "ISO/DIS 16703 Soil Quality – Determination og Mineral Oil Content by Gas Chromatography – 2001 February" /1/. Denne metode omhandler bestemmelse af olie i jord ved ekstraktion med acetone + heptan, florisiloprensning samt anvendelse af GC-FID til bestemmelse af kulbrinter fra C10 til C40. 9.2.1 MetodeprincipDen nye metode, "ISO/DIS 16703-Mod." er optimeret og videreudviklet til at opfylde de danske krav ved miljøundersøgelser og er forbedret således, at det er muligt at analysere totalkulbrinteindholdet i området fra benzen til og med n-alkan C40 med tilfredsstillende resultat. Ekstraktet fra samme prøveoparbejdning kan anvendes til bestemmelse af BTEX' er i prøven enten på GC-FID eller GC-MS-SIM. Desuden kan ved GC-MS-SIM på samme ekstrakt bestemmes de 16 EPA-PAH-forbindelser. Analyse af BTEX og PAH ved GC-MS kan udføres på samme ekstrakt. Den videreudviklede metode anvendes også til differentiering mellem kulbrinter af petrogen- og biogen oprindelse ved GC-MS kvantificering i SCAN-mode. I denne valideringsrapport er det ikke afprøvet, om der kan analyseres for chlorerede kulbrinter i pentanekstraktet fra den nye metode, men det er sandsynligt, at der kan udvikles en metode til bestemmelse af de chlorerede kulbrinter ved GC-ECD. Grundprincippet i denne nye optimerede metode, "ISO/DIS 16703-Mod." er; En kendt mængde af en jordprøve ekstraheres ved mekanisk rystning i 12-16 timer med acetone og pentan, hvorefter der tilsættes vand og det organiske lag separeres. En mængde af dette ekstrakt analyseres ved kapillar gaskromatografi med anvendelse af flammeionisationsdetektion, GC-FID eller massespektrometrisk detektion, GC-MS. Ekstraktet analyseres ved GC-FID til bestemmelse af totalkulbrinter og ved GC-MS til bestemmelse af PAH forbindelser, BTEX'er og særlige biomarkører. Den fuldstændige analyseforskrift for den nye metode, "ISO/DIS 16703-Mod." findes i appendiks A, Analyseforskrift. 9.2.2 MetodekravUd fra sammenligning med nuværende analysemetoder /2/ til bestemmelse af olier i jord er det forventeligt at resultaterne for metodevalideringen for "ISO/DIS 16703-Mod." lever op til nedenstående krav jf. tabel 9-1.
Tabel 9-1 Metodekrav til "ISO/DIS 16703-Mod." 9.2.3 ValideringsplanSelve valideringsforsøgene er foretaget som tilsætningsforsøg over to gange seks dage med hver 6 koncentrationsniveauer. Seks dage, hvor der er tilsat 6 forskellige koncentrationer af totalkulbrinter og seks dage hvor der er tilsat 6 forskellige koncentrationer af PAH og BTEX. Se bilag 5, for skema over valideringsplan. Desuden er der foretaget særskilte analyser i forbindelse med bestemmelse af detektionsgrænsen af totalkulbrinter. Valideringsrapporten består af en række delforsøg, som skal beskrive den endelige metode. Følgende emner er undersøgt;
9.2.4 Anvendte jordtyperDe jordtyper, der er anvendt til valideringsforsøgene, stammer fra aktuelle petrogene forureningslokationer, hvor to jordtyper er erhvervet igennem jordrensningsfirmaet Dansk Jordrens i Vemmelev samt en havekompost fra Jydske Haveselskab. Til bestemmelse af totalkulbrinter er der anvendt en forurenet blandejord, der består af lige dele kompost (naturlige kulbrinter), og jord forurenet med motorolie (petrogene kulbrinter). Desuden er der anvendt almindelig lerjord erhvervet fra en privat have i Bredstrup. På begge disse jordblandinger er der udført en teksturanalyse samt pH-bestemmelse, som kan ses i tabel 9-2. Der er også anvendt almindeligt rent sand til valideringen, men der er ikke foretaget nogen teksturbestemmelser herpå.
Tabel 9- 2. Teksturanalyse af de anvendte jordtyper. Til bestemmelse af BTEX og PAH forbindelser er der anvendt den almindelige lerjord som ovenover, samt sand. Den anvendte forurenede jord er en jord med en gammel motorolie- og tjæreforurening, jf. tabel 9-2. Med det tilhørende JB-nr. kan jordtyperne karakteriseres, som angivet i tabel 9-3.
Tabel 9- 3. Karakterisering af de anvendte jordtyper i henholdt til JB-nr. De anvendte jordtyper er neddelt og blandet, således at de fremstår som homogene jordblandinger. Se afsnit 3.2 for beskrivelse af homogenitetstest og homogenitetsanalyser. Desuden ses kromatogrammerne af disse jordprøver. Jordtypen med den gamle tjæreforurening er homogenitetstestet på samme måde som de øvrige jordtyper. 9.3 MetodespecificitetMetodens specificitet angiver, hvor godt de eftersøgte stoffer kan vurderes at være tilstede i de undersøgte jordprøver uden interferens fra andre stoffer i blandingen. Specificiteten skal med sikkerhed bevise, at det kun er de ønskede stoffer, der er i de udvalgte toppe. Desuden skal man kunne se og adskille markørtoppe/referencetoppe og kunne genkende dem i blandinger. 9.3.1 GC-FIDFor analyser af ekstrakter indeholdende totalkulbrinter på GC-FID kan identifikationen af olietypen være problematisk. Ofte ses blandingsforureninger eller for eksempel delvist nedbrudte forureninger. Problematikken kompliceres af, at der kan være tilstedeværelse af naturlige kulbrinter. Ved "rene" olieforureninger finder identifikationen sted ved at sammenligne kromatogrammerne for prøverne med kromatogrammer for rene olieprodukter. Her sammenlignes blandt andet kogepunktsintervaller samt tilstedeværelsen af pristan og n-alkan C17 samt phytan og n-alkan C18. Der er forudgående foretaget analyser af rene olieprodukter, således at strukturen af kendte olietyper kan identificeres. Eksempelvis ses der i figur 9-1 til 9-4 kromatogrammer af tre typiske olieforureninger samt en kompostjord. Figur 9-1 GC-FID-kromatogram af tjæreforurenet jord. Figur 9-1, viser kogepunktsfordelingen og det høje indhold af PAH'er, i en typisk tjæreforurenet jord. Figur 9-2. GC-FID-kromatogram af gasolieforurenet jord. Kulbrintefordelingen i figur 9-2 svarer til en relativt frisk gasolie, som ved nærmere eftersyn, kan ses af forholdene mellem phytan og C18 samt pristan og C17. De tydelige høje toppe for de lige n-alkaner er karakteristiske for en frisk gasolie. Figur 9-3. GC-FID-kromatogram af motorolieforurenet jord. Indholdet af kulbrinter i den tredje jordtype, som ses på figur 9-3, er overvejende højtkogende forbindelser i ét stort kontinuum, UCM (unresolved complex matter), og ud fra kogepunktsintervallet kan indholdet i overvejende grad karakteriseres som motor/smøreolie eller lignende. Der ses dog også indhold af de højtkogende PAH'er, fluoranthen, pyren m.fl., der ofte ses i forbindelse med forurening i byområder. Det vælges i denne sammenhæng at benævne indholdet som motor/smøreolie, da dette vurderes at være mest sigende for det aktuelle kogepunktsinterval. Figur 9-4. GC-FID-kromatogram af kompostjord. De ekstraherede forbindelser fra kompostjorden på figur 9-4 dækker et forholdsvist bredt kogepunktsinterval. Typisk ses et højt indhold af mange usystematiske enkeltkomponenter, hvilket sjældent ses i forbindelse med petrogene forureningskilder. Størstedelen af de ekstraherede kulbrinter befinder sig i fraktionen C25-C40. Til bestemmelse af om der er biogene kulbrinter i blandingen sammen med de petrogene, er der i projektrapportens del 2 en beskrivelse af en vejledning, som kan følges, til at identificere og fratrække en del af disse bidrag. 9.3.2 GC-MSFor analyser af PAH'er på GC-MS er specificitet et relativt lille problem, da analyse ved GC-MS er en meget sikker metode til identifikation af enkeltkomponenter. Her er det tydeligt, ud fra stoffernes massespektre, at bestemme et stofs identitet samt at få belyst, om der gemmer sig mere end ét stof under toppen i kromatogrammet. I figur 9-5 ses et kromatogram fra GC-MS, med PAH-er i det laveste koncentrationsniveau [11],der er undersøgt under valideringen. Figur 9-5. Kromatogram af tjæreforurenet jord. I figur 9-6 ses et kromatogram fra GC-MS med BTEX'er i det laveste koncentrationsniveau [12], der er undersøgt under valideringen. Figur 9-6. Kromatogram af BTEX'er i laveste koncentration. I bilag 6 findes en liste, hvor det fremgår, hvilke target- og kvalificeringsioner, der er brugt til kvantificeringen. Valget af targetionen og kvalificeringsionen for de enkelte komponenter er foretaget på basis af massespektra fra MS NIST [13] biblioteket samt scankørsler. Til bestemmelse af metodens genfindingsprocenter er der foretaget tilsætningsforsøg, hvor der er spiket med kendte mængder af forbindelser til jordprøver. Dette er med til at vise metodens specificitet. 9.4 RobusthedMetodens robusthed er afhængig af
Prøveudtagning Prøveudtagningen udføres uafhængigt af analyselaboratoriernes virke. Det er dog en kendt sag, at inhomogenitet, specielt i jordprøver ofte kan udgøre en større usikkerhed på det endelige resultat end selve analysen. I dag er der kommet mere fokus på prøvetagningsstrategier og -procedurer, idet disse er helt essentielle for resultatet. Prøvebehandling i felten (stikprøver eller blandingsprøver), opbevaring, emballage og transporttid er væsentlige parametre, og laboratorierne bør inddrages som rådgivere i forbindelse med at sikre disse aspekter. Ekstraktion Det ligger i metoden, at der skal ekstraheres i 12-16 timer, således at man er sikker på, at der er opnået ekstraktionsligevægt. I forbindelse med metodeudviklingen er der testet forskellige ekstraktionsmetoder med henblik på at dokumentere robustheden og derved sikre sammenlignelige resultater, jf. afsnit 5. Disse resultater har vist, at rystetiden har betydning for analyseresultatet og at resultaterne er robuste ved udrystning på mere end 12 timer. Oprensning Der foretages generelt ingen særlige foranstaltninger til oprensning på almindelige prøver ud over adskillelse af faserne med vand. Men er der mistanke om større mængder polære komponenter i prøverne, kan der eventuelt oprenses med vand og IST florisilsøjle. Kromatografisk performance. Kromatografering af kulbrinter op til n-alkan C40 stiller store krav til det anvendte udstyr især for de tungtkogende kulbrinter. Det er nødvendigt med en stor grad af vedligeholdelse af GC'en, specielt injektionsporten. Vedligeholdelsen indebærer regelmæssige liner-, nål- og septaskift samt rensning eller skift af guldplade. Desuden afskæring eller skift af kolonnen. Under projektforløbet er kravene for forholdet mellem C40 og C20 større end 80%, fra den oprindelige metode, blevet fulgt. Dette kan ofte svære svært at opretholde igennem en hel analysesekvens. Det er under projektforløbet vurderet, dog på et spinkelt grundlag, at en performance for C40/C20 > 60 % også producerer tilfredsstillende resultater, se afsnit 7.6.2. 9.5 Linearitet9.5.1 FormålMetodens linearitet beskriver metodens evne til at frembringe et signal, som er proportionalt med koncentrationen af det givne stof. Linearitet bestemmes primært ved analyse af en række standarder, som dækker hele det område, hvor der formodes at være linearitet. For at se om der er proportionalitet mellem respons og koncentration kromatograferes en række standarder med stigende koncentration og derefter udmåles top-arealerne. Hvis en grafisk afbildning af den fundne respons som funktion af den tilsatte koncentrationen giver en ret linie, er der proportionalitet. Hvis kurven begynder at krumme ved høje koncentrationer, er man i stand til at fastlægge, hvor høje koncentrationer man kan arbejde med uden at komme ud over det lineære område /6/. Lineariteten for totalkulbrinter er illustreret ved linearitetskurver for et rent olieprodukt i forskellige fortyndinger samt for olieforurenede jordprøver, der er spiket med olie i forskellige koncentrationer. Lineariteten af BTEX og de 16 PAH forbindelser er vist ved linearitetskurver for jordprøver, der er spiket med BTEX'er og PAH'er i forskellige koncentrationer. 9.5.2 Detektor- og metodelinearitet for kulbrinter på GC-FID9.5.2.1 Detektorlinearitet Til bestemmelsen af detektorlineariteten er der anvendt et rent olieprodukt, en tungtkogende motorolie (Motorolie Shell X-100). Heraf er der lavet en fortyndingsrække med et koncentrationsområde fra 16 – 8000 mg/L. Disse prøver er analyseret på GC-FID med 7 punkter i området. Der er foretaget lineær regression på resultaterne, og i tabel 9-4 ses det lineære område, liniens hældning, korrelationskoefficienterne og forklaringsgraden for den rene olie for hver fraktion samt det samlede totalkulbrinteindhold .
Tabel 9-4. Korrelationskoefficienter for oliefraktioner ud fra fortyndingsrække. Heraf ses, at korrelationskoefficienten, som beskriver, hvor godt de anvendte punkter ligger på regressionslinien, er yderst tilfredsstillende for den rene olie. Yderligere ses det, at den anvendte model beskriver 99,98 % af punktvariationen, der kan forklares i koncentrationsændringerne for den rene olie. Der gælder detektorlinearitet for totalkulbrinter i ren olieblanding fra 0 – 8000 mg/kg TS. Lineariteten for fortyndingsrækken af motorolien ses i figur 9-7.
Figur 9-7. Detektorlinearitetskurve for fortyndingsrække af motorolie. 9.5.2.2 Metodelinearitet Til bestemmelse af metodelineariteten for det totale indhold af kulbrinter er der anvendt rent sand, muldjord samt motorolieforurenet jord. Disse jorde er spiket med motorolie i forskellige koncentrationer, således at der er opnået en "fortyndingsrække" bestående af jordprøver med forskellige koncentrationer af kulbrinter. De anvendte jordtyper, som er beskrevet i afsnit 3.2.3, er alle blevet homogeniseret således, at indholdet af totalkulbrinter ligger inden for en usikkerhed på cirka 6-10 %. Indholdet af kulbrinter i muldjorden har i forskellige forsøg vist sig at ligge på mellem 102 og 110 mg /kg TS. Det rene sand har kun et indhold på 5-10 mg kulbrinter per kg TS, så det er så lavt, at det netop er svært at bestemme præcist, da det ligger under detektionsgrænsen. Se resultater fra homogenitetsforsøg i afsnit 8.3. Indholdet af den motorolieforurenede jord er bestemt ud fra middelværdien af de analyserede prøver. I tabel 9-5 ses indholdet i de tre jordtyper samt de forventede koncentrationer, efter at der er spiket med motorolie.
Tabel 9-5. Totalt kulbrinteindhold Til at bestemme lineariteten i måleområdet er der blandet 6 forskellige jordblandinger, således at der er koncentrationer i området ca. 100 til 5000 mg totalkulbrinter pr. kg tørstof. Der er foretaget dobbeltbestemmelser, men et af resultaterne på henholdsvis niveau 1 og 6 er udgået pga. uregelmæssig kromatografering. Der er foretaget lineær regression på resultaterne, og i tabel 9-6 ses det lineære område, liniens hældning, korrelationskoefficienterne og forklaringsgraden for ekstrakterne af den motorolieforurenede jord for hver fraktion og det samlede totalkulbrinteindhold .
Tabel 9-6. Korrelationskoefficienter for totalkulbrinteindholdet i jordprøver. Af den lineære regression ses det, at korrelationskoefficienten, som beskriver hvor godt de anvendte data ligger på regressionslinien er yderst tilfredsstillende for jordprøverne. Yderligere ses det, at den anvendte model beskriver 99,99 % af de variationer, der er i respons koncentrationen, der kan forklares i koncentrationsændringerne for det totale kulbrinteindhold i jordprøverne. Lineariteten for fortyndingsrækken af jordprøverne ses i figur 9-8. Figur 9-8. Metodelinearitet for totalkulbrinter med ISO/DIS-mod . Heraf ses, at der er liniaritet for bestemmelsen af total kulbrinter fra omkring 0 til 5000 mg/kg TS. Linien er ikke tvunget gennem (0,0). Da liniens skæring ligger meget tæt på nul, antages det, at der er liniaritet ned til detektionsgrænsen. 9.5.3 Metodelinearitet for BTEX og PAH på GC-MS9.5.3.1 Formål 9.5.3.2 Fremgangsmåde
Da muldjorden indeholder en lille mængde af PAH-er, er denne jord anvendt som laveste niveau, hvor fluoranthen forekommer i en middelkoncentration på 0,0474 mg/kg TS. Den motorolie- og tjæreforurenede jord indeholder også PAH'er, men i en noget større koncentration, og er anvendt til niveau 3 med en koncentration af fluoranthen på ca. 0,60 mg/kg TS. Koncentrationen af de øvrige PAH'er i disse to jordtyper ses i tabel 9-7. Det rene sand indeholdt ingen PAH'er før spiking.
Tabel 9- 7. Koncentration af PAH forbindelser. Ingen af jordtyperne indeholder BTEX'er, før disse bliver tilsat ved spiking. I tabel 9-8 ses de anvendte jordtyper og de spikede koncentrationer af BTEX'er og PAH'er , hvor PAH'erne er repræsenteret af fluoranthen. Der er fremstillet 6 forskellige koncentrationsniveauer. For BTEX er det i området fra 0,033 – 3,33 mg/kg TS [14] og for fluoranthen er det i området fra 0,047 – 3,33 mg/kg TS.
Tabel 9-8. koncentrationer af PAH og BTEX. 9.5.3.3 Linearitet på GC-MS Koncentrationen af PAH og BTEX er beregnet ud fra arealet af enkeltkomponenten beregnet overfor en PAH eller BTEX standard analyseret på samme sekvens. Til beregningen af BTEX er monobrombenzen anvendt som intern standard, mens PAH'erne er beregnet overfor de samme tre isotopmærkede [15] PAH'er jf. afsnit 8.10.2. Der er herefter foretaget lineær regression på resultaterne, og i tabel 9-9 og tabel 9-10 ses det lineære område, liniens hældning, korrelationskoefficienterne og forklaringsgraden for hver komponent, for henholdsvis BTEX'er og PAH'er.
Tabel 9-9. Korrelationskoefficienter for BTEX'er.
Tabel 9-10. Korrelationskoefficienter for PAH'er. Heraf ses, at korrelationskoefficienten, som beskriver, hvor godt de anvendte data ligger på regressionslinien, er tilfredsstillende for hver af de undersøgte forbindelser. Yderligere ses det, at den anvendte model beskriver mellem 99,09 % og 99,94 % af de variationer, der er i responskoncentrationen, og mellem 99,48 % og 99,88 for PAH-forbindelserne. Forklaringsgraden og korrelationskoefficienterne anses således for at være acceptable i de angivne måleområder, og der gælder metodelinearitet for BTEX'erne i koncentrationsområdet fra 0 – 3,33 mg/kg TS og metodelinearitet for PAH-forbindelserne i koncentrationsområdet fra 0 – 3,33 mg/kg TS. Lineariteten for indholdet af benzen og fluoranthen er vist i linearitetsplot i figur 9-9 og figur 9-10. Linearitetsplot for de øvrige BTEX'er og PAH'er udviser lignende linearitet.
Figur 9-9. Linearitetskurve for benzen.
Figur 9-10. Linearitetskurve for fluoranthen 9.5.4 KonklusionLinearitetsanalyserne viser, at der er fin linearitet i de undersøgte koncentrationsområder for både GC-FID og GC-MS, hvad angår de undersøgte parametre. Området, hvor der er metodeliniaritet, er for totalkulbrinteindhold i et ekstrakt af en motorolieforurenet jordprøve 0 – 5000 mg/kg TS. Der er metodeliniaritet for BTEX indhold i området 0 – 3,33 mg/kg TS. PAH indholdet er metodeliniært i området 0 – 3,33 mg/kg TS. 9.6 Detektionsgrænse9.6.1 FormålDet er målet at kunne overholde en forventet detektionsgrænse (DL), der opfylder de danske myndigheders krav om højest at komme op på 1/10 af acceptkriterier i jorden, f.eks. jordkvalitetskriterier, JKK /31/ og kriterierne ved klasseinddeling af jorden /5/, heunder klasse 1, ren jord som er et indhold på maksimum100 mg/kg TS. Dette svarer til, at der skal kunne opnås en detektionsgrænse for det totale kulbrinteindhold på ca. 10 mg/kg TS for fraktion C6-C35. Dog er det forventeligt, at DL for det totale kulbrinteindhold for den videreudviklede analysemetode, der her validers, bliver højere end de ovennævnte kriterier, da denne analysemetode måler det totale kulbrinteindhold fra C6 og helt op til C40. For BTEX'er og PAH'er er det målet at kunne være mindre end 1/10 af acceptkriterier og opnå de i dag anvendte detektionsgrænser på henholdsvis 0,01 mg/kg TS og 0,005 mg/kg TS. 9.6.2 Detektionsgrænse for kulbrinteindhold på GC-FIDDetektionsgrænsen er bestemt efter Miljøstyrelsens definition for miljømålinger, hvor detektionsgrænsen er den koncentration, som giver 99,5 % sandsynlighed for ikke at få falsk positivt resultat /6/. Detektionsgrænsen er således bestemt ved beregning på standardafvigelsen af 10 blindværdibestemmelser med en koncentration tæt på, det vil sige højst 5 gange over den forventede detektionsgrænse. Til forsøget er der således anvendt en naturlig prøve med en koncentration på ca. 100 mg/kg TS, da en naturlig prøve giver en større standardafvigelse og et mere sandt billede af detektionsgrænsen, end hvis der havde været anvendt en syntetisk prøve. Forsøget er foretaget under repeterbarhedsomstændigheder. De naturlige jordprøver, der er anvendt til dette detektionsgrænseforsøg, er den samme muldjord, der er anvendt i selve valideringsforsøgene. Den har et total indhold af kulbrinter på ca. 110 mg/kg TS. En beskrivelse af jordprøverne og homogenisering af jordmatrixen findes i afsnit 3.2.2. Prøverne er ekstraheret og analyseret som beskrevet i analysemetoden og detektionsgrænsen (DL) er beregnet udfra spredningen: DL = t0,995 (f) * s DL: Detektionsgrænsen s : Standardafvigelsen inden for samme serie. Analyseresultaterne i form af responsarealer er omregnet til indhold i mg/kg TS. I tabel 9-11 ses de beregnede detektionsgrænser for de enkelte fraktioner af kulbrinter samt de forventede detektionsgrænser, beregnet som 1/10 af acceptkriterierne fra JKK.
Tabel 9-11. Detektionsgrænseberegninger på de enkelte fraktioner. Heraf ses, at de forventede detektionsgrænser ud fra JKK fint overholdes for hver fraktion, samt at detektionsgrænsen for totalkulbrinter, for fraktionen C6-C35, ligger på 8,21 mg/ kg TS, altså under 10 mg/kg TS som 1/10 af acceptkriteriet på 100 mg/kg TS. I den nye videreudviklede analysemetode er totalkulbrinteindholdet imidlertid omfattet at fraktionen fra C6 og op til C40. Herved vil detektionsgrænsen for totalkulbrinter i dette område komme op på 18,8 mg/kg TS. Denne detektionsgrænse er forventelig i fraktionen fra C35 til C40. I den nye videreudviklede analysemetode beregnes fraktion C25-C35 og C35-C40 hver for sig, men de skal opgives samlet i analyserapporten. I tabel 9-12 ses de endelige detektionsgrænser for den nye videreudviklede analysemetode, bestemt ud fra de beregnede værdier. Her skal bemærkes, at detektionsgrænsen for det totale indhold af kulbrinter er en sum af detektionsgrænsen for de enkelte fraktioner. I analyserapporten kan der med fordel opgives "ikke påvist", når alle 3 fraktioner ligger under detektionsgrænsen. I de tilfælde, hvor der er påvist kulbrinter i en eller flere fraktioner, opgives summen af de enkelte fraktioner som totalkulbrinter.
Tabel 9-12. Sammenligning og bestemmelse af de nye detektionsgrænser. Heraf ses, at målet med at overholde de forventede detektionsgrænser, som opfylder de danske myndigheders krav er nået, og i den nye metode vil disse detektionsgrænser være gældende. 9.6.3 Detektionsgrænse for BTEX på GC-MSFor BTEX indhold målt på GC-MS er det målet at kunne overholde en detektionsgrænse på 0,01 mg/kg TS pr. komponent. Til bestemmelse af detektionsgrænsen er der anvendt de bestemmelser i valideringsserien der er foretaget i den laveste koncentration. Der er foretaget dobbeltbestemmelse over 6 dage, således at der er 12 bestemmelser pr. komponent. Da der er tale om bestemmelser, der er foretaget over flere dage, skal beregningen af detektionsgrænsen foretages med værdier af standardafvigelsen, hvor der er taget højde for afvigelserne inden for hver af de 6 serier. Detektionsgrænsen er således bestemt ved beregning på standardafvigelsen af 12 bestemmelser med en koncentration tæt på, det vil sige højst 5 gange over, den forventede detektionsgrænse. Til forsøget er der således anvendt en tom matrix (sand), hvortil der er tilsat en koncentration på ca. 0,033 mg/kg TS. Prøverne er ekstraheret og analyseret, som beskrevet i analyseforskriften, og detektionsgrænsen (DL) er beregnet udfra spredningen inden for serierne, Sw: DL = t0,995 (f) * Sw DL: Detektionsgrænsen Sw Standardafvigelsen inden for samme serie. Sw er estimatet for standardafvigelsen inden for serien og bestemmes ved: S2w=(d12+d22+d32+.....+dn2)/2n hvor d1, d2, d3..........dn er differensen mellem de enkelte prøvepars resultater med i alt n kontrolprøvepar (i alt 2n enkeltprøver) /7/. I tabel 9-13 ses beregningerne af detektionsgrænsen for de fem komponenter.
Tabel 9-13. Detektionsgrænseberegning for BTEX. Ved sammenligning med den forventede detektionsgrænse, er den nye metodes detektionsgrænse for BTEX'erne fastsat. Se tabel 9-14.
Tabel 9-14. detektionsgrænser for BTEX. Heraf ses, at detektionsgrænsen for benzen, toluen, ethylbenzen, m- og p-xylen og o-xylen ligger under 0,01 mg/kg TS. Detektionsgrænsen for hver af BTEX komponenterne fastsættes derfor til 0,01 mg/kg TS. 9.6.4 Detektionsgrænse for PAH på GC-MSDetektionsgrænsen for PAH'erne bestemmes ved at lave 12 bestemmelser på en naturlig prøve indeholdende PAH'er på lavt niveau. Da der tilstræbes en DL på 0,005 mg/kg TS, ville den ideelle prøve have et indhold på 0,025 mg/kg TS for hver PAH-forbindelse. Prøven har et indhold af fluoranthen på 0,047 mg/kg TS. Da fluoranthen erfaringsmæssigt er den dominerende PAH, vælges det at benytte denne prøve alligevel, da de øvrige PAH'er ligger på et mere passende niveau. Prøven indeholder ikke acenaphten og fluoren i detekterbart niveau. Prøverne er ekstraheret og analyseret som beskrevet i analysemetoden og detektionsgrænsen (DL) er beregnet udfra spredningen inden for serierne, Sw på samme måde som for detektionsbestemmelsen for BTEX'erne. Jf. afsnit 9.6.3. I tabel 9-15 ses de beregnede detektionsgrænser for de 15 PAH komponenter.
Tabel 9-15. Detektionsgrænseberegning for Pah forbindelser. Ved sammenligning med den forventede detektionsgrænse, som ses i tabel 9-16, er der fastsat en endelig detektionsgrænse for hver af PAH komponenterne. For acenaphten og fluoren vurderes, af erfaring, DL at være på samme niveau.
Tabel 9-16. detektionsgrænser for PAH forbindelser. Heraf ses, at detektionsgrænserne for alle PAH forbindelserne ligger under den forventede detektionsgrænse, og målene er overholdt. Der vil blive anvendt en detektionsgrænse på 0,005 mg/kg TS for hver PAH komponent. 9.6.5 KonklusionDetektionsgrænseberegningerne viser, at den undersøgte metode kan overholde de ønskede krav om detektionsgrænser både for totalkulbrinter på GC-FID og for BTEX og PAH-forbindelser på GC-MS. 9.7 Præcision/Repeterbarhed og reproducerbarhed9.7.1 FormålMetodens præcision er belyst ved analyser både indenfor og mellem serierne. Dette er foregået ved ;
9.7.2 FremgangsmådeDer er udført tilsætningsforsøg til forskellige jordtyper, hvor der er spiket med de pågældende stoffer for at opnå prøver med fire forskellige koncentrationsniveauer for at belyse repeterbarheden og reproducerbarheden. Der er spiket med olieprodukt i én serie, og i den anden er der spiket med BTEX'er og PAH'er på de fire niveauer. Repeterbarheden er beregnet som usikkerheden indenfor de seks dage, hvor standardafvigelsen er inden for serien, Sw og reproducerbarheden er givet ved Sb , standardafvigelsen mellem serierne. Repeterbarheden og reproducerbarheden bliver i det følgende udtrykt i procent af middelværdien. Den totale analyseusikkerhed ST er fundet ved; (ST)2=(Sw)2+(Sb)2 og
Hvor mv er middelværdierne af alle 2n analyseresultater. Repeterbarheden og reproducerbarheden er beregnet ved hjælp af StatGraphics som (Sw)2 og (Sb)2. Der er udført ensidet variansanalyse, hvorved Mean Square fremkommer inden for serierne og mellem serierne. Kvadratroden af disse giver henholdsvis repeterbarhed og reproducerbarhed i mg/kgTS. 9.7.3 Præcision for GC-FIDDer er udført tilsætningsforsøg som dobbeltbestemmelser og over 6 dage. Til jordtyperne er der tilsat kulbrinter på 4 niveauer svarende til et indhold på ca. 100 mg/kg TS, 568 mg/kg TS, 818 mg/kg TS og 998 mg/kg TS. Nogle af analyserne på det laveste niveau blev ikke analyseret tilfredsstillende og er blevet udelukket, derfor er der analyseret nogle ekstra prøver. Indholdet af totalkulbrinter er beregnet overfor n-alkan standard. I tabel 9-17 ses en oversigt over de udførte forsøg.
Tabel 9-17. Forsøgsoversigt. I tabel 9-18 er angivet de fundne værdier for repeterbarhed (angivet som Sw) standardafvigelse indenfor serien og reproducerbarhed (angivet som Sb)standardafvigelse mellem serierne. Samt den totale relative standardafvigelse RSDT for alle koncentrationsniveauerne.
Tabel 9-18. Repeterbarhed og relativ standard afvigelser for totalkulbrinter. Den totale analyseusikkerhed er som forventet på de fire niveauer. Usikkerheden på alle niveauer anses for acceptable. Usikkerheden på alle niveauer anses for acceptable. 9.7.4 Præcision for GC-MSTil bestemmelse af metodens præcision på GC-MS, er der udført tilsætningsforsøg som dobbeltbestemmelser over 6 dage. Til jordtyperne er der tilsat BTEX'er i et indhold svarende til ca. 0,033 mg/kg TS, 0,33 mg/kg TS, 0,67 mg/kg TS og 1,67 mg/kg TS, og der er tilsat fluoranthen, for at repræsentere PAH'erne i koncentrationer på ca. 0,047 mg/kg TS, 0,60 mg/kg TS, 0,93 mg/kg TS og 1,67 mg/kg TS. Indholdet af PAH'er og BTEX'er er beregnet overfor PAH og BTEX standardarder med en koncentration på 5 mg/kg. I tabel 9-19 ses en oversigt over de udførte forsøg.
Tabel 9-19. Forsøgsoversigt. I tabel 9-20 er angivet de fundne værdier for repeterbarhed (angivet som Sw) standardafvigelse indenfor serien og reproducerbarhed (angivet som Sb)standardafvigelse mellem serierne. Desuden ses den totale relative standardafvigelse RSDT for alle koncentrationsniveauerne.
Tabel 9-20. Præcision for BTEX. For BTEX'erne ses, at den relative standard afvigelse er rimelig for alle niveauer, da de ligger omkring 10 – 15 %, dog med en lidt høj tendens for et par stoffer i den lave koncentration, hvilket er forventeligt. I tabel 9-21 til 9-24 er repeterbarhed, reproducerbarhed og den relative standard afvigelse beregnet for PAH-forbindelserne, for de fire koncentrationsniveauer.
Tabel 9-21. Relative standardafvigelser af PAH for niveau 1.
Tabel 9-22, Relative standardafvigelser af PAH for niveau 3.
Tabel 9-23. Relative standardafvigelser af PAH for niveau 4.
Tabel 9-24. Relative standardafvigelser af PAH for niveau 5. Her skal først bemærkes, at det ikke var muligt at detektere de lave koncentrationer af acenaphten og fluoren. Den relative standardafvigelse er rimelig for alle niveauer, dog med en tendens til lidt højere afvigelser i den laveste koncentration. Den relative standardafvigelse i det laveste niveau ligger mellem 7,6 % til 49,9 %, hvorimod det ligger på mellem 8,4 – 12,9 % for det højeste niveau. Standardafvigelserne inden for serierne og mellem serierne viser sig også at være som forventet, med en lavere værdi for spredningen inden for serierne end mellem serierne. Metodekravet i afsnit 9.2.2. er opfyldt tilfredsstillende for de fleste parametre. 9.7.5 KonklusionDen samlede vurdering er, at metodens præcision både med hensyn til repeterbarhed og reproduktion for både GC-MS og GC-FID er acceptabel. 9.8 Nøjagtighed/Genfinding9.8.1 FormålDet er formålet at bestemme nøjagtigheden af metoden ved at vise, hvor tæt de målte værdier er på den sande værdi for prøven. Det vil sige, at vise overensstemmelsen mellem den målte værdi og den accepterede konventionelle værdi. Dette er bestemt ved genfindingsforsøg over seks dage. 9.8.2 Genfinding af totalkulbrinter på GC-FIDTil bestemmelsen af metodens nøjagtighed for indholdet af total kulbrinter i ekstrakter af jordprøver er der foretaget to slags genfindingsforsøg. Det ene er tilsætning af motorolie i forskellige koncentrationer direkte til rent sand. Det andet genfindingsforsøg er tilsætning af motorolie til en i forvejen motorolieforurenet jordprøve. I tabel 9-25 ses de anvendte jordtyper og koncentrationer, der er anvendt til genfindingsforsøgene.
Tabel 9-25. Jordrtyper og koncentrationer til genfindingsforsøg. Der er foretaget dobbeltbestemmelser på alle prøver, men på niveau 2 og niveau 6 er der kun foretaget bestemmelser over én dag. Datamaterialet fra disse to niveauer er således ikke så fyldestgørende, men ikke desto mindre er de medtaget for at vise, at der er fin genfinding på både lave og høje koncentrationsniveauer. I tabel 9-26 ses resultatet af genfindingsforsøgene ved tilsætning af motorolie til rent sand.
Tabel 9-26. Genfinding når tilsætning til sand. Til genfindingen af motorolie i en forurenet jordprøve ses der i tabel 9-27, at der er en fin genfindingsprocent. Her er middelværdien af niveau 3 (motorolieforurenet jordprøve) bestemt til den såkaldt "sande" koncentration.
Tabel 9-27. Genfinding når tilsætning til forurenetjord. Af ovennævnte analyser ses det, at der er en middelgenfindingsprocent på 105,1 % kulbrinteindhold målt på GC-FID på alle de målte koncentrationsniveauer, hvor de højeste koncentrationer ligger tættest på 100%. 9.8.3 Genfinding af BTEX og PAH på GC-MS9.8.3.1 Genfinding af BTEX Alle forsøg er foretaget over 6 dage med dobbeltbestemmelser. I tabel 9-28, ses de koncentrationer af BTEX'er, der er spiket med.
Tabel 9-28. Spikede BTEX koncentrationer. Resultatet af analyserne for genfinding af BTEX ses i tabel 9-29, hvor antal analyser, minimum og maksimumværdier samt genfindingsprocenterne ses. Heraf ses, at genfindingsprocenterne er fine, med en middel genfinding på 112 % og en range på 100,2 – 120,6 %.
Tabel 9-29. Genfindingsprocenter for BTEX. 9.8.3.2 Genfinding af PAH
Tabel 9-30. Spiket Fluoranthenkoncentration. I tabel 9-31 ses genfindingsprocenterne for sandprøverne, som er spiket med PAH i en koncentration på 1,67 mg/kg TS. Forsøget er foretaget over seks dage med dobbeltbestemmelser.
Tabel 9-31. Genfindingsprocenter for PAH forbindelser. Heraf ses, at genfindingsprocenterne for alle PAH forbindelser tilsat til almindeligt sand, ligger med en range på 96,7 – 108,2 % med en middel genfinding på 102,9 %. Desuden er genfindingen af PAH forbindelser i en tjæreforurenet jordprøve fundet. Middelværdierne af koncentrationsniveau 3 for hvert enkelt stof, anses for at være den sande værdi, der er spiket med PAH i en koncentration på 0,33 mg/kg TS. Disse koncentrationer og genfindingsprocenterne ses i tabel 9-32
Tabel 9-32. Genfindingsprocent for PAHforbindelser. Heraf ses, at genfindingsprocenterne for PAH forbindelserne ligger mellem 96,7 – 119,6 % og med en middelgenfinding på 110,7 %. 9.8.4 KonklusionKonklusionen af genfindingsforsøgene viser, at metoden har nogle pæne genfindingsprocenter, således at metoden kan anses for at være tilstrækkelig nøjagtig både for analyser af totalkulbrinter på GC-FID og analyser af BTEX og PAH forbindelser på GC-MS. Det bemærkes, at de lidt lave genfindinger der har været problemer med under udviklingen af metoden for PAH'er i afsnit 8.6 er blevet væsentligt forbedret ved brug af isotopmærkede interne standarder. De i afsnit 8.7 lave genfindinger for benzen gør sig ikke gældende under valideringen, idet der er taget forholdsregler for at forhindre afdampning. 9.9 Måleområde9.9.1 FormålMetodens måleområde er koncentrationsintervallet, i hvilket nøjagtighed, metodelinearitet og præcision er acceptable /6/. I praksis bestemmes dette område ved at anvende data fra linearitets og nøjagtighedsstudierne, der er foretaget tidligere i valideringsprocessen. 9.9.2 Vurdering af måleområdeNøjagtighed og præcision Nøjagtighedsundersøgelserne i afsnit 9.8 viser at der er en fin genfindingsprocent for alle de undersøgte stofgrupper op til og med den højest undersøgte koncentration. Præcisionsundersøgelserne fra afsnit 9.7 viser, at metodens præcision med hensyn til repeterbarhed for både indholdet af kulbrinter på GC-FID og for indholdet af BTEX og PAH-forbindelser på GC-MS er tilfredsstillende op til og med højeste undersøgte koncentration. Den højest undersøgte koncentration under valideringen er for totalkulbrinter 5000 mg/kg TS, for BTEX'er og PAH-forbindelser er den 3,3 mg/kg TS. Det undersøgte og dermed angivne øvre måleområde er disse koncentrationer. Måleområdet bliver således ikke så højt, men det er laboratoriets erfaring, at måleområdet ud fra yderligere liniaritetsforsøg, nøjagtighed og præsionsundersøgelser kan udvides til højere koncentrationer, specielt for enkeltkomponenter BTEX og PAH. Detektionsgrænse Detektionsgrænserne er fundet for alle komponenter med en tilfredsstillende nøjagtighed og præsion. Detektionsgrænsen er således startpunkt for måleområdet. 9.9.3 Metodens måleområdeKoncentrationsområdet inden for hvilket metoden er antaget at udvise tilstrækkelig linearitet, nøjagtighed og præcision, er givet i tabel 9-33. Detektionsgrænserne er den nedre koncentrationsgrænse. Den øvre grænse er begrænset af de koncentrationer, der har været anvendt til metode-valideringen.
Tabel 9-33. Det validerede måleområde. 9.10 KonklusionAf tabel 9-34, ses en oversigt over genfindinger og usikkerheder fundet ved valideringen af den nye metode: ISO/DIS 16703-Mod.
Tabel 9-34. Resultat af valideringen. Konklusionen på valideringen giver at:
Metodevalideringen af ISO/DIS 16703-Mod. Viser, at der ved anvendelse af denne metode kan opnås resultater, der er nøjagtige og præcise. Desuden udviser metoden tilfredsstillende detektionsgrænser for bestemmelse af både totalkulbrinter, BTEX- og PAH-forbindelser. ISO/DIS 16703-Mod. har under projektforløbet vist sig at være en praktisk anvendelig analysemetode på laboratoriet. Analysemetoden skal sendes ud i afprøvning i de danske miljølaboratorier inden den kan tages endelig i brug. I forbindelse med projektet er der lavet en detaljeret analyseforskrift, der kan følges direkte til udførelse af metoden. Den fuldstændige analyseforskrift for den nye videreudviklede og validerede analysemetode, ISO/DIS 16703-Mod. ligger i fuld udstrækning i appendiks A. 10 Verifikation af ekstraktions-metoderFor at verificere ekstraktionseffektiviteten af den nye validerede metode (ISO/DIS 16703-Mod.) sammenlignes ekstraktionseffektiviteten af kulbrinter i fraktionerne n-alkan C10-C40 og C25-C40 med pentan-ekstraktion, dichlormethan-ekstraktion samt ekstraktion i henhold til ISO/DIS 16703 med n-heptan som ekstraktionsmiddel. Fremgangsproceduren for disse tre metoder er allerede tidligere beskrevet i afsnit 3. Ekstraktfremstilling i henhold til ISO/DIS 16703 foretages i overensstemmelse med standardmetoden, idet råekstraktet vaskes med vand, som efterfølgende tørres ved tilsætning af vandfrit natriumsulfat, og afslutningsmæssigt oprenses med florisil. Til forsøget anvendes 4 forskellige jordtyper, som er kompostjord, motorolieforurenet jord, gasolieforurenet jord samt tjæreforurenet jord. De 4 jordtyper er tidligere anvendt til de sammenlignende ekstraktioner i afsnit 3. Det kvantitative indhold beregnes for hver ekstraktionsmetode og hver jordtype i mg/kg TS, og dataene sammenlignes vha. statistiske analyser. Mængden af prøve og opløsningsmiddel samt den samlede ekstraktionstid er fastsat i overensstemmelse med metodeforskrifterne. 10.1 Ekstraktion af jordprøverFor at vurdere de fire metoders ekstraktionseffektivitet mere generelt, ved ekstraktion af forskellige forureningstyper i forskellige jordmatricer, er ekstraktionerne udført på "rigtige" jordprøver, som forinden ekstraktion er homogeniseret efter samme metode, som beskrevet i afsnit 3.2.3. Der er udført 2 ægte bestemmelser for hver metode på hver jordtype. Forsøgsplanen kan ses i tabel 10.1:
Tabel 10.1. Forsøgsplan til sammenligning af de 4 ekstraktionsmetoder. Prøvematerialet er afvejet i 100 mL Duranglas, og der er ved samme lejlighed udtaget ca. 10 g prøve til tørstofbestemmelse efter DS 204. I forbindelse med udførelsen af ekstraktion i henhold til ISO/DIS 16703, er der anvendt 100 mL Duranglas til ekstraktion og vask, mens ekstraktet under tørring og oprensning har været overført til 35 mL reagensglas med skruelåg og PTFE (teflon)-indlæg. For alle metoder er der tilsat opløsningsmiddel med o-terphenyl som intern standard med en koncentration på 5 mg/L. 10.2 DatabehandlingKvantificeringen af kulbrinteindholdet er foretaget overfor en n-alkan standard på 5 mg/L (Restek # 31266), idet summen af n-alkan C12, C16, C20, C24 er anvendt til kvantificering af fraktionen C10-C25, mens summen af n-alkan C28, C30, C32 og C34 er anvendt til kvantificering af fraktionerne C25-C35 samt C35-C40. Alle beregninger er foretaget med intern standard på nær ISO/DIS 16703-metoden, og den bedst egnede blindprøve er anvendt til blindkorrektion. Det er valgt ikke at udføre beregningerne for ISO/DIS 16703-metoden med intern standard, idet gentagne resultater tyder på, at den interne standard (o-terphenyl) forsvinder markant under vask og oprensningstrin. Der er udført en flersidet variansanalyse med metode og jordtype som uafhængige faktorer og koncentration af kulbrinter (C10-C40) angivet i mg/kgTS som afhængig variabel. Bartletts test viser varianshomogenitet i datamaterialet og normalfordelte og statistisk uafhængige residualer kan antages. På figur 10.1 er resultaterne fra analysen angivet med 95%-konfidensintervaller.
Figur 10.1. Flersidet variansanalyse af data fra sammenlignende ekstraktionsforsøg for de 4 metoder for fraktionen C10-C40. Som det ses af variansanalysen, medfører ekstraktion med dichlormethan og ISO/DIS 16703-Mod. et relativt højt ekstraktionsudbytte på samme niveau. Ekstraktionsudbyttet for ISO/DIS 16703-Mod. er signifikant højere i forhold til både pentan- og ISO/DIS 16703-metoden. Den lave ekstraktionseffektivitet for ISO/DIS 16703-metoden kan skyldes aktiv fjernelse af prøvekulbrinter under den tidskrævende vaske- og oprensningsproces samt naturligvis den relativt korte ekstraktionstid. Analysen fører - i overensstemmelse med de tidligere resultater fra de sammenlignende ekstraktioner – til den konklusion, at der opnås den højeste ekstraktionseffektivitet ved anvendelse af dichlormethan-metoden eller ISO/DIS 16703-Mod., hvis der ses på summen af ekstraherede kulbrinter i området n-alkan C10-C40. På figur 10.2 ses de enkelte metoders ekstraktionsudbytte overfor de forskellige jordtyper repræsenteret ved en middelværdi med indtegnede 95%-konfidensintervaller. Det ser ud til, at ISO/DIS 16703 generelt er den dårligste metode ved ekstraktion af de 4 jordtyper. ISO/DIS 16703-Mod. ser ud til at være effektiv overfor samtlige jordtyper og ligger stort set på niveau med dichlormethan-ekstraktionen. Der ses dog en meget lav ekstraktionseffektivitet for dichlormethan-metoden for den gasolieforurenede jordtype. Det er ikke umiddelbart muligt at finde nogen forklaring herpå, men det vurderes, at dette ikke nødvendigvis er generelt gældende. Den gasolieforurenede jordtype har dog et relativt højt lerindhold og faktorer som inhomogenitet og specielle fysiske prøveegenskaber - som vanskeliggør en tilfredsstillende ekstraktion - kan være gældende.
Figur 10-2. De forskellige metoders ekstraktionseffektivitet for fraktionen C10-C40 for de forskellige jordtyper. Der er ligeledes udført en flersidet variansanalyse for summen af kulbrinter i fraktionen C25-C40, som kan ses på figur 10.3. De samme konklusioner kan drages herudfra.
Figur 10-3. Flersidet variansanalyse af data fra sammenlignende ekstraktionsforsøg for de 4 metoder for fraktionen C25-C40. Metodernes ekstraktionsudbytte for fraktionen C25-C40 for de 4 jordtyper kan ses af figur 10.4. Også her ses den største ekstraktionseffektivitet generelt for dichlormethan-metoden og ISO/DIS 16703-Mod.
Figur 10-4. De forskellige metoders ekstraktionseffektivitet for fraktionen C25-C40 for de forskellige jordtyper. 10.3 KonklusionSammenligning af ekstraktionseffektiviteterne for de 4 metoder har vist, at anvendelsen af dichlormethan-metoden eller ISO/DIS 16703-Mod. medfører det største ekstraktionsudbytte. Analyserne viser desuden, at anvendelsen af ISO/DIS 16703 medfører en signifikant lavere ekstraktionseffektivitet i forhold til de resterende tre metoder, hvilket sandsynligvis skyldes, at vaske- og oprensningstrinnene reducerer indholdet af petrokemiske kulbrinter i prøveekstraktet, samt at ekstraktionstiden er relativt kort 11 Petrogene biomarkører11.1 FormålFormålet med denne del af projektet er udfra eksisterende metoder bl. a. /10, 18/, at udvælge en række petrogene kulbrinter, der kan anvendes i forbindelse med vurderingen af, om der er kulbrinter af petrogen oprindelse dvs. stammende fra olie i jordprøver. Der er fokuseret på petrogene kulbrinter, der har betydning for tilstedeværelse af oliekulbrinter i højere grad en karakterisering af olietype og egenskaber. Det er målet, at begrænse antallet af kulbrinter af petrogen oprindelse, der er nødvendige i vurderingen. Der skal kvantificeres et acceptabelt antal, så metoden til vurdering af, om der er petrogene kulbrinter tilstede, ikke unødigt kompliceres og fordyres. 11.2 Olieprodukter og komponenterRåolie er et fossilt brændsel dannet af organisk materiale for millioner af år siden. Det består hovedsageligt af alifatiske kulbrinter og dels af mættede cykliske forbindelser som naphtener, cyclopentan og cyklohexaner med sidekæder. Råolien indeholder dog også mindre mængder af aromater som monoaromater og PAH. Raffinering af råolie medfører fraktionering i forskellige olieprodukter (tabel 11-1). Olieforbindelser kan desuden videreforarbejdes ved f. eks. "krakning", hvor der under høje temperaturer spaltes mindre og ofte umættede molekylære forbindelser fra højmolekylære forbindelser /23/. Tabel 11-1 viser typiske fraktioner i en destillationsproces:
Tabel 11-1. Raffinering af råolie /23/ 11.3 Forandring af olieprodukter - Forvitring"Weathering" er det ord, der anvendes til at beskrive den forandring, der sker med olie, når det udsættes for vind og vejr. Det kan oversættes med det danske ord forvitring. Forvitring kan opdeles i flere dele
Disse elementer er beskrevet i Nordtest Metoden /10/ og er vigtige til at kunne beskrive, hvad der sker med olie, når et tankskib med råolie springer læk eller af anden grund tømmer sine tanke ud i havet. Fordampning Ved fordampning forsvinder de lettere kogende komponenter fra olien. For benzin sker der eksempelvis fordampning af benzen, toluen og til en vis grad også xylener. De højere kogende oliekomponenter vil med tiden forekomme i større procentdel af det samlede indhold. Fordampning ses også som den forandring, der sker af og til ved kraftigt nedbrudt gasolie, hvor de lettere kogende komponenter er fordampet, og den uopløste bule, UCM (omtales i afsnit 11-5), har toppunkt senere i kromatogrammet. Opløsning Ved opløsning forstås opløsning af oliens komponenter i den omkringliggende vandfase. Dette kan ske både i den umættede zone i porevandet og i nedsivende regnvand samt i grundvandszone. Da olieprodukter typisk er lettere end vand, tilbageholdes et større oliespild som frifasen ovenpå grundvandsspejlet. I kontaktfasen mellem frifaseolie og grundvand sker opløsning af de vandopløselige komponenter. Graden af dissolution er afhængig af de enkelte komponenters opløselighed i vand. Typisk er heteroatom forbindelser (forbindelser som indeholder N, S eller O) mere opløselige end aromatiske forbindelser, som igen er mere opløselige end mættede forbindelser /10/. Indenfor de homologe serier af aromatiske forbindelser betyder antallet af substituenter også en stor del for vandopløseligheden, idet de ikke-alkylerede forbindelser er mere opløselige end de alkylerede. Bionedbrydning Ved bionedbrydning – nedbrydning ved hjælp af bakterier – forsvinder i første omgang de ligekædede forbindelser, typiske n-alkaner og senere de forgrenede forbindelser (iso-forbindelser). Mættede cykliske forbindelser er meget resistente overfor nedbrydning. Kemisk forandring og kontaminering Kemisk forandring og kontaminering er effekter, der er mere specielle, og er vigtig for vurdering af råolie på havoverflader, men i forbindelse med olie i jord har det ikke så stor betydning. Når olie spildes på jord, sker der umiddelbart en forandring som en følge af ovenstående effekter. En del komponenter forsvinder helt. Andre komponenter, der er mere resistente overfor nedbrydning, bevares og kan anvendes som specifik petrogen kulbrinte for, om der er petrogene kulbrinter tilstede i en prøve. 11.4 Acykliske isoprenoiderTypiske kulbrinter for olie er de acykliske isoprenoider (forgrenede forbindelser) /8/. Mest velkendt er pristan (C19, 19 carbonatomer, 11-1) og phytan (C20, 20 carbonatomer, 11-1) som fremgår af figur 11-1.
Figur 11-1. Eksempel på acykliske isoprenoider /8/ Ved isoprenoider forstås forbindelser med et skelet bestående af C5 isoprenenheder. Råolie er dannet af alger og planter i forhistorien. Pristan (C19) og phytan (C20) menes at være dannet i råolien udfra nedbrydningen af phytol, der er en sidekæde til chlorofyll. Forholdet mellem pristan og phytan i råolie fra Mellemøsten og Nordsøen er konstant, uanset om det er råolie, der er direkte oppumpet fra kilden, eller et behandlet olieprodukt som eksempelvis dieselolie. Visse råolier f. eks. fra Venezuela er meget ældre, og har et andet pristan/phytan forhold end olie fra Mellemøsten og Nordsøen. Pristan og phytan er tilstede i næsten alle mellem til højtkogende olieprodukter (petroleum, gasolie o. lign.), og er dermed gode indikatorer for mineralolie med kogepunkt større end 200°C. Pristan og phytan nedbrydes langsommere end de ligekædede n-alkaner. De er derfor tilstede i større eller mindre mængde ved selv stærk forvitrede olieforureninger, hvor der er olieindhold med kogepunkt større end ca. 200°C. Pristan og phytan skal ses i sammenhæng med norpristan og farnesan (se figur 11-2). Såfremt både farnesan, norpristan, n-alkan C17, pristan, n-alkan C18 og phytan er tilstede i en prøve, er der kraftig indikation for, at der er kulbrinter af petrogen oprindelse i prøven. Ved gaschromatografisk analyse (GC-FID) er det relativt enkelt at fastlægge de acykliske isoprenoider i selv meget små koncentrationer afhængig af forureningsart. GC-FID analysen anbefales dog ikke til beregning af indholdet af de enkelte komponenter, da der er alt for stor risiko for interferenser. Ved analyse på GC-MS kan ion 57 m/z eller 71 m/z eksempelvis vælges som kvantificeringsion (Target ion). Figur 11-2 viser et udsnit af et kromatogram med de acykliske isoprenoider. Figur 11-2. Udsnit af kromatogram med de acykliske isoprenoider. Det er valgt at udføre kvantificeringer af petrogene kulbrinter på data opsamlet af GC-MS i scan mode, da der her bevares flest informationer om prøvens indhold af kulbrinter. Efterfølgende beregninger er derfor alle udført ved GC-MS-scan. 11.4.1 Pristan/phytan forhold.Forholdet mellem isoprenoiderne pristan og phytan er unikt for enhver "frisk" råolie, og kan derfor være interessant i forbindelse med kildeopsporing af olieforurening. Dette forhold må ikke anvendes alene som olieindikator, men skal anvendes i sammenhæng med andre indikatorer for olie. Under iltrige (oxic) forhold ved den oprindelige oliedannelse dannes pristan (Pr), og under iltfri (anoxic, reduktive) forhold dannes phytan (Ph)/9/. Prøver med høj Pr/Ph ratio (>3,0) indikerer at olien er dannet under tilstedeværelse af terrestrisk organisk stof under iltrige forhold. Lave værdier (< 0,6) er typiske for olie dannet under iltfrie hypersaline miljøer. For olietyper med Pr/Ph forhold mellem 0,8 og 2,5 kan det ikke anbefales at anvende Pr/Ph forholdet til olieidentifikation og kildeopsporing alene. I Danmark er der hovedsalig anvendt olie fra Mellemøsten og Nordsøen, hvor Pr/Ph forholdet er 2,4 – 2,9. Nedbrydningen af pristan og phytan er forholdsmæssig langsom, og den effekt der kan påvirke dette er hovedsalig fordampning /10/. Dette kan have væsentlig betydning i forbindelse med "gamle" overfladenære olieforureninger i jord. Her kan der foregå en stor fordampning. Pr/Ph forholdet er i forbindelse med dette projekt beregnet for frisk dieselolie, samt en række referenceolier fra Restek med forskellig "weathering degree". Resultaterne for dette fremgår af tabel 11-2.
Tabel 11-2. Pr/Ph forhold i en række rene olier. Som det fremgår af tabel 11-2, er der nogen variation i Pr/Ph forholdet i forhold til nedbrydningsgraden for dieselolien. Dieselolien har været udsat for forskellig grad af forvitring. Umiddelbart ser det ud som om, at indholdet af pristan falder i forhold til phytan, men det er en relativ lille forskel, og er ikke skønnet som en signifikant ændring. Ændringen skyldes formentlig fordampning. For motorolien er indholdet af pristan lavt, hvilket skyldes det høje kogepunktsinterval (>350°C). Dette medfører derfor et lavt Pr/Ph forhold. Beregning af Pr/Ph forhold som olieindikator kan kun udføres, når der er tale om et mellem- til højtkogende olieprodukt som f. eks. petroleum, diesel- og fyringsolie. Letkogende olieprodukter som benzin indeholder en forholdsmæssig lille mængde af n-alkanerne C17 og C18, samt pristan og phytan. 11.4.2 Pristan/n-alkan og phytan/n-alkan forholdForholdet mellem pristan og n-alkan C17 samt phytan og n-alkan C18 kan anvendes i et vist omfang til aldersdatering ved hjælp af en GC-FID analyse eller evt. GC-MS /11/. Denne metode er dog forbundet med meget stor usikkerhed pga. af forskellige forhold mht. jordbund, vejrforhold og nedbrydningspotentiale. Gasolie (dieselolie, fyringsolie) vurderes som ikke forvitret (frisk), såfremt forholdet C17/pristan er større end 1 (1,0 – 1,5 empirisk), delvist forvitret, hvis forholdet ligger under 1 og kraftigt forvitret, hvis n-alkan C17 er helt nedbrudt /11/. Tilsvarende forhold er gældende for en hel række rå- og fuelolieprodukter. Beregning for en række rene olieprodukter fra Restek vist herunder i tabel 11-3. Beregningerne er foretaget ved udmåling af tophøjder i GC-FID.
Tabel 11-3. C17/pristan og C18/phytan forhold i en række rene olier. Som det fremgår af tabel 11-3, er forholdet mellem n-alkan C17 og pristan nær 1 for de fleste af de testede olier, og størrelsen af n-alkan C17/pristan er ikke signifikant høj. En regel om, at C17/Pr kan bruges i aldersdatering af olien, er derfor ikke at anbefale, specielt da der forekommer flere forskellige olieblandinger på det danske marked. Ved kvantificering af isoprenoiderne ved GC-FID anbefales det, at der benyttes tophøjde frem for respons, da dette hidtil har været almindelig praksis (/18/ og oplyst af A. B. Hansen, DMU). I beregning af isoprenoiderne i forbindelse med nærværende projekt er der hovedsaligt anvendt GC-MS-scan, og ion 57 m/z har været anvendt som kvantificeringsion. Ion 113 m/z er den ion, som anvendes af DMU i forbindelse med olieefterforskning, og i den endelige metode jf. analyseforskrift kapitel 10, er det da også denne, som anbefales i forbindelse med kvantificering af n-alkaner ved GC-MS. I forbindelse med beregning af indholdet af pristan og phytan ved GC-MS-scan med ion 57 m/z som kvantificeringsion er der forskellige responsfaktorer for disse i forhold til de n-alkaner, der anvendes til kalibrering. Derfor er det ikke muligt umiddelbart at kvantificere overfor nærmeste n-alkan. Det har været nødvendigt at foretage en korrektion af resultaterne, hvilket er sket i beregningerne. Følgende korrektionsfaktorer er opnået ved analyse af rene enkeltkomponenter (tabel 11-4, rene opløsninger fra Chiron). Korrektionsfaktorerne er fundet ved at dividere opløsningernes koncentration med responset af de enkelte komponenter ved GC-MS (se afsnit 13.6). I beregningen af selve koncentrationen af de enkelte komponenter er der kvantificeret overfor en referencestandard af n-alkan C-18, hvor responsfaktoren for denne er multipliceret med en korrektionsfaktor.
Tabel 11-4. Opnåede responsfaktorer for n-alkaner samt nor-pristan, n-alkan C17, pristan og phytan. Tilstedeværelsen af både farnesen, norpristan, n-alkan C17, pristan, n-alkan C18 og phytan er en kraftig indikator for tilstedeværelse af petrogene kulbrinter. Forholdene n-alkan C17/pristan og n-alkan C18/phytan kan derfor sammen med de øvrige beregninger give en indikation for, om der er petrogene kulbrinter i en given prøve. 11.5 Carbon Preference Indeks."Carbon preference indeks", giver som navnet antyder et udtryk for foretrukne kulstofforbindelser. De ulige n-alkaner stammer primært fra landplanter (n-alkan C25, C27, C29, C31 og C33). N-alkan C17 stammer hovedsagligt fra phytoplankton /12/. Der er flere metoder til beregning af CPI /9,13/, men som nævnt i "Interferenser ved bestemmelse af olie i jord"/12/, er den foretrukne metode til beregning som følger:
Ved beregning af CPI-indeks skal der udføres kvantitativ beregning af de enkelte alkaner for at eliminere fejl i forbindelse med diskriminering i injektionsport på GC-en samt faldende performance af n-alkanerne. I et mineralolieprodukt (fra Mellemøsten og Nordsøen) er CPI = 1, og fordelingen af de kvantitative indhold af lige og ulige n-alkaner er ens. Hvis CPI(25-33) > 1 er der bidrag fra ulige n-alkaner fra landlevende planter (epicuticular voks i højere planter /14/). Hvis CPI(15-21) > 1 er der bidrag fra forskellige marine og ferskvandsalger (n-alkan C17 kan være indikator for aquatiske planter /14/). Ved overfladenære prøver, hvor der er stort bidrag af kulbrinter fra planter, ses meget høje CPI. Herunder er vist CPI beregnet for de tidligere nævnte olieprodukter. Resultaterne fremgår af tabel 11-5. Det er kun relevant at undersøge CPI, i det kogepunktsinterval, hvor det faktiske olieprodukt befinder sig. Derfor er der ikke beregnet CPI(25-33) for dieselolie. Diesel Fuel #2 har CPI(15-21), der er højere end 1. Der er ikke nogen umiddelbar forklaring på denne afvigelse.
Tabel 11-5. CPI for en række rene olier. CPI vurderes til at være en potentiel indikator for, hvorvidt der er interferens fra naturlige kulbrinter i en jordprøve. Det er forholdsmæssigt let at identificere n-alkanerne, og de forekommer i et kontinuum, en blød bule, hvor der ikke diskrimineres mellem de enkelte toppe af n-alkaner. Ved bidrag fra organisk terrestrisk materiale, vil de ulige n-alkaner blive forholdsmæssig højere end de lige n-alkaner. Dette er vist i et eksempel fra en jordprøve i figur 11-3. Figur 11-3. Fordeling af n-alkaner (vist ved ionspor 57 m/z). Udfra højdeforskellen kan det ses, at der er bidrag fra terrestrisk organisk materiale. De ulige n-alkaner i prøver med bidrag fra naturlige kulbrinter er mere detaljeret beskrevet i afsnit 12.8. 11.6 Unresolved Complex MatterI forbindelse med både friske og forvitrede mineralolieprodukter forekommer der i GC-FID kromatogrammet en uopløst bule, som kaldes "unresolved complex matter" (UCM). Naturlige organiske kulbrinter vil i højere grad forekomme som kromatografisk separerede toppe /12/. UCM kan derfor være en indikator for, om der er et mineralolieprodukt tilstede. UCM beregnes enten kvantitativt eller som et forhold mellem opløste toppe (Resolved Peaks) i forhold til totalkulbrinte indholdet /15/. Der kan forekomme flere UCM i et kromatogram. UCM anvendes i vid udstrækning i litteraturen til vurdering af, om der er kulbrinter af petrogen oprindelse tilstede /16,17/. En beregning af UCM kan foretages, hvis det er vurderet, at der er andre indikatorer for indhold af kulbrinter af petrogen oprindelse i GC-FID kromatogrammet. Figur 11-4 viser et GC-FID kromatogram af dielselolie. Figur 11-4. Dieselolie ved GC-FID Figur 11-5 viser integrationer af opløste toppe i området n-alkan C8 - C40 i samme dieselolie, som vist i figur 11-4. Figur 11-5. Integration af opløste toppe i GC-FID af dieselolie UCM beregnes som differencen mellem total arealet og summen af de opløste toppe. UCM er i tabel 11-6 beregnet for en række rene olier i forskellige koncentrationsniveauer.
Tabel 11-6. UCM for en række rene olieprodukter Som det ses af tabel 11-6 er UCM høj, og for de fleste motorolier ligger den over 90%. For den undersøgte dieselolie er UCM væsentlig lavere, 73%. UCM kan give en indikation om, at der findes kulbrinter af petrogen oprindelse i prøven, såfremt UCM er en pæn veldefineret bule i kromatogrammet. 11.7 HopanerI råolie findes der triterpaner, herunder hopaner, og de er uundværlige i forbindelse med identifikation af olien. Disse komponenter forekommer i olieprodukter med et forholdsmæssigt højt kogepunkt > ca. 300°C. I højtkogende olieprodukter som motorolie og fuelolie vil hopaner nemt kunne identificeres ved GC-MS-scan med ion 191 m/z som kvantificeringsion. Ved GC-FID kan hopanmønstret i visse tilfælde identificeres, men der er stor risiko for interferens. I bilag 7 er vist en tabel over en række hopaner, som almindeligvis forekommer i råolie. De toppe, som er de mest karakteristiske er 5, 10, 12, 13, 15, 16, 19 og 20. Disse komponenter ses i alle højere kogende olieprodukter og kulbrinteprodukter af petrogen oprindelse. Disse komponenter nedbrydes langsomt, og specielt 17(H),21(H)-hopane (30ab, komponent nr. 10 i Bilag 7) er ekstrem resistent overfor nedbrydning. Denne komponent kan i visse tilfælde anvendes som intern standard (normeringsfaktor) /10,18/. Udvalgte hopaners kromatografiske respons i forhold til f. eks. n-alkan C18 er vist i bilag 8. Nedenfor i figur 11-6 er vist et GC-MS-scan kromatogram af motorolie som total ion kromatogram (GC-MS TIC). Hopanerne fremkommer med tydeligt mønster "ovenpå" den uopløste kromatografiske bule (UCM). Figur 11-6. Total ion kromatogram (TIC) af motorolie, hvor hopanerne tydeligt ses som toppe ovenpå den kromatografisk uopløste bule (UCM). TIC-et ved GC-MS-scan ligner i høj grad et kromatogram fra GC-FID. Indholdet af hopaner varierer fra olieprodukt til olieprodukt, så det er ikke umiddelbart muligt, at lave en korrelation mellem f. eks. 17(H),21(H)-hopane (30ab) og indholdet af olie i en given prøve. Tilstedeværelsen af hopaner, er i sig selv en indikation af, at der er petrogene kulbrinter tilstede. Som et minimumskrav for genkendelse af hopanmønstret skal Ts, Tm, 29ab, 30ab, 31abS og 31abR være synlige i kromatogrammet ved ionspor 191 m/z i GC-MS. Retentionstiderne for hopanerne kan nemt fastlægges ved at analysere en opløsning med f. eks. motorolie eller fuelolie i GC-MS Scan og ekstrahere ion 191 m/z. De omtalte hopaner har retentionstid ca. mellem n-alkan C28 (her retentionstid 20,83 min.) og n-alkan C35 (retentionstid 26,19 min.), se figur 11-7. Figur 11-7. Retentionstider for hopaner i forhold til n-alkaner i fuelolie. Nogen interferens kan dog ske på hopanerne (specielt 31abS og fremefter) ved et meget stort indhold af phytosteroler som vist herunder i figur 11-8. Det er formentlig sitosterol, som er sammenfaldende med 31 abR. Figur 11-8. Ionspor af 191 m/z af hhv. kompost tilsat motorolie (1000 mg/kg TS) – sort og kompost - grøn. Der ses interferens fra phytosteroler i hopanmønsteret i blandinger af olie og kompost. Indhold af hopaner ses som tidligere nævnt hovedsaligt i højerekogende olieprodukter som f. eks. motorolie (tung olie, Kp. ca. 400°C – 550°C). Da 30ab hopan er meget resistent overfor forvitring, er det en god indikator for tilstedeværelse af højtkogende petrogene kulbrinter. I nedenstående figur 11-9 er sammenhængen mellem indholdet af 30ab hopan i en række højtkogende olieprodukter vist.
Figur 11-9. Indhold af 30ab hopan i forhold til indhold af totalkulbrinter Som det fremgår af figur 11-9 er indholdet af 30ab hopan i en prøve tilsat motorolie svarende til 100 mg/kg TS mellem 0,01 og 0,04 mg/kg TS i de undersøgte olier. I en prøve tilsat 500 mg/kg TS Shell SAE 20W-50 motorolie er indholdet af 30ab hopan ca. 0,12 mg/kg TS. 11.8 Steraner og diasteranerEn anden gruppe af karakteristiske petrogene kulbrinter for olie er steranerne og diasteranerne. Steraner forekommer som regel samtidig med hopaner, men mønsteret for steraner og diasteraner er mere kompliceret end for hopanerne. Steranerne anvendes ligesom hopanerne til olieidentifikation, idet steranmønsteret er helt unikt for hver råolie. I nærværende rapport er det skønnet, at petrogene og naturlige kulbrinter kan adskilles ved brug af hopaner i højere grad end ved steranerne. Mønsteret for hopanerne er noget mere tydeligt end for steranerne. Steranerne er derfor ikke vurderet yderligere i rapporten. Steraner kan ses i et kromatogram fra GC-MS-scan ved ionspor 217 m/z og diasteraner ved ionspor 218 m/z. 11.9 PAH forbindelserI tidligere undersøgelser er det påpeget, at en række særlige PAH-forbindelser har stor værdi for verifikation af indhold af kulbrinter af petrogen oprindelse bl. a. /10,12/. Nedenfor i figur 11-10 er vist en opsummering af fordeling PAH-forbindelser i en række udvalgte olier /10/. Methyleringsgraden er anført som hhv. C0 for moderkomponenten – uden methylgrupper på, C1 for mono-methyleret, C2 for to methylgrupper og så fremdeles. Indenfor hver af grupperne C1, C2 og C3 og så videre er der en hel række isomerer. Under selve oliedannelsen bliver alle isomerer dannet, og isomerernes indbyrdes forhold kan anvendes til at karakterisere en råolie. Disse forhold kan sammen med nogle af de andre petrogene kulbrinter bl. a. sige noget om, hvilket moderfjeld (source rock) olien oprinder fra. I destillerede olieprodukter ses de PAH serier, som ligger indenfor destillationsintervallet. Sædvanligvis ses alle isomererne i de "friske" olieprodukter, og ofte ses de homologe rækker som "bølger" i GC-MS, når der eksempelvis udtrækkes de karakteristiske ioner fra total ion kromatogrammet. Dette kan anvendes i forbindelse med at afgøre, om der i en given prøve er tale om petrogen eller naturlige kulbrinter. Figur 11-10. Indhold af naphthalener, phenanthrener, dibenzothiophener, fluorener samt chrysener i en række udvalgte olier /10/. Det er derfor relevant at undersøge hvorvidt disse forbindelser kan anvendes i forbindelse med lokalisering og detektion af olieforureninger. En række af disse komponenter er som vist i figur 11-10 meget dominerende i de rene olieprodukter, men der sker en fordampning og nedbrydning af disse komponenter, således at fordelingen forandres med tiden. Det er ikke muligt at identificere disse komponenter udfra GC-FID, idet de ligger gemt i den kromatografisk uopløste bule. Disse komponenter kan derfor kun identificeres ved GC-MS. 11.9.1 NaphthalenerIndholdet af naphthalener kan anvendes som indikator for petrogene kulbrinter i forbindelse med olieprodukter med et kogepunktsinterval, der er mindre end n-alkan C25. Letkogende olieprodukter som benzin og gasolie indeholder naphthalener. Ved analyse af rene olieprodukter ved GC-MS-scan, fremkommer der karakteristiske "mønstre", der kan anvendes til dokumentation af tilstedeværelsen af kulbrinter af petrogen oprindelse. I bilag 9 kan de relative retentionstider samt amount/area for de enkelte naphthalen forbindelser ses. Mønstrene for naphthalenerne kan ses af nedenstående udtræk af kromatogrammer af olieprodukterne fra scan-kørslerne (figur 11.12 – 11.14). For alle ionspor er o-terphenyl (intern standard, IS) vist ved ion 230 m/z, så de relative retentionstider kan vurderes (grøn kurve). Indledningsvis er vist et GC-MS-scan kromatogram af indhold svarende til 50 mg/kg TS gasolie tilsat kompost (Figur 11.11). Figur 11-11. GC-MS-scan (TIC) af gasolie 50 mg/kg tilsat homogeniseret kompost. Figur 11-12 viser samme prøve, hvor naphthalens karakteristiske ion 128 m/z er udtrukket. Der ses kun spor af naphthalen ved retentionstiden 10,2 minut. Figur 11-12. Naphthalen, som ses ved retentionstiden 10,2 min. Ionspor af ion 128 m/z. Methylnaphthalenerne, 2- og 1-methylnaphthalen, ses ved hhv. retentionstid 11,2 og 11,4 minut i figur 11-13. Figur 11-13. Methylnaphthalener. Ionspor af ion 142 m/z. (hhv. 2- og 1-methylnaphthalen med Rt. 11,2 min og 11,4 min). Dimethylnaphthalenerne ses herunder i figur 11-14. Figur 11-14. Dimethylnaphthalener. Ionspor af ion 156 m/z. Tri- og tetramethylnaphthalener forekommer i et forholdsvis lavt indhold i gasolie, og de er svære at identificere udfra GC-MS-scan. Såfremt det er muligt at finde det mønster, der fremkommer, når der udtrækkes ionspor på de karakteristiske ioner 128 m/z, 142 m/z og 156 m/z, er det overvejende sandsynligt, at der er petrogene kulbrinter i prøven. Naphthalenerne er ikke specielt kendetegnende for gasolie og benzin. De forekommer i benzin i en relativ lille mængde. Som et lille forsøg, er benzin inddampet til næsten tørhed, for at fjerne så stor en del af de flygtige komponenter som muligt. Hovedfraktionen i frisk benzin har kogepunktsinterval fra ca. 40° C (Rt. ca. 5 minutter) til ca. 130° C (Rt. ca. 10 minutter). Selv ved meget kraftig inddampning er der fortsat indhold af de flygtige komponenter som benzen, toluen og xylenerne. Det ses udfra nedenstående kromatogram, figur 11-15, at der nu ses indhold af kulbrinter, der strækker sig helt op til 15 minutter i kromatogrammet. Undersøges om der er naphthalener tilstede ved at udtrække ionspor af de karakteristiske ioner, giver det et meget fint udslag i kromatogrammerne som vist i figur 11-16 – 11-19. Toppen der ses ved retentionstiden 15,8 minut er den interne standard o-terphenyl. Figur 11-15. Kromatogram af benzin, der er inddampet på rotationsfordamper til næsten tørhed. Der ses fortsat et stort indhold af flygtige forbindelser med retentionstid ca. 5 min. til 10 min. Figur 11-16. Inddampet benzin. Naphthalen ionspor 128 m/z. Naphthalen har retentionstid 9,85 min. Ion 230 m/z viser hvor den interne standard o-terphenyl kommer. Figur 11-17. Inddampet benzin. Methylnaphthalener. Ionspor 142 m/z. 2-methylnaphthalen har retentionstid 11.0 min. og 1-methylnaphthalen 11,2 min. Figur 11-18. Inddampet benzin. Dimethylnaphthalener. Ionspor af ion 156 m/z. Retentionstid fra ca. 11,9 – 12,6 min.
Figur 11-19. Inddampet benzin. Trimethylnaphthalener. Ionspor af ion 170 m/z. Retentionstid fra ca. 12,6 – 14,0 min. Naphthalenerne forekommer kun i ringe mængde i motorolie og lignende produkter med kogepunktsinterval > n-alkan C25. Olieprodukter som f. eks. rå- og fuelolie, der har et kogepunktsinterval, der spænder over det meste af det kromatografiske område har derimod naphthalenerne rigt repræsenteret. Naphthalenerne forekommer ikke i samme grad eller slet ikke i rene jordprøver dvs. prøver, hvor der kun er kulbrinter, der stammer fra naturlige kulbrinter. Indholdet af naphthalener varierer afhængigt af olietype og destillationsinterval. I forbindelse med forvitring af aromater i olie i naturen, forsvinder naphthalenerne først, således at kompositionen af de homologe rækker af hhv. naphthalener, phenanthrener, dibenzothiophener, fluorener og chrysener forandres /9/. Dette gør, at i stærkt forvitrede gasolier er naphthalenerne stort set væk. Naphthalenerne ses derfor kun i forholdsmæssig frisk olie, og ikke i komplet nedbrudt eller forvitret olie. Tilstedeværelsen af disse i en jordprøve indikerer, at der er kulbrinter af petrogen oprindelse. Indholdet af naphthalener (sum af naphthalen, methylnaphthalen, di- og trimethylnaphthalener) i forhold til mængden af totalkulbrinter er afbildet i nedenstående figur 11.20.
Figur 11.20. Indhold af naphthalener (sum) i forhold til totalkulbrinter. Naphthalenerne er hovedsaligt dominerende i kulbrinteprodukter med et kogepunktsinterval svarende til gasolie (let olie, Kp. ca. 170°C – 400°C). Shell dieselolie har et samlet indhold af naphthalener på 1,0 mg/kg TS i en prøve, hvor der er tilsat gasolie svarende til 50 mg/kg TS. 11.9.2 PhenanthrenerPhenanthrenerne udgør som naphthalener en homolog række, og såfremt der er indhold af de methylerede phenanthrener i en given jordprøve indikerer dette tilstedeværelsen af kulbrinter af petrogen oprindelse. Phenanthren i sig selv forekommer i stor mængde i forskellige vegetabilske olie (specielt i olivenolie, rapsolie mm. egne analyser), og indhold af phenanthren i sig selv er ikke en indikator for indhold af kulbrinter af petrogen oprindelse. Methyl- og dimethylphenanthrener er derimod mere karakteristiske for petrogene kulbrinter, og disse kan anvendes som indikation af indhold af petrogene kulbrinter. Fordelingen af methylphenanthrenisomererne anvendes bl.a. til kildeopsporing, og forholdet mellem visse af isomererne anvendes som modenhedsindikatorer bl. a. /16,18/. Phenanthren identificeres i GC-MS-scan ved ionspor 178 m/z, methylphenanthrener ved 192 m/z, dimethylphenanthrener ved 206 m/z og trimethylphenanthrener ved 220 m/z. Højere methylerede phenanthrener er ikke undersøgt i nærværende projekt. Figur 11-21 viser ionspor af de karakteristiske ioner i en olieforurenet jordprøve. Figur 11-21. Ionspor af hhv. methylphenanthrener (sort), dimethylphenanthrener (grøn) og trimethylphenanthrener (blå). Responsfaktorerne fremgår af bilag 10. Responsfaktorerne for de forskellige grupper er beregnet, og forskellen i respons er anvendt i forbindelse med beregninger af indholdet i de senere afsnit af rapporten. Indhold af de homologe rækker af phenanthrener indikerer kraftigt, at der er petrogene kulbrinter i en prøve. Phenanthrener (sum af phenanthren, methylphenanthren, di- og trimethylphenanthrener) er afbildet som funktion af indholdet af totalkulbrinter i figur 11.22 for en række eksempler af mineralolie.
Figur 11.22. Indhold af phenanthrener (sum) i forhold til total kulbrinter. Indholdet af summen af phenanthrener i forhold til total kulbrinter i de testede olier er mere lig hinanden i forhold til sammenhængen mellem naphthalenerne og totalkulbrinter. Phenanthrenerne i en jordprøve med ikke-forvitret Shell dieselolie svarende til 50 mg/kg TS er ca. 0,15 mg/kg TS. For 100 mg/kg ikke-forvitret Shell dieselolie er indholdet af phenanthrener ca. 0,40 mg/kg TS. 11.9.3 Dibenzothiophener, fluorener og chrysenerDibenzothiophen mønsteret er som phenanthren forholdsmæssig let genkendelig, såfremt den pågældende olie har kogepunktsinterval i området omkring disse. Hvis der er phenanthrener tilstede, er der meget stor sandsynlighed for, at dibenzothiophenerne også er tilstede. Dibenzothiophenerne kan anvendes som biomarker i forbindelse med olieidentifikation. Figur 11-23 viser mønster for dibenzothiophener. Figur 11-23. Eksempel på GC-MS af methyldibenzothiophener /26/. Til venstre ses øverst monomethyldibenzothiophener (ionspor 198 m/z), midt i dimethyldibenzothiophener (ionspor 212 m/z), og nederst trimethyldibenzothiphener (ionspor 226 m/z). Til højre er øverst vist monomethyldibenzothiphen ionsporet af fuelolie, samt 3 forskellige olier. Methyldibenzothiophener er i vid udstrækning anvendt som specifik petrogen kulbrinte for råolie, da fordelingen mellem isomererne er forholdsmæssig resistent overfor forvitring /16,18,26/. Det er valgt ikke at kvantificere dibenzothiophenerne, idet disse forekommer i meget små mængder, og det kræver oprensning og inddampning af analyseekstraktet for at kunne opnå de ønskede lave detektionsgrænser (ca. 0,01 mg/kg TS). I forbindelse med dette projekt er der analyseret i scan mode på GC-MS. Dette er valgt for ikke at miste information om de enkelte prøver. Ved GC-MS-scan er følsomheden forholdsmæssig lav, og svarer nogenlunde til den følsomhed, som opnås ved GC-FID. En detektionsgrænse for enkeltkomponenter ved GC-FID er typisk 0,1 mg/kg TS. Hvis analyserne var udført i selected ion mode (SIM), hvor kun enkelte ioner undersøges, ville følsomheden være ca. 50-100 gange bedre, og det ville da være muligt at beregne indholdet af dibenzothiophenerne. Laboratoriet har i forbindelse med fastlæggelse af responsfaktorer også analyseret hhv. dibenzothiophen, methyldibenzothiophener, dimethyldibenzothiophener, og trimethyldibenzothiophener, og responsfaktorerne forholder sig nogenlunde som de alkylerede phenanthrener (methyl-, dimethyl- etc.). Fluorener og chrysener er også anvendt som specifikke petrogene kulbrinter, og i forbindelse med identifikation af råolie i en lang række artikler bl. a. /16,18/. De anvendes også i forskellige "diagnostic ratios" vedr. råolie og efterforskning af oliespild. Indholdet af disse komponenter er relativt lavt i olierne, og det er nødvendigt med en vis prøveoparbejdning af jordekstraktet for at kunne detektere disse komponenter. Sædvanligvis vil det være tilstrækkeligt med at undersøge prøverne for de øvrige nævnte specifikke petrogene kulbrinter. I specielle tilfælde kan det være nødvendigt at analysere prøver i SIM for at afgøre, hvorvidt der er kulbrinter af petrogen oprindelse i prøven. Dibenzothiophener, fluorener og chrysener kan detekteres i SIM ved deres respektive ionspor, og tilstedeværelsen af disse er i høj grad en indikator for kulbrinter af petrogen oprindelse. 12 Naturligt forekommende kulbrinter12.1 FormålFormålet med denne del af projektet er at kortlægge indholdet af naturligt forekomne kulbrinter, der giver bidrag til totalkulbrinte indholdet i forbindelse med analyse af jordprøver for indhold af olie. Udfra en undersøgelse af jordprøver fra udvalgte lokaliteter er det hensigten at identificere enkeltkomponenter og mønstre, der karakteriserer de naturlige kulbrinter. Disse kan anvendes i forbindelse med identifikation og vurdering af, hvorvidt der er naturligt forekomne ekstraherbare kulbrinter i en jordprøve. Det er målet at finde en metode til at kunne fratrække bidraget fra de naturligt forekomne kulbrinter helt eller delvist fra et indhold af totalkulbrinter. Det er målet, at kunne skelne indhold af petrogene kulbrinter fra naturlige kulbrinter i prøver, hvor der er indhold af både petrogene kulbrinter og bidrag fra naturlige kulbrinter. Dette skal danne grundlag for vurdering af indholdet af kulbrinter af petrogen oprindelse, og dermed hvorledes jorden skal håndteres. 12.2 MetodeDer er udvalgt en række lokaliteter, som repræsenterer forskellige biotoper. De giver et bredt billede af hvilke bidrag af naturlige kulbrinter, som kan forventes at give interferens ved olieanalyser. På disse lokaliteter er udtaget prøver i forskellige dybder for at kunne kortlægge typiske kulbrintesammensætninger og eventuelt nedbrydnings- og komposteringsstadier. Der er udført dobbeltbestemmelse for at illustrere den variation, som kan forventes i disse typer af prøver. Nedbrydningen af blade, grannåle, græs, fæces fra dyr etc. sker kontinuert, og denne undersøgelse giver kun et øjebliksbillede af nedbrydningsstadierne. Nedbrydningsgraden er i høj grad afhængig af årstid, jordbundsforhold og mange andre faktorer, og det ligger udenfor omfanget af denne redegørelse, at kortlægge indflydelsen af de enkelte faktorers indvirkning. For at få et billede af, hvordan omsætningen af naturlige kulbrinter i de øverste jordlag sker, er der også foretaget en række "skrab" i de øverste jordlag. Der er meget stor inhomogenitet i "skrabeprøverne", og det er ikke muligt at udføre en fuldstændig homogenisering af prøverne. Det er derfor valgt for disse prøver kun at udføre enkeltekstraktioner, for at få et indtryk af kulbrintesammensætningen. For alle prøver er udført tørstofbestemmelse, og alle resultater er anført i mg/kg TS. Der er desuden udført bestemmelse af glødetab for alle prøverne, idet denne værdi er en god indikator for mængden af organisk materiale i en prøve. 12.3 Valg af lokaliteterLokaliteterne er udvalgt efter en målsætning om, at få en så stor variation imellem jordtyper som muligt. I alt er undersøgt 9 lokaliteter: Agerland-sand, agerland-ler, eng, granskov, tørvemose, bøgeskov, indvundet landområde, strandeng og diffust forurenet lokalitet. Lokaliteterne er udvalgt efter forslag fra ingeniør Per Kjemtrup, Bascon, Århus efter anvisning fra cand. scient. Karen Halling, AnalyCen. 12.4 PrøveudtagningPrøveudtagningen blev udført i oktober måned 2002. Ved prøveudtagningen blev båret handsker og boreudstyret blev rengjort mellem hver lokalitet. Boredybder samt jordtyper (farve, lugt mm.) blev noteret. Prøverne er udtaget med håndbor, ca.10 cm ad gangen ned til 40-50 cm. Derudover er udtaget overfladeprøver samt "skrabeprøver" 2-3 cm ad gangen ned til 10 cm, ved nogle lokaliteter. Skrabene er så vidt muligt delt i jordlaget, hvor der kunne ses en naturlig overgang mellem jordlagene. Prøverne er udtaget i en duobag pose, og herfra er der efter manuel homogenisering udtaget prøver i forvejede Red-cap glas til efterfølgende ekstraktion. Dette er gjort i felten. 12.5 AnalysePrøverne fra Red-cap glas er ekstraheret efter den videreudviklede ISO/DIS 16703 metode jf. kapitel 10. Ekstrakterne er tappet i 4 GC-vials; 2 til umiddelbar analyse ved hhv. GC-FID og GC-MS og to til opbevaring i fryser til evt. senere brug. Den resterende prøve i duogas posen benyttes til geologisk bestemmelse, tørstof samt glødetab. De geologiske bestemmelser af jordtyperne og jordlagene illustreres i boreprofiler af lokaliteterne. Boreprofilerne er vedlagt i bilag 12. 12.5.1 Lokalisering af egnede lokaliteter til prøveudtagningDe "upåvirkede" lokaliteter er udvalgt udfra diverse kortmaterialer, primært fremstillet af Århus Amt, samt ved kontakt til Århus Amt, Natur og Miljø, Jonna Mosgård. Lokaliteternes anvendelighed i det pågældende projekt er ikke vurderet på forhånd ved besigtigelse. Lokaliteterne er, af hensyn til de praktiske forhold i forbindelse med prøveudtagningen, forsøgt udvalgt indenfor et snævert geografisk område, som overvejende ligger umiddelbart syd for Århus. Alle lokaliteter er undersøgt for forureningsregistreringer via Århus Amts fortegnelse over kortlagte forureninger. I det efterfølgende er en nærmere beskrivelse af lokaliteterne. H1: Agerland-sand Udbredelsen af områder med kvartære (istidsaflejringer) sandede aflejringer af moræneler eller smeltevandssand er forholdsvist begrænset, set i forhold til moræneaflejringer, som i Århus Amt er den altdominerende terrænnære aflejringstype. Området er udvalgt udfra DGU's jordartskort over Danmark. Kortet angiver jordarten i den øverste meter. Lokaliteten er beliggende NØ for Odder. H2: Agerland-ler Den dominerende aflejring umiddelbart under dyrkningslaget består de fleste områder i Århus Amt af moræneler. Placeringen af lokaliteten er derfor kun udvalgt udfra hensynet til de øvrige lokaliteter. Det foreslåede område, er udvalgt udfra DGU's jordartskort over Danmark. H3: Eng Det udvalgte område er en ferskvandseng, beliggende langs en bæk mellem Mariendal og Neder Fløjstrup. Kildematerialet er Århus amts kort over Naturbeskyttelsesloven §3-områder. H4: Granskov Det udvalgte område er en ældre nåletræsplantage, beliggende i Storskoven syd for Marselisborg. Årsagen til en ældre beplantning er valgt er et ønske om et tykt muldlag. Desuden sprøjtes ældre nåletræsbeplantninger ikke. Området er udvalgt udfra flyfotos. H5: Tørvemose/højmose Århus Amt har videregivet oplysninger om en aktiv højmose (fortsat vækst af Sphagnum) beliggende ca. 15-20 km NV for Århus. H6: Bøgeskov I de afgrænsende østlige og vestlige områder til granbeplantningen findes gammel bøgeskov, som antages at være uden behandling af sprøjtemidler. Området er udvalgt udfra flyfotos. H7: Indvundet landområde/kunstigt afvandet område Området med indvundet land, hvor der kan ske påvirkning fra marine sedimenter er valgt udfra DGU's jordartskort, samt ved kontakt til Århus Amts afdeling for Landskab. Området er beliggende umiddelfart syd for Nordsminde Fjord. Områdets aflejringer er marine og vil ifølge DGU's jordartskort overvejende bestå af sandede aflejringer med et varierende indhold af skaller umiddelbart under dyrkningsarealet. Inddæmningen vurderes at være ældre end 100-150 år, da opslag på generalstabskort fra sidste halvdel af 18-tallet viser det inddæmmede område. Området benævntes tidligere Frederiksdal Inddæmningen. H8: Strandeng Området med strandeng er beliggende umiddelbart nord for det kunstigt afvandede område ved Norsminde Fjord. Området er lokaliseret udfra Århus Amts kortmateriale om NBL §3-områder. Under et eventuelt muldlag kan forventes samme aflejringer som beskrevet under H7, indvundet landområde. H9: Diffus forurenet lokalitet Lokaliteten er udvalgt i samarbejde med Århus kommunale værkers Affaldskontor, Inger Gregersen og Bo Utoft og er beliggende i Århus på ejendommen Eugen Warmings Vej 10. Ejendommen er dokumenteret ved forureningsundersøgelse med kemiske undersøgelser af jorden1. Lokaliteten er kommunalt ejet og er beliggende i Botanisk Have, som ikke er industripåvirket. Den pågældende jord er forventet at være eksponeret før 1940'erne. Der er fundet forhøjet indhold af bly og PAH'er. Fyldjord til mindst 0,5 m. 12.5.2 GC-MS-scan analyseSelve detektionen ved GC-MS-scan er foretaget ved et Agilent 5973MSD quadropol instrument. Den gaschromatografiske metode og scanparameter instillinger fremgår af bilag 11. Der er foretaget sammenligning mellem fundne komponenter og MSNist databasen, som er en database med over 100000 forskellige komponenters massespektre. Databasen undersøger sammenfaldsprocenten mellem den aktuelle komponents massespektrum i prøven og med databasen. Dette "hit" er anført i procent sammenfald. Et hit på 80% og derover anses for en tilfredsstillende sammenfaldprocent. Den komponent, som databasen foreslår, er ikke nødvendigvis korrekt. Det er nødvendigt, at vurdere den aktuelle kemiske stofgruppe, som databasen foreslår; er det sandsynligt, at den foreslåede komponent kan forekomme i ren jord. Samtidig er det nødvendigt at vurdere, om den foreslåede komponent har et kogepunkt, der svarer til den retentionstid, som den er kromatografieret ved i den gaskromatografiske analyse. Ved GC-MS-scan analyse af råekstrakterne ses kun de komponenter, der dels er ekstraherbare i acetone/pentan blandingen (analysemetoden i kap. 10), og dels kan kromatografieres ved gaskromatografi. Der er mange komponenter i planter, og når der ikke indledningsvis er foretaget en fraktionering af jordekstraktet med henblik på f. eks. polaritet, er det en meget kompleks blanding at undersøge ved GC-MS-scan. De opnåede massespektre af hver enkelt top ved GC-MS-scan analyse af topprøverne (0 – 10 cm) er indledningsvis undersøgt. Der er i bilag 13 vist total ion kromatogrammet (Total Ion Chromatogram = TIC) af den ene af de dobbeltbestemte topprøver. Der ses en række sammenfaldende træk i forbindelse med identifikationen af enkeltkomponenter i alle lokaliteter. Der er fundet en gruppe stoffer med retentionstid fra ca. 15 til 18 minutter, som kan karakteriseres som "syrer". Det er en blanding af flere isomerer, men hovedsagligt er der tale om estre af umættede fedtsyrer n-C14:0, n-C16:0 og n-C18:0 syrer, men også forskellige mættede fedtsyrer. Det er svært at få et fornuftigt hit i MSNist, da mange af disse komponenter er sammenfaldende i kromatogrammet, og de opnåede massespektre ikke er "rene". Fedtsyrerne er omtalt under afsnit 12.6. Der ses en gruppe af stoffer med retentionstid fra ca. 22 minutter til 28 minutter. Denne stofgruppe karakteriseres som phytosteroler, og er omtalt under afsnit 12.7. De ulige n-alkaner som forefindes i stort set alle prøver med terrestrisk organisk bidrag er omtalt under afsnit 12.8. For at kunne vurdere nedbrydningspotentialet af udvalgte komponenter, er de enkelte kromatogrammer integreret manuelt udfra TIC kromatogrammet. Udfra retentionstiden er de enkelte komponenters indhold fulgt dels gennem boreprofilprøverne og dels i skrabeprøverne. For at vurdere ændringen i de enkelte komponenters indhold i forhold til totalkulbrinteindholdet, har det været nødvendigt at beregne totalkulbrinteindholdet ved GC-MS-scan. Dette er udført overfor n-alkaner, og resultatet for totalkulbrinteindholdet er kun en approksimation. Indholdet af totalkulbrinter i prøverne ved GC-FID er også beregnet og fremgår at tabeller i bilag 14. Enkeltkomponenterne er anført i procent i forhold til totalkulbrinteindholdet. Denne værdi er ikke eksakt, da der ikke er foretaget en egentlig kvantificering af disse overfor referencestoffer. Følgende tilnærmelser er anvendt;
Resultaterne af beregningerne fremgår af tabellerne i bilag 14. 12.6 Fedtsyrer.12.6.1 ForekomstFedtsyrer forekommer hovedsagligt i planter i bunden form, esterificeret til glyceroler, som fedt eller lipider. Disse lipider kan udgøre op til ca. 7% af tørvægten i blade i højere planter, og er også et vigtigt indholdsstof i cellemembraner, chloroplast og mitochondria i planter /19/. Lipider forekommer også i højt indhold i frø eller frugter fra mange planter, og udgør et lager af energi til brug ved senere spiring. Visse plante frø har et meget stort indhold af lipider f. eks. oliven, raps mm., og dette udnyttes kommercielt i forbindelse med fødevarer. En lille del af fedtsyrerne forekommer som estre, hvor der f.eks. sidder en methylgruppe på syregruppen. Denne gruppe er tilstrækkelig upolær til at de ved ekstraktion overføres til pentanfasen. Ved GC-MS-scan detekteres disse ved retentionstider fra ca. 15 minutter til 18 minutter. Fedtsyrefordelingen er meget plantespecifik, og i nærværende rapport er dette emne ikke omhandlet. 12.6.2 Identifikation – 73 m/z – retentionstidFedtsyrerne kan lokaliseres ved GC-MS-scan ved at "ekstrahere" ion 73 m/z. Figur 12-1 og 12-2 viser hhv. TIC og ionspor af 73 m/z.
Figur 12-1. TIC af H19 fra lokalitet H3 eng.
Figur 12-2. Ionspor af ion 73 m/z af H19 (0-10 cm). Toppe ses der, hvor fedtsyrer kromatografieres. Typisk ses en lille gruppe af toppe umiddelbart før den interne standard o-terphenyl, der her har retentionstiden 15,89 minutter, og umiddelbart efter ses igen to grupper ved ca. 16,2 minutter og 17,3 minutter. 12.6.3 Fedtsyrers samlede indhold i forhold til totalkulbrinterFedtsyrernes indhold i forhold til totalkulbrinteindholdet fremgår af bilag 14. Indholdet er anført i procent som "syrer". Dette skyldes, at der ikke altid har været en entydighed i det massespektrum som MSNist har anført, og ikke alle toppe er medregnet i syreindholdet. Indholdet af syrer ligger relativt lavt i forhold til totalkulbrinter. Indholdet er højest i skrabeprøverne, hvor der er græs tilstede f. eks. fra ferskvandsengen – lokalitet H3, hvor indholdet af syrer i profilprøverne i 0 – 10 cms dybde er hhv. 17,7% og 18,4%. Indholdet af syrer er forholdsmæssigt højt ved denne lokalitet helt ned i 30 cms dybde, hvor der er gennemsnitligt 8,6% "syrer". Indholdet af "syrer" er bestemt i en prøve af græs fra H1, agerlandsand. Her er indholdet af syrer 10,6%. Der skal dertil nævnes, at græsprøven ikke er findelt til analysen, men at der er ekstraheret på helt materiale. Resultaterne er derfor kun et fingerpeg af, hvad man kan forvente at finde. 12.6.4 NedbrydningspotentialeSyrerne nedbrydes hurtigt i forhold til totalkulbrinteindholdet, og forekommer kun i højt indhold (> 10%), hvor der er synlige planterester. Syrerne synes også hovedsagligt at være tilknyttet områder, hvor der er græsser på overfladen. Det må derfor konkluderes, at syrernes indhold kun har en indflydelse på totalkulbrinteindholdet i de prøver, hvor der ses synlige planterester. 12.7 PhytosterolerPhytosteroler er triterpenoider, der dannes i planter via terpenbiosyntesen, som vist i figur 12-3
Figur 12-3. Terpenoid biosyntesevej i planter /20/. Terpener er dannet udfra isopren enheder, der er C5 enheder. Eksempler på forskellige triterpener er vist i nedenstående figur 12-4.
Figur 12-4. Strukturer af en række triterpenoider /20/. 12.7.1 ForekomstPhytosteroler forekommer i stort set alle planter, og har forskellig aktivitet i planterne. Phytosteroler er i høj grad planteartsspecifikke, men der er visse phytosteroler, der forekommer i faktisk alle de undersøgte lokaliteter. Tre phytosteroler forekommer i alle planter; sitosterol, stigmasterol og campesterol. De er alle essentielle komponenter i plante cellemembraner, og de forekommer frit i planterne, eller som simple 3-glucosider. Indholdet af disse varierer i planterne afhængigt af hvilken plante, der er tale om. Andre phytosteroler kan f. eks. være alpha-spinasterol, der er en isomer af stigmasterol, som findes i spinat og lucerne, brassicasterol, der er en plante celle membran komponent, og fridelin som findes i en lang række lichener mv./19/.
12.7.2 Identifikation – 93 m/zDa phytosteroler oprindeligt er dannet udfra isopren-enheder, følger fragmentering ved massespektrometri et nogenlunde ensartet mønster. Det har vist sig, at der for stoffer dannet udfra isopren-enheder(herunder terpener) forekommer et karakteristisk fragment på 93 m/z. Dette fragment forekommer i stort set alle phytosteroler. Figur 12-5 viser et eksempel på fragmentering af terpenen b-myrcen. Figur 12-5. Eksempel på fragmentering af terpener /22/ Lokalisering af hvor phytosteroler forekommer i kromatogrammet, kan foretages ved at udtrække ionspor 93 m/z i et GC-MS-scan kromatogram. Indholdet af ion 93 m/z svinger meget fra stof til stof, og kan ikke anvendes som en egentlig kvantificeringsion. 12.7.3 Sitosterol og sitostenonSitosterol og sitostenon forekommer i stor set alle de undersøgte lokaliteter. Typisk ser et kromatogram ud ved både GC-MS-scan og GC-FID som nedenstående figur 12-6, hvor sitosterol er den store top ved retentionstiden ca. 24 minutter og sitostenon kommer ud med retentionstiden ca. 25 minutter. Figur 12-6. Lokalitet H6: Bøgeskov 0-10 cm. Den interne standard o-terphenyl har retentionstiden 15,9 minutter. Udtrækkes ionspor 93 m/z ses udslag netop der, hvor der forventes at være phytosteroler. Dette er illustreret i figur 12-7. Figur 12.7. Udsnit af kromatogram fra prøve udtaget i bøgeskov, 0-10 cm. Ionspor af ion 93 m/z. Sitosterol ses ved retentionstiden ca. 24 minutter. Massespektrum at sitosterol er vist i figur 12.8. Figur 12.8. Massespektrum af sitosterol. (Match quality 90%). Indholdet af sitosterol er beregnet for alle de rene lokaliteter i forhold til totalkulbrinter ved GC-MS. Resultaterne fremgår af tabellerne i bilag 14. Indholdet ligger nogenlunde konstant i forhold til indholdet af totalkulbrinter. Der ses dog en forskel i indholdet fra lokalitet til lokalitet. For eksempel er indholdet af sitosterol i overfladeprøverne (0 – 10 cm) for agerland-sand gennemsnitligt 1,0% af totalkulbrinteindholdet, og tørvemosen er indholdet helt oppe på 16,8%. Det er derfor ikke umiddelbart muligt at lave en korrelation mellem indholdet af sitosterol og indholdet af naturligt forekommende kulbrinter. Idet sitosterol forekommer i stort set alle prøver, kan den fint anvendes som markør for, hvorvidt der er bidrag fra naturlige kulbrinter. Sitosterol er også den phytosterol, som forekommer i det højeste procentvise indhold af alle de øvrige phytosteroler. 12.7.4 StigmasterolStigmasterol er som sitosterol en markør, der må forventes at kunne findes i stort set alle prøver, hvor der er indhold af naturligt forekommende kulbrinter. Retentionstiden for stigmasterol er ca. 23,5 minutter (sitosterol ca. 24 min), og indholdet er forholdsmæssigt mindre end sitosterol (ca. 1/3). Indholdet af stigmasterol er kun med sikkerhed identificeret i 2 af de undersøgte lokaliteter og det procentvise indhold er beregnet i disse. 12.7.5 Andre phytosteroler (Campesterol, brassicasterol, fridelin, m. fl.)En hel række andre phytosteroler er identificeret i de forskellige lokaliteter, og nedenstående tabel 12-1 viser hvilke komponenter, der er identificeret og beregnet i de forskellige lokaliteter
Tabel 12-1. Oversigt over indhold af udvalgte phytosteroler, der er undersøgt. Campesterol forventedes identificeret i alle prøver, men er kun med sikkerhed identificeret i H2; Agerland-ler og H8; Strandeng, hvilket indikerer, at denne komponent primært er tilknyttet lokaliteter med forskellige græsser. Campesterol findes i alle højere plantearter, og er et strukturstof i plante celle membraner /19/. Brassicasterol er som stigmasterol en komponent i plante cellemembraner. Denne komponent er ikke identificeret med sikkerhed i nogle af lokaliteterne, og hvis det er brassicasterol, så kun i et meget lille indhold. Retentionstiden er ca. 22,8 minutter. Fridelin findes i et forholdsmæssigt højt indhold i tørvemosen, lokalitet H5. Desuden er der fundet et forholdsmæssigt højt indhold i bøgeskoven, lokalitet H6. Indholdet er her mellem 0,8% (H9, 20 – 30 cm) og 2,3% (B17, 0-10 cm) i forhold til det samlede totalkulbrinteindhold. Til gengæld finder man ikke fridelin i skrabeprøverne, hvilket indikerer, at indholdet af fridelin her må stamme fra svampe, lichener mm. der er den del af komposteringsfaunaen i en bøgeskov. 12.7.6 Phytosterolers samlede indhold i forhold til totalkulbrinterDet samlede indhold af phytosteroler er skønnet til at være alle enkelttoppe med retentionstid fra og med sitosterol (Rt. Ca. 23,9 min) og fremefter. I forbindelse med de indledende beregninger, er alle enkelttoppe i TIC-et (resolved peaks) fra og med sitosterol summeret, og indholdet beregnet i forhold til totalkulbrinteindholdet (GC-MS-scan). Resultaterne af denne approksimation fremgår af tabellerne i bilag 14 (phytosteroler, total %). Indholdet af phytosteroler varierer noget fra den ene lokalitet til den anden. Det højeste indhold af phytosteroler ses i tørvemosen, lokalitet H5, 40 – 50 cm, bestemmelse 1 (A3) hvor indholdet udgør 40% af totalkulbrinte indholdet. Sitosterol i denne prøve udgør i alt 23,9% af totalkulbrinte indholdet (i alt 55 mg/kg ved GC-MS scan bestemmelse). Tørvemosen er lidt speciel, hvad angår indholdet af totalkulbrinter og arten af disse. Der ses et meget stort indhold af organisk stof i alle prøver, hvilket er udtrykt ved glødetabsbestemmelserne, der varierer fra 93,4% (vegetationsprøve) til 12,5% (40 – 50 cm). Sitosterol er den helt dominerende phytosterol med indhold varierende fra 11,4% (A4, 40 – 50 cm) til 40% (A3, 40 – 50 cm). 12.7.7 NedbrydningspotentialeBetragtes det procentvise indhold af phytosteroler i forhold til den dybde, hvori prøverne er taget ses et svagt fald i indhold ned gennem boreprofilen for en række af lokaliteterne, intet fald for andre, og en stigning i igen andre (tabel 12-2).
Tabel 12-2. oversigt over ændring i indhold af total indhold af phytosteroler i forhold til totalkulbrinter Nedbrydningen af phytosterolerne vurderes til at være afhængig af, dels hvor meget organisk materiale der tilføres jorden, dels hvor fugtig jorden er, og desuden om der sker en bearbejdning af jorden. Landbrugsarealerne H1 og H2 har et forholdsmæssigt konstant indhold af phytosteroler. Her sker der ca. 2 gange årligt en pløjning af jorden, således at det organiske materiale er jævnt blandet. I granskoven, bøgeskoven og strandengen er der et meget stor tilførsel af organisk materiale fra blade og nåle fra træerne. Derfor ses en stor omsætning fra de øverste lag og nedefter i dybden. I Botanisk Have er der græs, der bliver klippet nogle gange årligt, og den samlede tilførsel af organisk materiale er begrænset. Phytosterol indholdet ses derfor at være rimelig konstant i dybden. Tørvemosen har et konstant indhold af phytosteroler. Dette må skyldes, at der fortsat er et meget stort uomsat indhold organisk materiale selv i 50 cms dybde. Det inddæmmede område har også et rimeligt konstant højt indhold af phytosteroler. Dette må også forklares ved, at der er et stort indhold af organisk ikke omsat materiale. Glødetabsbestemmelsen for det inddæmmede område viser, at indholdet af organisk materiale er jævnt stigende fra 10,2% (C14, 0 – 20 cm) til 16,3% (100 – 120 cm), hvilket kan forklare det høje indhold af phytosteroler. 12.8 Ulige n-alkaner12.8.1 ForekomstI olieprodukter forekommer n-alkanerne altid med en ligelig fordeling. Dette er gældende selv ved raffinerede produkter som terpentin, petroleum, gasolie og fuelolie. Plante voks/paraffiner indeholder kulbrinter, specielt lang-kædede n-alkaner med overvægt af molekyler med et ulige antal af kulbrinter /21/. Tilstedeværelse af ulige n-alkaner, som stammer fra planter, giver sig udtryk i en ulige fordeling af n-alkanerne. Dette er omtalt under beregning af CPI jf. afsnit 11.4, som netop beskriver dette. N-alkanerne kan derfor give en indikation om bidrag fra terrestrisk organisk plantemateriale. 12.8.2 Identifikation – 57 eller 71 m/zEn forholdsvis enkel metode at identificere n-alkanerne på, er ved GC-MS-scan at udtrække ionspor 57 m/z eller ion 71 m/z, der er fragmenter af hhv. CH3(CH2)3+ eller CH3(CH2)4+. Ved GC-FID kan n-alkanerne også identificeres relativt nemt, hvis der ses spor af den regelmæssige forekomst af n-alkanerne, som fremkommer pga. deres lineære kogepunktsforløb (se f. eks. /10/). 12.8.3 NedbrydningspotentialeNedbrydningspotentialet for de ulige n-alkaner er som for phytosterolerne beskrevet i en generel tabel, tabel 12-3, hvor hver enkelt lokalitet er omtalt for sig.
Tabel 12-3. Oversigt over ændring i indhold af total indhold af de ulige n-alkaner i forhold til totalkulbrinter (* sidste 4 prøver er stort set rene). Tabellen viser, at indholdet af n-alkaner ændres med dybden, og ændringen er meget forskellig fra lokalitet til lokalitet. Der er stigning i det procentvise indhold af n-alkaner i prøver med stort bidrag fra plantemateriale; H3, Ferskvandseng, H4, Granskov og H6, Bøgeskov. Resultaterne kan tolkes som om, at n-alkanerne i disse lokaliteter ikke nedbrydes så hurtigt i forhold til de øvrige indholdsstoffer i jorden. 12.8.4 Indhold af ulige n-alkaner i forhold til totalkulbrinterIndholdet af de ulige n-alkaner i de enkelte rene lokaliteter beregnet i forhold til totalkulbrinter ved GC-MS ses af bilag 14. Summen af indholdet af n-alkan C25, C27, C29 og C31 er beregnet. Fordelingen af n-alkanerne i de forskellige lokaliteter er ikke ens. Der anes en tendens til, at der er overvægt af de lettere kogende ulige n-alkaner i de prøver, hvor der er et stort bidrag af blade og nåle som f. eks i H4; Granskov og H6; Bøgeskoven. Indholdet er højest i lokalitet H6, bøgeskoven i 30 – 40 cms dybde (F7) med et indhold på 28,7%.Generelt ses laveste indhold i lokalitet H8, strandeng (gennemsnitligt 1,6%). Indholdet af de ulige n-alkaner, der stammer fra plantemateriale giver et ikke ubetydeligt bidrag til det samlede totalkulbrinteindhold i prøverne fra de rene lokaliteter. 12.9 UCM for udvalgte lokaliteter.Totalkulbrinte indholdet og UCM er beregnet for alle de udvalgte lokaliteter ved hhv. GC-FID og GC-MS-scan. Resultaterne fremgår af bilag 14. I forbindelse med beregningerne af enkeltkomponenter i de udvalgte lokaliteter, er der foretaget en manuel integration af alle enkelttoppe i TIC kromatogrammet. Summen af disse er anvendt til beregning af UCM ved GC-MS-scan. UCM beregnet ved GC-MS-scan er stort set konstant ned gennem boreprofilerne for alle lokaliteterne og de gennemsnitlige UCM for både GC-MS-scan og GC-FID er beregnet. Tabel 12-4 viser den forskel, det giver at beregne ved de to teknikker.
Tabel 12-4. Oversigt over gennemsnitlig UCM for de forskellige lokaliteter beregnet ved hhv. GC-MS-scan og GC-FID. Som det fremgår af ovenstående figur, ses der forholdsmæssig stor forskel på UCM beregnet fra GC-MS-scan eller GC-FID. Principielt er beregningen den samme, men det ser ud som om, UCM er større ved GC-FID end GC-MS-scan. Dette tyder på, at de cykliske mættede forbindelser og andre, som danner UCM, ikke giver så stort respons ved GC-MS-scan som ved GC-FID. I forhold til olierne beskrevet under afsnit 11.5 ses et forholdsmæssigt lavere gennemsnitlig UCM. Lavest er H5; Tørvemosen med 74% UCM (FID), og højeste er H1; Agerland sand med 92% (FID). Der vil således altid vil være en kromatografisk uopløst bule uanset, om der er tale om petrogene eller naturlige kulbrinter. Det er ikke umiddelbart muligt at opklare indholdet af denne. Som en generel regel vil man kunne sige, at hvis der ikke er andre indikationer på, at der er petrogene kulbrinter tilstede i en given prøve, og de sædvanlige naturlige kulbrinter er tilstede samtidig med, at UCM er under 75%, er der med meget stor sandsynlighed tale om, at prøven udelukkende består af naturlige kulbrinter. 12.10 Specielle lokaliteter12.10.1 Gytje fra Københavns havn (opfyldte arealer)I en lang række analyserede prøver fra Københavns indre havn, er der fundet meget store indhold af kulbrinter med kogepunktsinterval fra ca. 280°C til ca. 350°C. Området hvor disse karakteristiske kulbrinter er fundet har hovedsagligt været indenfor Refshalevej, figur 12-9.
Figur 12-9. Kort over Dokøen og omkringliggende øer. Prøverne er typisk fra ca. 4-5 meters dybde nær den oprindelige havbund i et lag af gytje. Et karakteristisk kromatogram for denne type af kulbrinter er vist i figur 12-10 i et GC-MS-scan af ekstrakt af en udvalgt prøve. Indholdet af kulbrinter i denne prøve er bestemt ved GC-FID til 13000 mg/kg TS. Figur 12-10. Kromatogram af prøve fra Nyholm, ca. 3,0 – 4,2 mut., Københavns havn. Området fra ca. 15,0 minutter til 19,0 minutter er forstørret op herunder i figur 12-11. Figur 12-11. Samme prøve som figur 12-9, dog er tidsaksen forstørret i området 15,0 – 19,0 minut. Den interne standard o-terphenyl ses ved retentionstiden 15,67 minut. Det er forsøgt at identificere de enkelte toppe i kromatogrammet, og en række sandsynlige komponenter er foreslået ved sammenligning med MS Nist spectral database. Følgende komponenter vist i figur 12-12 – 12-15 er identificeret med stor sandsynlighed (high match quality): Figur 12-12. Top Rt. 16,55 min. 4b,5,6,7,8,8a,9,10-octahydro-4b,8-dimethyl-2-isopropylphenanthren. Øverst ses massespektrum af komponenten i den aktuelle prøve, og nederst den foreslåede komponent fra databasen. Overensstemmelse (Match quality) er på 90%, hvilket anses for en tilfredsstillende overensstemmelse. CAS nr.: intet anført. Figur 12-13. Top med Rt. 17,06 min. Bis(1-methylethyl)-1,1'-bisphenyl. Match quality 90%. CAS nr.: 069009-90-1 Figur 12-14. Top med Rt. 17,82 min. 1-Methyl-7-(1-methylethyl)-phenanthren. Match quality 90%. CAS nr.: 483-65-8 De øvrige komponenter i kromatogrammet har det ikke været muligt at identificere med sikkerhed. Den top, der forekommer med retentionstiden 17,82 min, 1-methyl-7-(1-methylethyl)phenanthren, også kaldet reten, er tidligere set i forbindelse med forbrænding af nåletræ /23/. Reten kan ikke anvendes som en specifik indikator for denne type kulbrinter, da reten også forekommer i olieprodukter /18/.Sammenlignes kromatogrammet fra figur 12.10 med et kromatogram af træskibstjære, fremstillet efter originale procedurer [20] , ses der en del sammenfaldende toppe i kromatogrammerne figur 12-15, GC-FID). Figur 12-15. GC-FID kromatogram af trætjære. GC-MS-scan kromatogrammet af træskibstjære er vist i nedenstående figur 12.16. Figur 12.16. GC-MS-scan Kromatogram af træskibstjære. Toppene med retentionstid 17,2 min.og 17,7 min. er identiske med toppene ved hhv. 16,6 min. og 17,1 min. i prøven fra Nyholm. Da der er monteret ny GC-kolonne, er retentionstiden rykket. Reten ses ved retentionstiden 18,5 min. Den store top ved retentionstiden 19,1 min. er formentlig nedenstående komponent vist i figur 12.17. Figur 12.17. Massespektrum af komponent ved retentionstiden 19,1 min. i træskibstjære Match quality 96%. CAS Nr.: 1235-74-1, Methyl dehydroabietat. Denne komponent findes også i den undersøgte prøve, dog er match quality ikke særlig høj, og indholdet er væsentligt lavere end i den friske træskibstjære. En delprøve af den undersøgte prøve er sendt til undersøgelse ved Nationalmuseet Marinarkæologisk afdeling. Prøven, der dels bestod af hele stumper af træ, blev vurderet til at bestå af kerneved fra moseeg. De historiske begivenheder i netop dette område bekræfter, at der har været stor aktivitet i havneområdet med fund af flere skibsvrag af træ. (Information fra Nationalmuseets Marinearkæologiske Afdeling). Kulbrinteindholdet kan derfor evt. stamme fra impregnering af skibe, pæle el. lign. med trætjære. 13 Tilsætning af olie til kompost13.1 FormålFormålet med denne forsøgsrække er at undersøge beregningsteknikken vedr. de naturlige kulbrinter, samt hvor stor indflydelse indholdet af phytosteroler har på totalkulbrinteindholdet. Det er hensigten at bevise, at indholdet at phytosteroler med stor præcision kan fratrækkes indholdet af totalkulbrinter, selvom der er petrogene kulbrinter med stort set samme retentionstid som disse. 13.2 Oprindelse af kompostenDen udvalgte kompost, som er anvendt til tilsætningsforsøgene stammer fra en anlægsgartner ved Kolding. Komposten består udelukkende af forskelligt plantemateriale uden tilsætning af slam mv. Det er hovedsagligt plantedele fra forskellige buske og træer (hovedsagligt stammer) og uden græsser og tilsvarende mindre frøplanter. Plantedelene er indledningsvis findelt med en flis maskine, og efterfølgende har stakken komposteret passivt. Kompoststakken var ca. 50 m x 5-7 m x 2,5 m. Stakken er vendt hver 2. måned, og komposten er ca. 1 år gammel. 13.3 Homogenisering og analyseKomposten er først blevet sigtet over et fintmasket soldevæv (maskestørrelse ca. 2 mm) for at fjerne større sten, grene m.v. Jorden har herefter gennemgået en omhyggelig sammenblanding. 4 delprøver blev udtaget til analyse for totalkulbrinter jf. analyseforskrift kapitel 10, og de opnåede resultater er vist i tabel 13-1.
Tabel 13-1. Resultat af homogenitetstest af kompost. Tørstofindholdet er bestemt til 79,25. Inhomogenitet udtrykt ved RSD på i alt 5% anses for at være acceptabelt. 13.4 Identifikation af indholdsstoffer i kompostenTo kompostprøver er undersøgt ved GC-MS-scan (figur 13-1), og de enkelte indholdsstoffer i komposten er forsøgt fastlagt. Figur 13-1. GC-MS-scan kromatogram af ren kompost. For identifikation af ulige n-alkaner er der udtrukket ionspor 71 m/z og fedtsyrerne er identificeret udfra massespektre og ionspor af ion 73 m/z. Phytosterolerne blev identificeret ved hjælp af massespektre, referencestoffer, og dels ved ionspor af ion 93 m/z. 13.4.1 Indhold af fedtsyrerIndholdet af fedtsyrer i komposten er ubetydeligt. Komposten har en alder af ca. 1 år, og der var ikke synlige græsrester i komposten. Sandsynligheden for, at der er fedtsyrer tilstede jf. tidligere beskrevet, er dermed meget lille. 13.4.2 Indhold af phytosterolerIndholdet af phytosteroler i den "rå" kompost er beregnet til hhv. 72,6 mg/kg TS og 75,2 mg/kg TS, dvs. 34,2%. Indholdet af sitosterol udgør 7% af den samlede mængde phytosteroler. 13.4.3 Indhold af ulige n-alkanerIndholdet af de ulige n-alkaner er beregnet til hhv. 0,15 mg/kg TS og 0,37 mg/kg TS. Dette er et relativt lavt indhold. Kompoststakken er 1 år gammel, og den er hovedsagligt lavet udfra træflis. Dette indikerer, at når der ikke har været brugt blade og græs til at fremstille komposten, forekommer der ikke så stort et indhold af de ulige n-alkaner. Dette er også indlysende, da de ulige n-alkaner hovedsaligt er tilknyttet de grønne plantedele – blade, stængler mm. 13.5 Genfindingsforsøg13.5.1 FormålFormålet med disse genfindingsforsøg er at fastlægge nogle værdier for udvalgte specifikke petrogene kulbrinter for rene olier samt beregne de forskellige indeks på flere koncentrationsniveauer af olie. 13.5.2 Tilsætning af gasolieSom det fremgår af nedenstående tabel 13-2, forløber genfindingen af gasolie (dieselolie fra Shell, bilag 4) således, at de høje koncentrationer har en relativ lav genfindingsprocent (89% og 81%), og de lave koncentrationer har for høj en genfindingsprocent (194% og 172%). Dette afvigelse kan umiddelbart ikke forklares, og som det fremgår af valideringen af beregningsmetoden (afsnit 9), burde der ikke være genfindingsproblemer.
Tabel 13-2. Beregnet genfinding af tilsat gasolie ved GC-FID, udfra beregning af totalkulbrinte indhold i prøverne og totalkulbrinte indhold i den "rene" kompost jord. Beregning af indhold af PAH'er, hopaner, isoprenoidforhold og naturlige kulbrinter fremgår af nedenstående tabel 13-3.
Tabel 13-3. Beregninger udført for kompost tilsat gasolie. #Indhold kvantificeret ved GC-MS-scan med korrektionsfaktor 1,7. Som det fremgår af ovenstående tabel 13-3, er indholdet af naphthalenerne og phenanthrenerne som forventet højt. Naphthalener og phenanthrenerne ses tydeligt selv ved 50 mg/kg gasolie tilsat.Indholdet af disse følger proportionalt mængden af tilsat gasolie. Der ses som forventet ikke spor af hopaner, idet hopaner ses forholdsvis i de højere kogende olieprodukter som motorolie. n-Alkan C17/pristan forholdet er nogenlunde konstant og gennemsnitligt 1,1 med en spredning på 7,7%. n-Alkan C18/phytan er ligeledes lavt, og pristan/phytan forholdet er tilsvarende lavt, og som forventet rimelig konstant. Det lave forhold indikerer, at gasolien formentlig er begyndt at forvitre. CPI (15 – 21) ligger pænt nær 1 (gennemsnitligt 1,06 med en spredning på 2,4%). CPI (25 – 33) viser nogen interferens fra naturlige kulbrinter idet værdien ligger over 1. Phytosterol indholdet er gennemsnitligt fundet til 92,8 mg/kg TS med en spredning på 7,1%. Indholdet af de ulige n-alkaner er lavt, og det indhold, der er identificeret i de højeste koncentrationer, skyldes formentlig forskellene i n-alkaner (se beregningsmetode afsnit 13-6) 13.5.3 Tilsætning af motorolieSamme kompostprøve som ovenfor er anvendt til tilsætning af motorolie (Shell X-100 Motor Oil, bilag 4). Følgende resultater er opnået (tabel 13-4).
Tabel 13-4. Beregnet genfinding af tilsat motorolie ved GC-FID, udfra beregning af totalkulbrinte indhold i prøverne og totalkulbrinte indhold i den "rene" kompost jord. Beregning af indhold af PAH'er, hopaner, isoprenoidforhold og naturlige kulbrinter fremgår af nedenstående tabel 13-5.
Tabel 13-5. Beregninger udført for kompost tilsat motorolie. #indhold kvantificeret ved GC-MS-scan med korrektionsfaktor 1,7. Der ses ingen naphthalener og phenanthrener i motorolien, men hopanerne kan detekteres selv ned til 100 mg/kg TS motorolie. Disse prøver er analyseret i GC-MS-scan. Følsomheden ville kunne forbedres væsentligt, hvis der detekteres i SIM (selected ion mode). Detektionsgrænsen kan formentlig forbedres med en faktor 10 – 50 afhængig af, hvor mange ioner, der sættes ind i vinduet, hvor hopanerne detekteres. Beregningerne, hvor isoprenoiderne og CPI-indexerne indgår, er behæftet med stor usikkerhed, da toppene er meget små, og specielt n-alkanerne, der kromatografieres oveni den uopløste bule i motorolie, er svære at identificere. Det gennemsnitlige indhold af phytosteroler er 82,1 mg/kg TS med en spredning på 14%. 13.5.4 Tilsætning af fuelolieFuelolie (Shell Fuelolie 77, bilag 4) er tilsvarende tilsat homogeniseret kompost. Resultaterne for genfindingsforsøget er vist i tabel 13-6.
Tabel 13-6. Beregnet genfinding af tilsat fuelolie ved GC-FID, udfra beregning af totalkulbrinte indhold i prøverne og totalkulbrinte indhold i den "rene" kompost jord. Beregning af indhold af PAH-er, hopaner, isoprenoidforhold og naturlige kulbrinter fremgår af nedenstående tabel 13-7.
Tabel 13-7. Beregninger udført for kompost tilsat fuelolie. #indhold kvantificeret ved GC-MS-scan med korrektionsfaktor 1,7. Som det fremgår af ovenstående tabel identificeres stort set alle de komponenter, der ledes efter. Naphthalener og phenanthrener identificeres i forholdsmæssigt store mængder. Hopanerne ses også, dog er indholdet lavere end for eksempelvis motorolien. Isoprenoidforholdene vidner om, at der er tale om et "friskt" olieprodukt. CPI (15 – 21) er pænt nær 1, som forventet for et "rent" olieprodukt. CPI (25 – 33) vidner om bidrag fra naturligt forekommende kulbrinter. Det gennemsnitlige indhold af phytosteroler er 81,6 mg/kg TS, med en spredning på 6,7%. 13.6 Beregningsmetode og GC-MS-scan metodeBeregningerne, der ligger til grund for de enkelte kvantitative tal, er givet herunder. Beregning af indhold af enkeltkomponenter;
Hvor Apr : Responset for prøven I de tilfælde, hvor der er benyttet korrektionsfaktorer, er standardens responsfaktor ganget med den pågældende korrektionsfaktor. Responsfaktorerne er beregnet, som følger:
Korrektionsfaktorer anvendes f. eks. ved beregning af isoprenoiderne ved GC-MS-scan, hvor ion 57 m/z er anvendt som kvantificeringsion. I alle analyseserier er der analyseret n-alkan C18, men ikke nor-pristan, n-alkan C17, pristan og phytan. n-Alkan C18 er derfor brugt som kalibreringsstandard til hhv. n-alkan C17, pristan og phytan med korrektionsfaktor i nærværende rapport. I følgende tabel 13-8 ses hvilke komponenter, der eksempelvis kan anvendes som kalibreringsstandarder, og hvilke ioner, der kan anbefales ved kvantificering ved GC-MS-scan.
Tabel 13-8: Valg af kvantificeringsstandarder og masser til kvantificering *= ion 113 m/z er her valgt pga., at denne ion anvendes i forbindelse med olieefterforskning /25/. I undersøgelserne i rapporten er der kvantificeret ved brug af ion 57 m/z. 13.6.1 Beregning af PAH:Indholdet af de methylerede grupper integreres som summer over hele "isomerbunken". Eksempler er vist i figur 13-2 og 13-3.
Figur 13-2. Eksempel på enkeltvis integration af methylphenanthrener. Alle 4 toppe skal integreres samlet. Ionspor 192 m/z.
Figur 13-3. Eksempel på integration af dimethylphenanthrener. Ionspor 206 m/z. 13.6.2 Beregning af indholdet af de ulige n-alkanerUlige n-alkaner = (C25-C24)+(C27-C26)+(C29-C28)+(C31-C30)+(C33-C32) 13.6.3 Beregning af indhold af phytosterolerPhytosterolerne integreres som en sum fra og med sitosterol og til der ikke ses flere phytosteroler. Dette kan gøres både ved GC-FID og GC-MS-scan. Et eksempel på integration af phytosteroler ved GC-FID er vist i figur 13-4. Figur 13-4. Eksempel på automatisk (øverst) og manuel (nederst) integration af phytosteroler i GC-FID Integrationen foretages ved enten at trække basislinie umiddelbart under phytosteroltoppene (ikke til basisliniebund). Phytosteroler ligger ovenpå UCM. Alternativt kan alle opløste toppe fra og med sitosterol og fremefter lægges sammen. Ved GC-FID kvantificeres indholdet af phytosteroler overfor de standarder, der anvendes i det pågældende kogepunktsinterval. Det er blevet undersøgt, om indholdet af phytosteroler kvantificeret på GC-MS-scan er proportionalt med GC-FID, eller om der eventuelt kan anvendes en omregningsfaktor mellem GC-MS-scan og GC-FID. Formålet ville være, at identificere phytosteroler ved ionsporet i GC-MS SIM (ion 93 m/z) og umiddelbart derefter at integrere indholdet i totalionsporet. Herved er phytosterolerne dels lokaliseret meget præcist i GC-MS-scan kromatogrammet, og den efterfølgende integration bliver udført med stor præcision på samme injektion på gaschromatografen. Indledningsvis er responset for en række phytosteroler undersøgt ved hhv. GC-MS-scan og GC-FID som vist i tabel 13-9 og 13-10.
Tabel 13-9. Udvalgte phytosterolers responsfaktorer ved GC-MS-scan
Tabel 13-10. Udvalgte phytosterolers responsfaktorer ved GC-FID. Beregnet som konc./areal. Som det fremgår heraf, ses nogen forskel på responset i GC-MS-scan af de enkelte phytosteroler, og det vil være nødvendigt at udføre yderligere undersøgelser af flere forskellige phytosteroler for at kortlægge flere responsforskelle mellem de forskellige phytosteroler. GC-FID viser bedre overensstemmelse mellem de forskellige phytosterolers responsfaktorer. Ved tilsætningsforsøgene med hhv. gasolie, motorolie og fuelolie, er indholdet af phytosteroler beregnet på hhv. GC-MS-scan og GC-FID, som vist i tabel 13-11.
Tabel 13-11. Phytosteroler. Sammenligning mellem GC-MS-scan og GC-FID. *udelukket Phytosterolerne er ved GC-MS-scan beregnet overfor n-alkan C30 i total ion kromatogrammet (TIC-et). Som det fremgår heraf, er det muligt, at foretage en ensartet integrering og kvantificering ved hhv. GC-MS-scan og GC-FID. Forskellen i indhold er gennemsnitlig en faktor 1,7. For at undersøge, om der er en sammenhæng mellem GC-MS-scan og GC-FID, der kan udtrykkes ved en faktor mellem de to kvantificeringsmetoder, er indholdet af phytosteroler i prøverne fra 0 – 10 cm fra de rene lokaliteter kvantificeret ved hhv. GC-MS-scan og GC-FID. Nedenstående tabel 13-12 viser de fundne indhold og forholdsmæssige forhold (faktor).
Tabel 13-12. Kvantificering af phytosteroler ved hhv. GC-MS-scan og GC-FID. Som det fremgår af ovenstående tabel 13-12 er der stor forskel på beregning ved hhv. GC-MS-scan og GC-FID for de forskellige lokaliteter. Det er således ikke muligt at finde en faktor, der med rimelig stor præcision kan omregne indholdet af phytosteroler beregnet ved GC-MS-scan til samme indhold fundet ved GC-FID. Forskellen beror på detektionsteknikken, hvor responset ved GC-FID er rimelig proportionalt med antallet af kulstof i et molekyle, hvorimod responset ved GC-MS-scan er proportionalt med summen af fragmenter i et givet molekyle. Indholdet af phytosteroler og kompositionen af disse er meget forskellig fra lokalitet til lokalitet. Udfra undersøgelserne vedrørende de rene lokaliteter kan følgende konklusioner drages vedrørende beregningsmetode til phytosteroler:
På baggrund af dette vurderes det som mest korrekt at udføre et eventuelt fradrag for phytosteroler fra indholdet af totalkulbrinter ved GC-FID, ved analyse af olie i jord, hvor der er mistanke om interferens fra naturlige kulbrinter. 13.7 Beregning af usikkerhed ved kvantificeringFor beregning af usikkerheden ved kvantificering ved GC-MS-scan er der udført 8 bestemmelser af to forskellige prøver, der indeholder henholdsvis naturlige kulbrinter og spor af olie. Resultaterne er vist i nedenstående tabel 13-13 (prøve 1) og 13-14 (prøve 2), og usikkerheden er udtrykt som CV%. Tabel 13-13. Prøve 1 Resultater markeret med * er udelukket fra beregningen. # beregning udført ved GC-MS-scan med korrektionsfaktor 1,7. Tabel 13-14. Prøve 2. # Beregning er udført ved GC-MS-scan med korrektionsfaktor 1,7. Den samlede usikkerhed er beregnet som den gennemsnitlige variationskoefficient i procent i tabel 13-15:
Tabel 13-15. Gennemsnitlig usikkerhed for 2 prøver med minimum 8 fold bestemmelse. Usikkerheden ved kvantificering vurderes til at være tilfredsstillende. Phytosterolerne er her kvantificeret ved GC-MS-scan, men den samme usikkerhed forventes at kunne opnås ved GC-FID. 13.8 Fastlæggelse af detektionsgrænserDet anbefales, at de enkelte laboratorier selv fastlægger detektionsgrænserne, afhængig af om der vælges analyse ved GC-MS-scan eller GC-MS SIM. Detektionsgrænserne i nærværende projekt, hvor der er analyseret ved GC-MS-scan, er skønnet udfra signal/støjforhold. Signalstørrelse er ca. 3 gange støjen på basislinien. Tabel 13-16 viser den skønnede detektionsgrænse for de enkelte grupper.
Figur 13-16. Skønnede detektionsgrænser ved GC-MS-scan 13.9 Delkonklusion petrogene kulbrinter vs. naturlige kulbrinter.Som det fremgår af kapitel 11 – 13 kan tilstedeværelse af de petrogene kulbrinter; naphthalener, phenanthrener og hopaner give en indikation om tilstedeværelse af kulbrinter af petrogen oprindelse i prøver, hvor der samtidig er stort bidrag af kulbrinter af naturlige kulbrinter. Tilstedeværelse af isoprenoiderne; farnesan, norpristan, pristan og phytan er sikker indikator for tilstedeværelse af petrogene kulbrinter. Disse kan med stor præcision kvantificeres ved GC-MS-scan. Isoprenoidforholdene n-alkan C17/pristan, n-alkan C18/phytan, Pristan/Phytan er i sig selv ikke indikator for petrogene kulbrinter, og er kun velegnede som kildeopsporing. CPI kan vise, om der er tilstedeværelse af naturlige kulbrinter (plantemateriale etc.) i en jordprøve, og hvor der forekommer ulige n-alkaner. UCM forekommer både i prøver med indhold af kulbrinter af petrogen og naturlige kulbrinter, og kan i sig selv ikke anvendes som indikation af tilstedeværelse af kulbrinter af petrogen oprindelse. Indhold af phytosteroler giver indikation om tilstedeværelse af naturligt forekommende kulbrinter. Sitosterol kan anvendes som indikator for, hvorvidt der er naturligt forkommende kulbrinter i en jordprøve. I helt rene jordprøver, hvor totalkulbrinteindholdet er højt pga. bidrag fra naturligt forekommende kulbrinter, forekommer der ikke indhold af de ovenstående petrogene kulbrinter, som også er nærmere omhandlet i kapitel 11. 14 Svensk standard til beregning af totalkulbrinter ved GC-MS14.1 FormålFormålet med dette afsnit er at beskrive en alternativ metode til kvantificering af totalkulbrinter. Denne metode anvendes i Sverige. Metoden kan ikke umiddelbart implementeres i Danmark, idet vurderingsgrundlaget for totalkulbrinter principielt er forskelligt fra den danske metode til kvantificering af totalkulbrinter. Metoden muliggør i højere grad en differentiering mellem kulbrinter af mineralolieoprindelse (petrogene kulbrinter) og naturlige kulbrinter (naturlige kulbrinter). 14.2 Definitioner af petrogene kulbrinterFlygtige kulbrinter, såsom benzin og dieselolie, kan jf. "Naturvårdsverkets instruktion om förorenade bensinstationer", rapport 4889, 1998, detekteres med massespektrometer (MS) eller flammeionistationsdetektor (FID) efter gaschromatografisk separation med kapillarkolonne (GC). Massespektrometer anbefales af Naturvårdsverket pga. sin højere specificitet og lavere detektionsgrænse med f. eks. 57 m/z for alifater og 91 m/z for aromater. SPIMFAB (den svenske pendant til Oliebranchens Miljøpulje) har i den svenske kvalitetsmanual /24/ beskrevet en metode til kvantificering af kulbrinter ved GC-MS ved at anvende udvalgte enkeltkomponenter. Bilag 15 viser uddrag af kvalitetsmanualen, samt hvilke komponenter, der anvendes til kvantificering. Kulbrinterne opdeles i alifater og aromater, med fraktionsdeling inden for hhv. aromaterne og alifaterne:
Der optages GC-MS fullscan i området 35 m/z til 200 m/z til bestemmelse af de flygtige forbindelser ved headspace teknik og GC-MS. Der optages også GC-MS fullscan i området 35 m/z til 350 m/z til bestemmelse af alifatiske og aromatiske mellemkogende forbindelser ved GC-MS. Der udføres GC-MS-SIM til bestemmelse af enkeltkomponenter i grupperne >C8-C10, >C10-C35 og til de carcinogene PAH/øvrige PAH. 14.3 Princip til beregning af totalkulbrinterDer kvantificeres mod en n-alkan standard indeholdende de lige n-alkaner fra C8 til C40 (DRH-004S-5x; Accu standard). Fra en fullscan kørsel af standarden udtrækkes ionspor af ion 57 m/z. Responsfaktorer (Rf) for alle n-alkanerne fra C8 til C34 i referencestandarden beregnes ved ion 57 m/z. Middelværdien af responsfaktorerne indenfor de respektive grupper beregnes (koncentration i standard/integreret topareal). De integrerede toppe i kromatogrammet for prøven multipliceres med de respektive responsfaktorer eller responsfaktormiddelværdier. Alifater >C5-C8 kvantificeres mod n-alkan Rf af C8 Såfremt der forekommer UCM, integreres helt til basislinien (og ikke "valley to valley"). Der er indført nogle korrektionsfaktorer for summen af aromater; Aromater >C8 – C10 multipliceres med en faktor 4,0 For flygtige og mellemflygtige kulbrinter gælder det, at enkeltkomponenter kvantificeres mod de respektive komponenter. 14.4 Metodik til at beregne fradrag af phytosterolerDen metode, som er patenteret af A/S AnalyCen, udnytter det faktum, at ion 93 m/z stort set altid er tilstede ved phytosteroler, og at denne ion ikke optræder i nævneværdig grad i olieprodukter. Den metode, som skal anvendes til integration i MS, er integration af enkelttoppe ved ion 71 m/z med split helt ned til basislinien, der trækkes i kromatogrammet på samme måde, som det bliver gjort i eksempelvis ISO/DIS 16703. Samme integration udføres for ion 93 m/z. De toppe, som har sammenfaldende retentionstider, og hvor ion 93 m/z udgør mere end f. eks. 40% af ion 71 m/z medregnes ikke til summen af toppe, der anvendes til at beregne alifater. Hermed fratrækkes phytosterolbidraget til alifaterne. Denne sammenligning og beregning kan udføres automatisk ved brug af regneark. 14.5 Beregning af indhold af totalkulbrinter i tilsætningsforsøgVed den ovennævnte teknik, har det vist sig, det ikke umiddelbart er muligt at genfinde hele den tilsatte mængde af olie. Ekstrakterne fra tilsætningsforsøgene (afsnit 13) er blevet undersøgt ved den svenske metode. Følgende resultater i nedenstående tabel 14-1 blev opnået ved beregning af indhold af alifater.
Tabel 14-1. Alifatfraktion og indhold af phytosteroler Som det fremgår af resultaterne er indholdet af alifater meget lavt. Indholdet af alifater i komposten er også lille i forhold til den mængde kulbrinter, der beregnes ved GC-FID. Dette er også at forvente, idet der kun ses efter indhold af kulbrinter ved ionspor 71 m/z. Ved at anvende dette princip medkvantificeres kun en lille del naturlige kulbrinter. 14.6 Vurdering af metoden i relation til danske forholdDen svenske metode og definition af totalkulbrinter i jord adskiller sig væsentligt fra den danske metode, men princippet vedrørende ion 93 m/z er forsøgt overført til danske forhold. De svenske grænseværdier/kvalitetskrav er således også defineret anderledes end de danske kvalitetskrav. Vedlagt i bilag 16 ses de svenske kvalitetskrav. Idet prøverne altid analyseres i GC-MS i scan, er der stor mulighed for at undersøge kromatogrammet yderligere. En svaghed ved metoden er dog, at der ikke foretages scan på høje masser >350 m/z. Dette betyder, at man for højtkogende komponenter ikke har en molvægt, og det kan være svært at identificere ukendte komponenter ved hjælp af massespektrum. Ved denne metode er UCM meget lille, hvilket muliggør integrationer ned til en basislinie, der er trukket "vandret og lige" fra umiddelbart efter injektionstop til slut. Beregningsarbejdet med at sammenligne retentionstider, responsforholde og fradrag er en meget stor beregningsopgave, såfremt det skal udføres manuelt. Dette kan løses ved hjælp af små makroer i f.eks. Excel. 15 Vejledning15.1 FormålDet er formålet, at udarbejde en trinvis procedure til evaluering af kromatogrammer af jordprøver ved GC-FID, for derved at ensarte den metodik, der anvendes til vurdering af indholdet af kulbrinter i en jordprøve. Det er hensigten, at den trinvise procedure skal give laboratorierne et bedre beslutningsgrundlag for oprindelsen af kulbrinteindholdet i en jordprøve. Såfremt det ikke er muligt at tilordne indholdet af kulbrinter til en bestemt gruppe af olieprodukter (f. eks. gasolie eller motorolie), er det nødvendigt at foretage en mere detaljeret undersøgelse af specifikke oliekulbrinter ved GC-MS-scan. Herved opnås ikke en tilordning af kulbrinterne til et bestemt olieprodukt, men de specifikke petrogene kulbrinter jf. kapitel 12 kan anvendes som vurderingsgrundlag. Der er udarbejdet en analysestrategi i afsnit 15.2. Afsnit 15.3 beskriver den trinvise procedure, og der er refereret til bogstavindeks benævnt i beslutningstræ illustreret i afsnit 15.3.1.10. 15.2 AnalysestrategiVed identifikation af kulbrinter af petrogen oprindelse udføres indledningsvis screeningsundersøgelse af jordekstraktet på GC-FID. Totalkulbrinteindholdet beregnes, og herved dannes et overordnet billede af prøvens indhold af kulbrinter. Det er muligt at få et overblik over, hvorvidt prøven indeholder petrogene/naturlige kulbrinter. Ved GC-FID analysen kan
Er der uvished om, hvorvidt indholdet af naturlige kulbrinter bidrager væsentligt til det samlede kulbrinteindhold, eller om der er tale om petrogene kulbrinter, kan prøveekstraktet efterfølgende analyseres ved GC-MS-scan. Resultaterne af GC-MS-scan evalueres med hensyn til;
Såfremt et eller flere af de første 3 punkter er erkendt, er der med stor sandsynlighed indhold af petrogene kulbrinter i prøven. 15.3 Trinvis evaluering af resultaterErfaringerne viser, at en trinvis evaluering af analyseresultaterne er en velegnet metode til at afgrænse omkostningerne vedrørende analyse af jordprøver. Desuden er GC-FID screeningen ofte tilstrækkelig, til vurdering af prøvens indhold og arten af kulbrinter set i relation til klassifikationsgrænserne. Trin 1 GC-FID screening, kvantificering og evaluering (Kap. 15.3.1). 15.3.1 Trin 1: GC-FID analyseIndledningsvis foretages en kvalitativ vurdering af kogepunktsintervallet for kulbrinteindholdet. Denne tilordning forholder sig som hovedregel til, hvorvidt indholdet af kulbrinter vurderes til at være af petrogen oprindelse. Såfremt der er tale om flere kulbrinteprodukter af petrogen oprindelse i samme prøve, hvilket i nogle tilfælde fremstår som flere UCM i samme kromatogram, anbefales det indledningsvist at vurdere hver UCM særskilt, og sluttelig samle de enkelte vurderinger i en fælles konklusion. 15.3.1.1 1. Gren. Figur 15.1. Kromatogram af benzin. Ved forvitring af benzin sker der oftest en fordampning af de lettere kogende kulbrinter, benzen, toluen etc., og fraktionen af kulbrinter i kogepunktsintervallet omtrent mellem n-alkan C9 og C12 (ca. 7,0 – 11,0 min) bliver mere dominerende. Det er ikke altid muligt at afgøre, hvilken type af olieprodukt, der er tale om, samt oprindelsen af disse. Som hovedregel vil BTEX-erne samt C9 og C10 aromaterne [21] være dominerende i indhold (op til ca. 65% /32/) i ikke forvitret benzin, men i princippet kan der være tale om mange forskellige olieprodukter. Såfremt kulbrinter i prøven hovedsageligt er fordelt som letkogende kulbrinter med kogepunktsinterval mellem n-alkan C5 og C10, og der samtidig er højt indhold af BTEX-er (>50%), vurderes indholdet af kulbrinter i relation til kvalitetskriteriet for kulbrinter med kogepunktsinterval mellem n-alkan C5 – C10 [A] /31/. Såfremt kulbrinterne i kromatogrammet ikke kan beskrives ved ovenstående, følges pilen mod [B] og videre mod 2. gren.
Figur 15.2.1. Gren i beslutningstræet 15.3.1.2 2. Gren
Figur 15.3.2. Gren i beslutningstræet Ved 2. gren i beslutningstræet undersøges det, om kulbrinterne hovedsaligt forkommer med kogepunktsinterval mellem n-alkan C7 og C12 (ca. 5,0 – 11,0 min.), og at der samtidig ses en afgrænset UCM [C]. Mineralolieprodukter som f. eks. ikke-forvitret terpentin har et kromatogram med dette udseende. Figur 15.4. Kromatogram af terpentin. Såfremt dette ikke er tilfældet følges pilen mod [D], og 3. gren i beslutningstræet. 15.3.1.3 3. Gren
Figur 15.5.3. Gren i beslutningstræet. Såfremt der forekommer andre kulbrinter i kromatogrammet, som ikke tidligere er nævnt, følges pilen mod [F]. 15.3.1.4 4. Gren. Samtidig vurderes indholdet af BTEX-er. I f.eks. frisk petroleum er indholdet af BTEX-er forholdsmæssigt lavt (1-5%, egne erfaringer), og indholdet vokser ikke ved ældning. I et kromatogram af frisk petroleum ses ofte UCM, som vist på nedenstående kromatogram figur 15.7 af petroleum. Såfremt kulbrinteindholdet i kromatogrammet vurderes til at ligne nedenstående kromatogram følges pilen mod [G]. Det er ikke muligt eentydigt at bestemme arten af olieproduktet, men indholdet kan vurderes i relation til jordkvalitetskriterierne /31/.
Figur 15.6. 4. Gren i beslutningstræet. Hvis ikke indholdet af kulbrinter kan tilordnes ovenstående beskrivelse, følges pilen mod [H]. Figur 15.7. Kromatogram af petroleum. 15.3.1.5 5. Gren Figur 15.8. Kromatogram af gasolie. Hovedfraktionen, dvs. 70 – 100% af kulbrinterne, skal befinde sig i dette kogepunktsinterval. Det vurderes visuelt, om isoprenoiderne; farnesan, norpristan, pristan og phytan er tilstede, og om der evt. også ses n-alkaner i en jævn række. Såfremt dette er tilfældet, er der meget stor sandsynlighed for, at kulbrinterne stammer fra gasolie eller tilsvarende kulbrinteprodukt. Pilen følges mod [I], og indholdet vurderes i relation til kvalitetskriteriet for ren jord /31/.
Figur 15.9. 5. Gren i beslutningstræet. Hvis isoprenoiderne og n-alkanerne ikke er tilstede visuelt, eller kogepunktsintervallet er højere, følges pilen mod [J] i beslutningstræet. 15.3.1.6 6. Gren.
Figur 15.10. 6. Gren i beslutningstræet. Hvis indholdet af kulbrinter ikke umiddelbart kan tilordnes i hht. til ovenstående følges pilen mod [L]. 15.3.1.7 7. Gren Figur 15.11. Kromatogram af motorolie. Figur 15.12. Kromatogram af fuelolie. Figur 15.13. Kromatogram af bunkerolie. Det er ikke altid muligt, at tilordne kulbrinteprodukter til et specifikt olieprodukt, og især for de højtkogende motorolier og tilsvarende produkter, er det ikke muligt at differentiere mellem produkter. Som en fællesnævner er kogepunktsintervallet oftest mellem n-alkan C25 og C35, men ved høje indhold strækker kogepunktsintervallet sig ofte op i fraktionen n-alkan C35 til C40 og af og til højere endnu. Toppunktet for UCM er oftest placeret mellem n-alkan C25 og C35. Hvis dette er gældende, følges pilen i beslutningstræet mod [M], og indholdet af kulbrinter vurderes i relation til kvalitetskriteriet for 3. fraktion. Nogle olieprodukter, som f. eks. bunkerolie (figur 15.13), har et kogepunktsinterval, der strækker sig betydeligt ud over intervallet (> n-alkan C40). Indholdet af kulbrinter i en jordprøve med denne type olie vil derfor være underestimeret. Det er ikke muligt ved de almindeligt anvendte kromatografibetingelser, som anvendes til olie i jord, at få et præcist indhold af olie i jorden. Det vil i disse tilfælde være nødvendigt, at foretage injektion af jordprøveekstraktet med andre injektions- og kromatografieringsbetingelser.
Figur 15.14. 7.gren i beslutningstræet. Hvis indholdet af kulbrinter ikke kan tilordnes efter ovenstående følges pilen mod [N] i beslutningstræet. 15.3.1.8 8. Gren. Hvis UCM fremstår som ikke veldefineret (<50-80%), og der er mange opløste toppe, herunder PAH-mønster, som det f.eks. fremgår af nedenstående kromatogram i figur 15.15, kan indholdet tilnærmelsesvis karakteriseres som "tjære/asfalt". Figur 15.15. Kromatogram af "tjære/asfalt". Prøve med diffust forurenet jord. Indholdet af kulbrinter er svært at tilordne til et petrogent kulbrinteprodukt, men typen af kulbrinter kan karakteriseres som "tjære/asfalt", diffust forurenet byjord eller tilsvarende, da kromatogrammet kvalitativt ser ud som en række tjæreprodukter. Hvis PAH indholdet (er ofte også kvantificeret) overstiger kvalitetskriteriet på 1,5 mg/kg TS for summen af 7 udvalgte PAH-er [23], følges pilen mod [O] i beslutningstræet, og der foretages en vurdering i forhold til klassifikationskriterierne med henblik på PAH-indholdet /31/.
Figur 15.16. Gren 8 i beslutningstræet. Hvis PAH indholdet er under 1,5 mg/kg TS følges pilen mod [R] i beslutningstræets gren 9. 15.3.1.9 9. Gren.
Figur 15.17. 9. Gren i beslutningstræet. Dette er specielt gældende for prøver, hvor der er indhold af kulbrinter, der stammer fra naturligt forekommende kulbrinter, der kan skjule bidrag fra petrogene kulbrinter. 15.3.1.10 Samlet beslutningstræ for GC-FID analysen. Figur 15.18. Beslutningstræ trin 1. 15.3.2 Trin 2: GC-MS-scan analyseVed GC-MS-scan analysen opnås som regel et kromatogram, der i høj grad ligner kromatogrammet ved GC-FID. Udvalgte toppes massespektrum kan undersøges nærmere med henblik på en eventuel identifikation. Enkeltkomponenter kan kvantificeres udfra deres karakteristike ionspor. For at undersøge, om prøven indeholder kulbrinter af petrogen oprindelse, kvantificeres indhold af de n-alkaner, isoprenoider, de homologe rækker af naphthalener og phenanthrener. 30ab hopan kvantificeres samtidig med, at det karakteristiske hopanmønster bekræftiges. Dette foretages i én arbejdsgang, og alle resultater er tilgængelige, til vurdering af hvorvidt der er indhold af petrogene kulbrinter i prøven. I samme arbejdsgang på GC-MS, lokaliseres et evt. indhold af phytosteroler og fedtsyrer. Resultaterne opstilles i en tabel f. eks. i Excel, og de forskellige indeks beregnes. Et eksempel er vist nedenfor i tabel 15.1.
Tabel 15.1. Eksempel på resultat fra GC-MS-scan analysen. # ingen enhed. 15.3.2.1 Evaluering af resultater af GC-MS-scan analysen. Indholdet omregnet til mg/kg TS jordprøve (60 gram jord, 20 ml opløsningsmiddel) i en række forskellige olieprodukter er vist i nedenstående tabel 15.2.
Tabel 15.2. Eksempler på indhold af specifikke petrogene kulbrinter i en række udvalgte olieprodukter (i.d. = ikke detekteret, i.b. = ikke beregnet). Indhold af n-alkan C17 og C18 samt isoprenoiderne farnesan, norpristan, pristan og phytan viser også, at der er indhold af petrogene kulbrinter tilstede i prøven. N-alkan C17 og C18 kan som følge af forvitring være forsvundet. CPI indeks over 1 viser, at der er bidrag fra naturlige kulbrinter i prøven. Indholdet af phytosteroler beregnet ved GC-MS-scan analysen giver yderligere et fingerpeg om, hvorvidt der er bidrag fra naturlige kulbrinter i prøven. Det præcise kvantitative indhold af phytosterolerne bør kvantificeres ved GC-FID. Indholdet af de ulige n-alkaner, som er bidraget fra de naturlige kulbrinter, kan kvantitativt fratrækkes totalkulbrinteindholdet ved GC-FID. 15.3.2.2 Kvantitativ beregning af totalkulbrinter efter trin 2. Totalkulbrinter ved GC-FID = Total(FID)–phytosteroler(FID)–ulige n-alkaner(MS*) –fedtsyrer(FID) Phytosteroler skal identificeres ved GC-MS-scan og beregnes ved GC-FID. De ulige n-alkaner beregnes ved GC-MS-scan. *Såfremt der forekommer både lige og ulige n-alkaner og CPI > 1, beregnes indholdet af de ulige n-alkaner som differensen mellem den lige n-alkan, der forekommer umiddelbart før den ulige n-alkan, og den pågældende ulige n-alkan. 15.3.3 Trin 3: GC-MS-SIM analyseHerunder søges mere specifikt efter petrogene kulbrinter. Detektionsgrænsen er lavere. Hopaner og evt. også steraner og diasteraner kan nøjere identificeres og kvantificeres. Indholdet af dibenzothiophener, chrysener og fluorener kan evt. også undersøges. 15.3.4 Eksempel på anvendelse af beslutningstræetI forbindelse med udvikling af beslutningstræet, har det været nødvendigt at afprøve træet på prøver med indhold af rene olier, blandede prøver med tilsat olie til kompost eksempelvis samt prøver fra "rene" lokaliteter. Nedenfor i figur 15.19 er vist GC-FID kromatogram af ren kompost tilsat et indhold af gasolie svarende til 50 mg/kg TS. Figur 15.19. Kromatogram af kompost tilsat gasolie svarende til 50 mg/kg TS. Trin 1: Kromatogrammet viser ingen pæn veldefineret UCM. Der ses kulbrinter i hele det kromatografiske område. Der ses ikke det karakteristiske PAH-mønster for fluoranthen, pyren, benz(a)anthrancen, chrysen osv. Der er tegn på indhold af naturlige kulbrinter (phytosteroler) ved 25 – 30 min. Følges beslutningstræet for trin 1, ender man i sidste gren 9 ved [S]. Totalkulbrinteindholdet er beregnet til 340 mg/kg TS [24]. Trin 2: Resultaterne af GC-MS-scan analysen fremgår af tabel 15.3.
Tabel 15.3. Trin 2. Resultat af GC-MS-scan. #Korrigeret med en faktor 1,7 (se kapitel 13.6.3). Der ses homolog række af naphthalener, nogen phenanthrener, n-alkanerne C17 og C18 samt isoprenoiderne. Dette viser, at prøven indeholder kulbrinter af petrogen oprindelse. Isoprenoidforholdene viser, at der er relativt ikke forvitret gasolie tilstede, idet n-alkan C17/pristan er større end 1. Der ses ingen hopaner (<0,01 mg/kg TS). Dette viser, at der formentlig ikke er tungtkogende petrogene kulbrinter tilstede i prøven (motorolie etc.). Total ionsporet er vist i figur 15.20. Figur 15.20. Total ionspor af eksemplet. Prøven viser ved GC-MS-scan analysen, at prøven indeholder naturlige kulbrinter, idet der er fundet et foreløbigt indhold af phytosteroler på 96 mg/kg TS ved GC-MS-scan. Ved GC-MS-scan ionspor 93 m/z ses kromatogrammer som vist i figur 15.21. Figur 15.21. Ionspor 93 m/z i eksemplet. I den følgende figur 15.22 er vist phytosterolernes placering i totalionkromatogrammet og integration af disse. Figur 15.22. Total ionspor af phytosteroler. Sitosterol forekommer med retentionstiden 24,2 minut. Toppens massespektrum bekræfter, at der er tale om sitosterol . Indholdet af phytosteroler integreres i GC-FID kromatogrammet som illustreret i figur 15.23, og beregnes overfor n-alkaner tilhørende fraktionen C25 – 35. Figur 15.23. Integration af phytosteroler ved GC-FID. Indholdet af phytosteroler er ved GC-FID beregnet til 79 mg/kg TS. Der ses kun et beskedent bidrag fra ulige n-alkaner på 0,19 mg/kg TS. CPI(25 – 33) indekset på 6,65 viser, at der er et bidrag fra ulige n-alkaner til totalkulbrinteindholdet. Indholdet af syrer ved ionspor 73 m/z er undersøgt. I GC-MS-scan ses toppe med retentionstiden ca. 16,5 min. (figur 15.24). Figur 15.24. Ionspor 73 m/z (sort) i eksemplet. Ionspor 230 m/z (grøn) viser placering af den interne standard o-terphenyl ved 16,0 min. Disse toppe integreres ved GC-FID som vist i figur 15.25. Indholdet er vurderet til at være behæftet med meget stor usikkerhed pga. interferens fra andre kulbrinter i prøven. Det er derfor valgt ikke af medregne bidraget fra syrer i dette eksempel. Figur 15.25. Integration af syrer. Det korrigerede indhold af totalkulbrinter, hvor der er fradrag for phytosteroler og ulige n-alkaner er: Total kulbrinter = 340 mg/kg TS – 79 mg/kg TS – 0,16 mg/kg TS = 260 mg/kg TS [25] Ved anvendelse af klassifikation jf. trin 1 vurderes jorden til kraftigt forurenet (klasse 4 jord i /5/). I dette tilfælde var der begrundelse for at fortsætte analysen til trin 2. Her findes indhold af naphthalener, n-alkaner og isoprenoider, hvilket viser som forventet, at prøven indeholder petrogene kulbrinter. Derudover findes indhold af phytosteroler, der viser, at prøvende indeholder bidrag fra naturlige kulbrinter. Idet indholdet af phytosteroler fratrækkes totalkulbrinteindholdet opnås en lavere forureningsgrad bedømt udelukkende udfra totalkulbrinteindholdet. Det tilsatte indhold på 50 mg/kg TS svarer til klassifikationsgrænsen for ren jord forurenet med let olie (C10 – C25) /5/. Det forventes, at indhold af petrogene kulbrinter på netop dette niveau (50 mg/kg TS), kan klassificeres som ren jord. Det er ikke muligt fuldstændigt at frikende prøvens indhold af kulbrinter for indhold af petrogene kulbrinter på basis af GC-MS-scan, men det er muligt, at reducere totalkulbrinteindholdet med 23%, som er bidraget fra de naturligt forekommende kulbrinter. 15.4 DelkonklusionDen udviklede trinvise metode til vurdering af indholdet af kulbrinter i en jordprøve, hvor der samtidig er indhold af naturligt forekommende kulbrinter, har vist sig at være anvendelig i det undersøgte eksempel. Det er lykkedes at "nedklassificere" totalkulbrinteindholdet fra at være kraftigt forurenet, der formentlig ville være anvist til rensning til lettere forurenet jord, som kan genanvendes i for eksempel bygge- og anlægsarbejder. 16 Metodeafprøvning16.1 FormålFormålet med dette afsnit er, at eftervise GC-FID og GC-MS-scan metoden på en række prøver med kendt indhold af petrogene kulbrinter og naturlige kulbrinter. Formålet er at verificere, at blandinger af jord med kendt indhold af olie kan genfindes, samt at tilsatte mængder af ren olie til jord kan genfindes. 16.2 Fremgangsmåde4 typer jordprøver er udvalgt til afprøvning; N1 Supermuld. Ren kompost. Der er indledningsvis foretaget grundig findeling, sigtning og homogenisering, og udført 4-fold analyse på de enkelte prøver. Følgende blandinger er fremstillet af jordene;
Tabel 16.1. Blandinger af jord til afprøvning. Blandingerne er fremstillet ved at afveje præcist 30 gram homogeniseret jord af hver. Der er udført dobbeltbestemmelser dvs. 2 udvejninger for hver. Desuden er der foretaget tilsætningsforsøg med kendt mængde olieblanding af opløsning fra BAM (BAM KS 5004, blanding af gasolie og motorolie) til jord med indhold af naturlige kulbrinter, N1 og N2. Der er tilsat en 6,0 ml af en standard med koncentrationen 5000 mg/L i pentan (der er ved beregning korrigeret for volumenændring). Dette indhold svarer til 500 mg/kg TS olie. Indholdet af totalkulbrinter er kvantificeret, og ved GC-MS-scan analysen er indholdet af de specifikke petrogene kulbrinter kvantificeret. Desuden er indholdet af phytosterolerne og de ulige n-alkaner kvantificeret ved hhv. GC-FID og GC-MS-scan. Resultaterne fremgår af bilag 18. Bilag 18 viser også kromatogrammer fra GC-FID af de enkelte jordtyper. 16.3 Resultat af de enkelte jordtyper.De homogeniserede jordtyper har opnået tilfredsstillende resultater ved GC-FID både med hensyn til totalkulbrinter og phytosteroler, hvilket fremgår af RSD. For totalkulbrinter varierer den mellem 3,5% og 10,4% og for phytosteroler mellem 0,8% og 18,7%. Som det fremgår af bilag 18, er indholdet af petrogene kulbrinter beregnet ved GC-MS-scan i N1 og N2, som forventet lavt eller helt fraværende. Disse jorde karakteriseres som helt "rene" kompostjorde. De petrogene kulbrinter beregnet ved GC-MS-scan i de olieforurenede jordtyper O1 og O2 er også som forventet forholdsmæssigt høje set i relation til suppelerende klassifikationskrav omtalt i konklusionen afsnit 18.2.2. Der ses tilfredsstillende usikkerhed udtrykt ved RSD%. 16.4 Resultat af blandeforsøgetBlandeforsøget viser, at det er muligt at genfinde indholdet af totalkulbrinter samt phytosteroler ved GC-FID. Dette fremgår af genfindingsprocenten, der for totalkulbrinter ligger gennemsnitligt på 96% og for phytosterolerne på 92%. Indholdet af udvalgte petrogene kulbrinter fremgår af nedenstående tabel 16.2. Tabel 16.2. Indholdet og genfindingen af udvalgte petrogene kulbrinter i blandeprøverne.*lavt niveau – ikke kommenteret. Som det fremgår af tabel 16.2, er genfindingen af de petrogene kulbrinter tilfredsstillende med genfindingsprocenter variende fra 53% (N1+O2, Phenanthrener sum) til 126% (N2+O2, norpristan). 16.5 Resultat af tilsætningsforsøgetTilsætningsforsøget viser, at den tilsatte mængde BAM standard genfindes tilfredsstillende med genfindingsprocenter fra 68,3% til 75,2%. Indholdet af phytosteroler ved GC-FID er gennemsnitligt beregnet til 53,3 og 53,8 mg/kg TS. Dette svarer til en genfinding på hhv. 78,6% og 88,3% i forhold til de rene jordtyper N1 og N2. Genfindingen af de petrogene kulbrinter ved GC-MS-scan er beregnet overfor et teoretisk indhold i en kontrolprøve af BAM standarden. Genfindingen af de petrogene kulbrinter er særdeles tilfredstillende. Tabel 16.3 viser udvalgte petrogene kulbrinter.
Tabel 16.3. Genfinding af udvalgte petrogene kulbrinter i tilsætningsforsøg ved GC-MS-scan. 16.6 DelkonklusionVed dette kapitel er det eftervist, at blandinger af jord med kendt mængde olie kan genfindes med en tilfredsstillende nøjagtighed. Det er desuden vist, at de petrogene kulbrinter genfindes tilfredsstillende i de blandede jordtyper. Det er også muligt, at genfinde indhold af phytosteroler i prøver, der er blandet udfra kendt mænde kompostprøve og kendt mængde prøve med indhold af olie. Ved tilsætningsforsøg til de to "rene jordtyper" er det vist, at både indhold af totalkulbrinter samt de petrogene kulbrinter kan genfindes med rimelig stor nøjagtighed. 17 Analyse af prøver49 forskellige prøver er ekstraheret som dobbeltbestemmelser efter den foreskrevne metode. Indholdet af totalkulbrinter er beregnet, og der er foretaget en kvalitativ vurdering af kromatogrammet med henblik på vurdering af, om der er petrogene eller naturlige kulbrinter tilstede i prøveekstraktet (Trin 1). UCM er beregnet for at afgøre hvor stor en del af kulbrinterne, der formentlig ikke kan tilordnes. Egentlig kunne proceduren stoppe her for en række af prøverne, hvor der er et meget stort indhold af petrogene kulbrinter tilstede. Det er valgt at udføre trin 1 (GC-FID) og 2 (GC-MS-scan) af analysen på samtlige prøver. Resultaterne fremgår af bilag 17, hvor den kvalitative vurdering af prøvens indhold af kulbrinter også fremgår. Kromatogrammerne er ikke vist. 17.1 ResultaterResultaterne for dobbeltbestemmelserne af totalkulbrinter ved GC-FID, indholdet af phytosteroler og ulige n-alkaner samt det korrigerede totalkulbrinteindhold fremgår af tabel 17.1. Tabellen viser også oprindelig klassifikation jf. /5/ og dernæst ny klassifikation, hvor der er fradrag for de naturligt forekommende kulbrinter.
Tabel 17.1. Totalkulbrinter med og uden korrektion for indhold af naturlige kulbrinter. 3 ud af 49 prøver (6,1%) har det været muligt at "omklassificere" til en lavere forureningsklasse jf. /5/ (fremhævet). I de med gråt markerede prøver, er indholdet af de specifikke petrogener lavt, og under de i afsnit 18.2.2 supplerende klassifikationsgrænser. Disse udgør i alt 24% (12 ud af 49). 17.2 DelkonklusionDen udviklede trinvise procedure har vist sig anvendelig på udvalgte prøver. GC-MS-scan af prøverne er en enkelt metode til at afgøre, hvorvidt der er petrogene kulbrinter tilstede i en jordprøve. 18 Konklusion18.1 Metodeudvikling og valideringFormålet med projektets første del, har været at finde en egnet gaskroma-tografisk analysemetode til bestemmelse af mineralolie i jord, som opfylder de danske krav ved miljøundersøgelser, og som kan anvendes af alle danske laboratorier med ensartede resultater. Der er et udbredt ønske om, at visse analysemetoder bør udsluses, da der anvendes ekstraktionsmidler, der er belastende for både arbejdsmiljøet og vores omgivende miljø. Desuden er der fra international side kommet nogle nye metoder frem, som det vil være fordelagtigt at anvende, forudsat at de er tilpasset danske forhold. Som udgangspunkt i forbindelse med dette projekt, er der derfor blevet afprøvet forskellige analysemetoder, der i dag anvendes på danske laboratorier samt nogle nye metoder for at sammenligne metodernes effektivitet. Herfra er det blevet klart, at analysemetoden, "Soil Quality – Determination and Mineral Oil Content by Gas Chromatography, Draft International Standard ISO/DIS 16703, - 2001 February." /1/, her kaldet ISO/DIS 16703, som er en ny officiel ISO standard, der er i høring, har været den mest oplagte metode til at bygge undersøgelserne videre på. Ekstraktionsmetoden ISO/DIS 16703 er herefter blevet optimeret på forskellige områder, således at den nye modificerede metode, ISO/DIS 16703-Mod. som er udviklet i forbindelse med dette projekt, er langt mere effektiv end den oprindelige metode. Ekstraktionsudbyttet er væsentlig forbedret, samtidig med at selve ekstraktionproceduren er blevet mere simpel, hvilket dermed har lettet arbejdsgangen i laboratoriet. Desuden er det med den nye metode ISO/DIS 16703-Mod. muligt at analysere totalkulbrinteindholdet i området fra benzen til og med n-alkan C40 med tilfredsstillende resultater. Ekstraktet fra samme prøveoparbejdning kan anvendes til bestemmelse af BTEX' er i prøven enten på GC-FID eller GC-MS-SIM. Desuden kan der ved GC-MS-SIM bestemmes de 16 relevante polycycliske aromatiske hydrocarboner (EPA-PAH-er) på samme ekstrakt. Analyse af BTEX og PAH ved GC-MS kan udføres på samme ekstrakt. Den videreudviklede metode kan også anvendes til differentiering mellem petrogene og naturligt forkommende kulbrinter ved GC-MS-Scan. Efter genfindingsforsøg og en omfattende validering af den nyudviklede metode ISO/DIS 16703-Mod. står det klart, at der er tale om en pålidelig og effektiv analysemetode, der fuldt ud lever op til forventningerne for analyser, der anvendes i forbindelse med miljøundersøgelser. Det anbefales derfor, at ISO/DIS 16703-Mod. erstatter de nuværende analysemetoder til bestemmelse af mineralolie i jord, der i dag anvendes på danske laboratorier. 18.2 Petrogene kulbrinter vs. naturlige kulbrinter, trinvis procedure.18.2.1 Begrænsning i forhold til de nuværende metoderVed vurdering og håndtering af forurenet jord anvendes i dag klassifikationsgrænserne som nævnt i de forskellige vejledninger /5,27,31/. Ved disse grænser er der ikke taget højde for et eventuelt bidrag fra naturlige kulbrinter. Disse kan medføre for højt beregnet totalkulbrinteindhold, og ved fejlvurdering af kromatogrammet fra GC-FID kan det betyde evt. fejlhåndtering af jorden. Eksempelvis vil phytosterolerne give et væsentligt bidrag til totalkulbrinter i fraktionen >C25 – C40. Det er ikke muligt fuldstændigt at skelne mellem petrogene og naturlige kulbrinter ved GC-FID. Dette skyldes, at der for både de petrogene og naturlige kulbrinter forekommer en uopløst bule (UCM), som består af mange forskelligartede kulbrinter, der ikke kan skelnes fra hinanden. Det er ligeledes ikke muligt at lave en generel metode til omregning fra en udvalgt naturlig kulbrinte til total indhold af naturlige kulbrinter. For prøver, der indeholder alene petrogene kulbrinter, jf. trin 1, afsnit 15.2.1, er der ingen problem i henhold til nuværende klassifikationsgrænser. Problemerne opstår ved blandinger af petrogene og naturlige kulbrinter. Til at varetage en korrekt klassificering er det nødvendigt at opstille et supplement til grænseværdier, som er tilknyttet direkte til en række analyseparametre og en trinvis procedure. 18.2.2 Supplerende klassifikationskrav for ren jordVed indikation af tilstedeværelse af naturlige kulbrinter i trin 1 (GC-FID), og hvor det ikke umiddelbart er muligt at afgøre olietype udføres trin 2 (GC-MS-scan), der kan klarlægge indholdet af specifikke petrogene kulbrinter, "syrer", ulige n-alkaner, phytosteroler og evt. andre. Indholdet af totalkulbrinter ved GC-FID genberegnes med fradrag fra de identificerede naturlige kulbrinter. Såfremt indholdet af kulbrinter i området C25 – C40 fortsat overstiger de gældende kvalitetskrav, anbefales det, at der er et andet sæt regler for indhold af petrogene kulbrinter, der træder i kraft. Det er ikke muligt at opstille en klassificering af en blanding af petrogene og naturlige kulbrinter for kulbrinter med kogepunktsinterval mellem n-alkan C10 og C40 alene på baggrund af GC-FID analysen. Der bør derimod stilles krav til specifikke petrogene kulbrinter, selvom indholdet af disse varierer mellem olieprodukter. For eksempel kan der stilles krav til et maksimumindhold af hopaner, idet indholdet af hopaner i sig selv er en tydelig indikation for tilstedeværelse af petrogene kulbrinter. Desuden vil det være hensigtsmæssigt at sætte krav til naphthalener og phenanthrener. Følgende klassifikationskrav anbefales som supplement for klassifikation af ren jord med henblik på specifikke petrogene kulbrinter i trin 2 (tabel 18-1):
Tabel 18-1. Supplement til klassifikationsgrænser for specifikke petrogene kulbrinter. Med betegnelsen sum menes alle de homologe isomerer jf. afsnit 11.9. Ovenstående supplement til klassifikationsgrænserne er udarbejdet på basis af de i dette projekt testede olietyper. Det anbefales, at der foretages flere undersøgelser af disse værdier, med henblik på eksakt fastsættelse af grænseværdierne samt eventuelt en toksikologisk vurdering. 19 Referencer/1/ ISO/DIS 16703 Soil Quality – Determination of Mineral Oil Content by Gas Chromatography –2001 February, Draft International Standard /2/ Bestemmelse af olie i jord, gaschromatografisk metode, DHI, 1998 /3/ Dybdahl, H.P., Bestemmelse af tungtflygtige kulbrinter i jord, Miljøstyrelsen, Notat, December 2000. /4/ Miljøstyrelsen, "Jordforureningsloven" – lov nr. 370. Af 2. juni 1999 om forurenet jord /5/ Vejledning i håndtering af forurenet jord på Sjælland, Juli 2001, Amterne på Sjælland og Lolland/Falster samt Frederiksberg og København. /6/ Lund U., Andersen K. og Sørensen P.S. Håndbog i metodevalidering for miljølaboratorier, VKI sag nr.: 404444/910, 1994. /7/ Krav til analysekvalitet for specialparametre i NOVA 1998 – 2002, Udkast, Miljøstyrelsen 1999. /8/ MacKenzie, A. S. and Jülich, K. F. A. (1984) Application of Biological Markers, Academic Press, London. /9/ Peters, K. E. and Moldowan, J. R. (1993) The Biomarker Guide, Englewood Cliffs, New Jersey. /10/ Nordtest Method: Oil Spill Identification NT CHEM 001. 2. 1991. /11/ Christensen L.B. and Larsen T.H. Method for Determining the Age of Diesel Oil Spills in the Soil. GWMR Fall, 142-149. 1993. /12/ Dybdahl, H. P. Interferenser ved bestemmelse af olie i jord. Teknisk Rapport Nr. 2. 1. 2001. Miljøstyrelsen. /13/ Revill, A. T., Volkman, J. K., O'Leary, T., Summons, R. E., Boreham, C. J., Banks, M. R., and Denwer, K. Hydrocarbon biomarkers, thermal maturity, and depositional setting of tasmanite oil shales from Tasmania, Australia. Geochimica et Cosmochimica Acta 58(18), 3803-3822. 1994. Pergamon. /14/ Hostettler, F. D. A Record of Hydrocarbon Input to San Francisco Bay as traced by Biomarker Profiles in Surface Sediment and Sediment Cores. Marine Chemistry 64, 115-127. 1999. /15/ Latimer, J. S. and Quinn, J. G. Aliphatic Petroleum and Biogenic Hydrocarbons Entering Narragansett Bay From Tributaries under Dry Weather Conditions. Estuaries 21(1), 91-107. 1998. /16/ Wang, Z., Fingas, M., and Page, D. S. Oil Spill Identification a Review. 843, 369-411. 1999. Journal of Chromatography A, Elsevier. /17/ Volkman, J. K., Holdsworth, D. G., Neill, G. P., and Bavor Jr, H. J. Identification of natural, anthropogenic and petroleum hydrocarbons in aquatic sediments. The Science of the Total Environment 112, 203-219. 1992. Amsterdam, Elsevier Science Publishers B. V. /18/ Daling P.S., Faksness L.-G., Hansen A.B., and Stout S.A. Improved and standardised methology for oil spill fingerprinting. 2002. /19/ Harborne, J. B. and Baxter, H. Phytochemical Dictionary, A Handbook of Bioactive Compounds from Plants. Taylor & Francis Ltd. 1993, London. /20/ Harborne, J. B. Phytochemical Methods, A Guide to Modern Techniques of Plant Analysis. Halsted Press, New York, 1973. /21/ Tissot, B. P.and Welte, D. H, Petroleum Formation and Occurrence. 2.nd. ed. 1984, Springer-Verlag, Berlin Heidelberg New York Tokyo. /22/ Silverstein R.M., Bassler G.C. and Morrill T.C., Spectrometric ientification of organic compounds, Fifth Edition 1991, John Wiley & Sons, Canada. /23/ Falkenberg J. A., Hjuler H., Grøn C., Dybdahl H.P., Broholm K. og Østfeldt P., Miljøprojekt 728, Teknologiudviklingsprogrammet for jord- og grundvandsforurening. Kilder til jordforurening med tjære herunder benzo(a)pyren i Danmark, Miljøstyrelsen, 2002. /24/ SPIMFABs Kvalitetsmanual, Avsnitt 3.6, Version 02/05 /25/ Wrang P., European Crude Oil Identification System, Users Manual, National Evironmental Research Institute, Denmark, 1992 /26/ Wang, Z. and Fingas, M. Use of Methyldibenzothiophenes as Markers for Differentiation and Source Identification of Crude and Weathered Oils. Environ.Sci.Technol. 29, 2842-2849. 1995. /27/ Vejledning fra Miljøstyrelsen. Vejledning om prøvetagning og analyse af jord. Nr. 13 1998. /28/ Bøwadt, S., Dybdahl, H.P., Bennetzen, S. og Vejbøl, J. Miljøstyrelsens Jordforureningskontor. Bestemmelse af PAH i jord. Februar 2000 /29/ Statgraphics® Plus 5, Manugistics 2000 /30/ Landbrugsrådgivningscentret, Håndbog i plantedyrkning, Landbrugsforlaget, 2002. /31/ Miljøstyrelsen. 2002, Liste over kvalitetskriterier i relation til forurenet jord. Opdateret 1. oktober 2002. /32/ Miljøprojekt nr. 223, Benzin- og dieselolieforurende grunde. Miljøstyrelsen 1993. Fodnoter[1] DS/EN ISO 9377-2:2001. Vandundersøgelse - Mineralolie (hydrocarbon olieindeks) – Del 2: Væskeekstraktion og gaskromatografi. [2] Florida TRPH standard, Restek Cat.#31266. [3] I bilag 1. ses analyseforskrift af metoden i den form, den er anvendt i de sammenlignende ekstraktionsforsøg. [4] Som følge heraf vil ekstraktionsmetoden som forsøgsvariabel kovariere med analysedagen. [5] 4. fraktion indgår ikke i de sammenlignende ekstraktioner pga. det lave indhold af kulbrinter. [6] F3 =Jordtype 3 (motorolieforurenet jord), A3 = Behandling 3 bestemmelse A, jf. bilag 3. [7] Resultatet udgået pga. utæt glas. [8] Resultatet er en outlier og er udelukket. [9] Total Petroleum Hydrocarbons. [10] Med undtagelse af naphtalen. [11] Fluoranthenindhold på 0,047 mg/kg TS. [12] 0,033 mg/kg TS. [13] National Institut of Standards and Teknologi. Massespektralt bibliotek. [14] m-p-xylen koncentration 0,066-6,66 mg/kg TS. [15] De tre interne standarder er phenanthren-d10, fluoranthen-d10 og benzo(a)pyren-d12. [16] Både acetone og pentan IS er tilsat med kanyle gennem membran. [17] Lavt niveau = niveau 1 (5*DL) [18] Højt niveau = middelværdier for niveau 3, 4 og 5. [19] For PAH er der ikke foretaget tilsætning på niveau 1. [20] Venligst doneret af Vikingeskibsmuseet i Roskilde. [21] Cumen, hemimelliten, pseudocumen, mesitylen, duren, isoduren, p-cymen og tetralin. [22] Doneret af Asger Hansen, DMU. [23] Fluoranthen, Benz(b+j+k)fluoranthen, Benzo(a)pyren, Indeno(1,2,3-cd)pyren og Dibenz(a,h)anthracen. [24] Svarer til kvalitetsklasse 4 i "Vejledning i håndtering af foreurenet jord på Sjælland" /5/. [25] Svarer til kvalitetsklasse 2 i "Vejledning i håndtering af foreurenet jord på Sjælland" /5/. Appendiks A Analyseforskrift
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
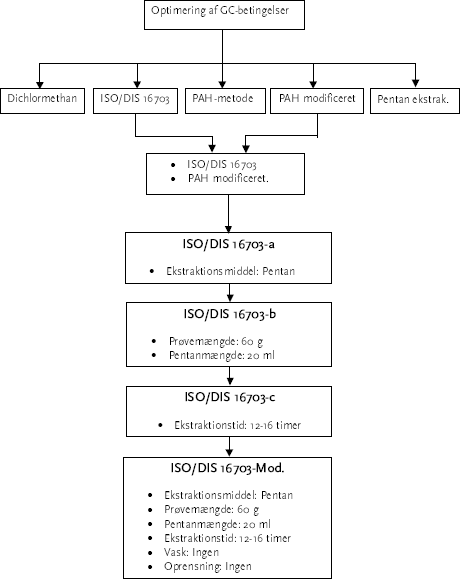
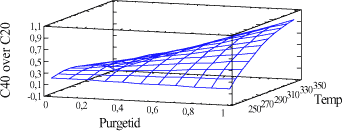
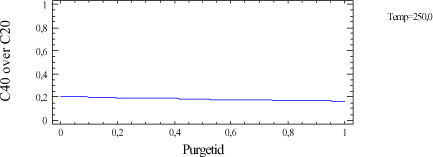
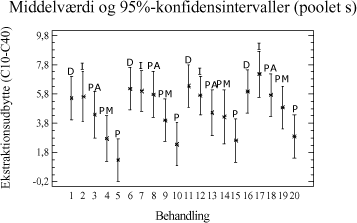
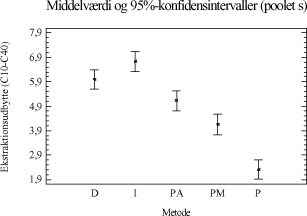
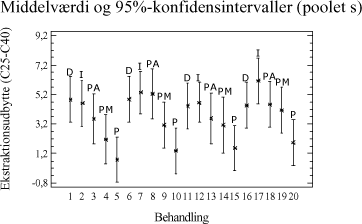
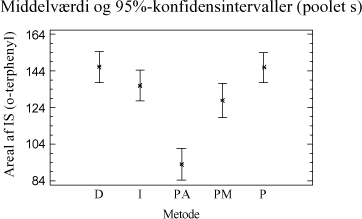
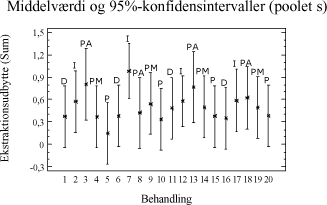
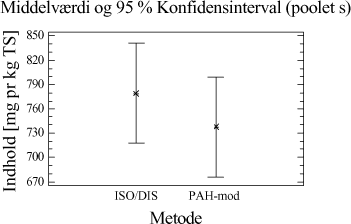
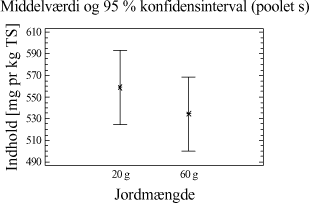
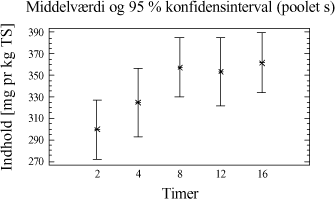
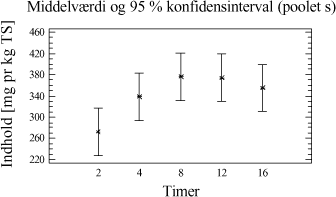
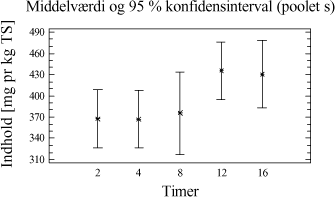
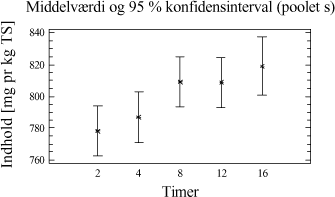
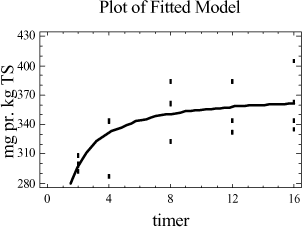
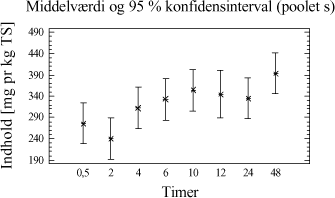
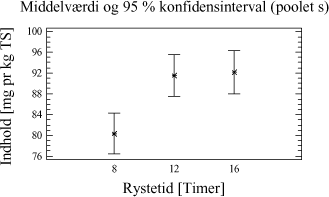
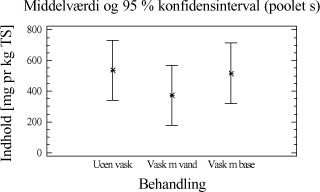
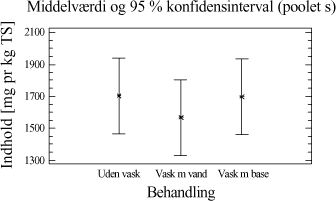
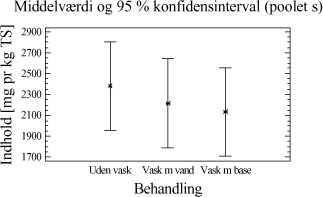
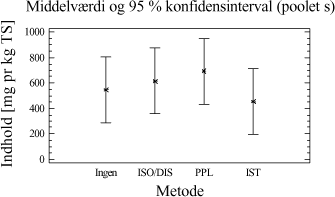
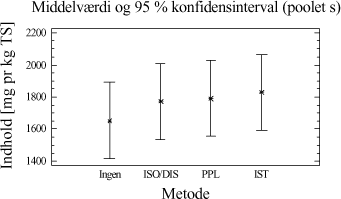
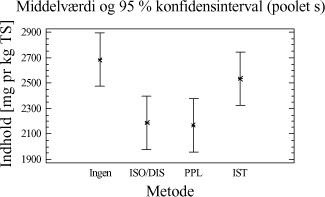
![Figur 6-8. plot af middelværdi [mg/kg TS]. Hvor 1 = Før vask, 2 = Efter vask, 3 = Efter PPL søjle, 4 = Efter IST søjle, 5 = Efter florisil](images/s077a.gif)
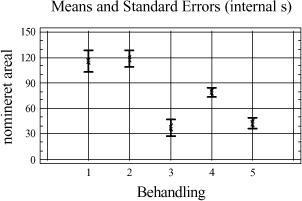
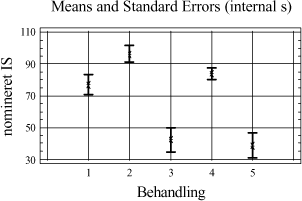
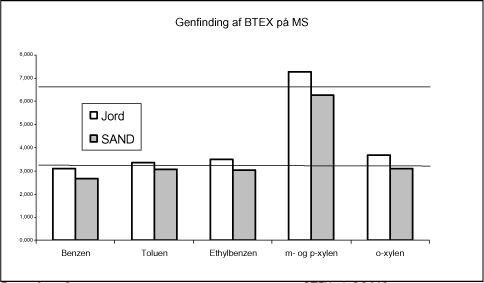
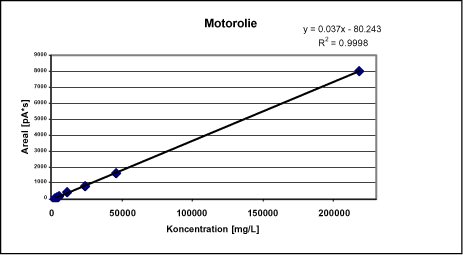
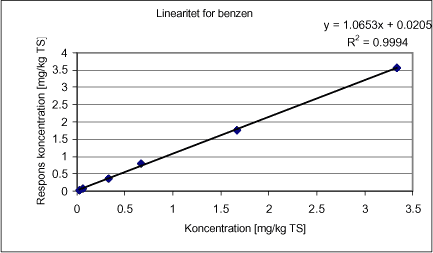
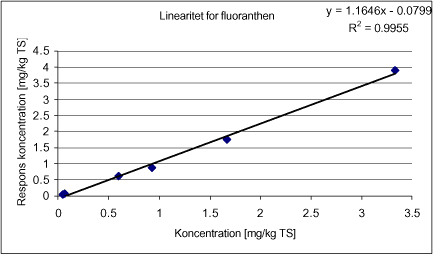
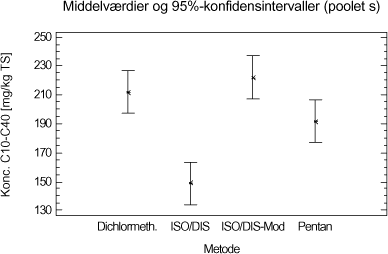
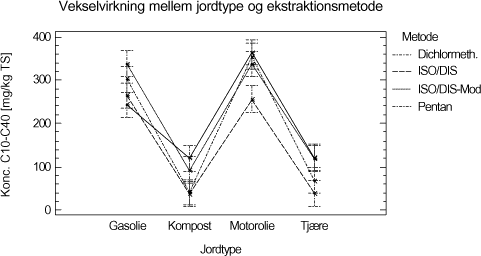
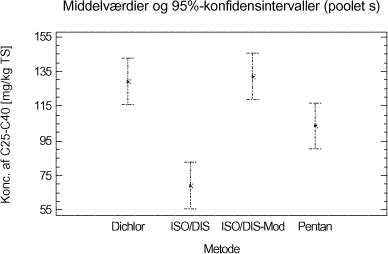
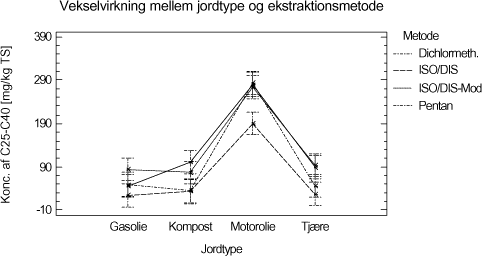
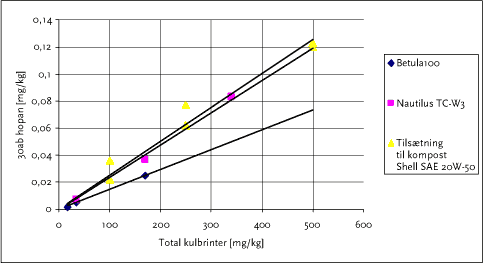
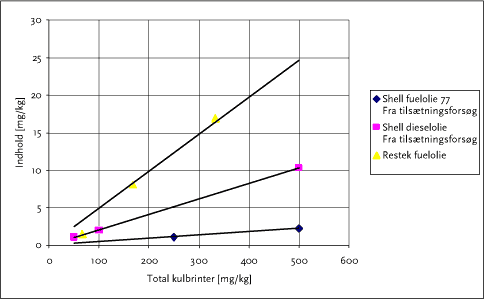
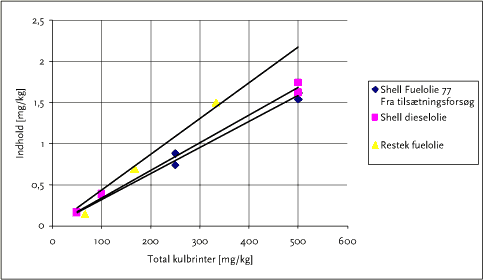
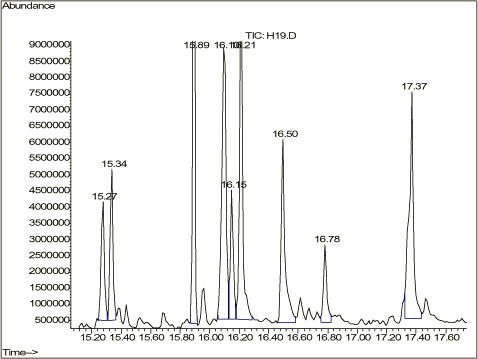
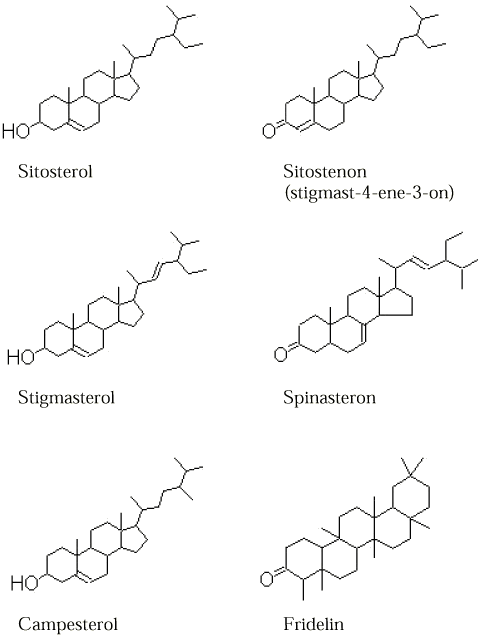
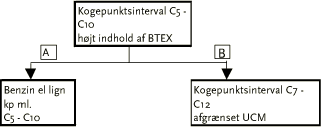
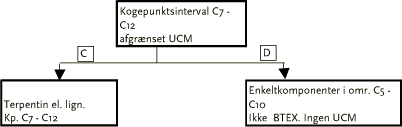


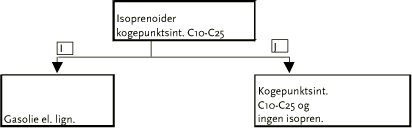
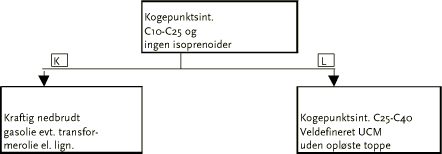


| C6 – C10: | <2,5 - 5000 mg/kg TS |
| C10 – C25: | <5 - 5000 mg/kg TS |
| C25 – C 35: | <10 - 5000 mg/kg TS |
| C35 – C40: | <15 - 5000 mg/kg TS |
| Samlet: | <20 – 5000 mg/kg TS |
| BTEX: | <0,01 – 3,33 mg/kg TS |
| PAH: | <0,005 – 3,33 mg/kg TS |
2 Reference
”ISO/DIS 16703 Soil Quality – Determination of Mineral Oil Content by Gas Chromatography – 2001 February”, Draft International Standard. /1/
3 Anvendelsesområde
Metoden kan benyttes til at bestemme indholdet af totalkulbrinter, BTEX og PAH-forbindelser i jord samt petrogene kulbrinter og naturlige kulbrinter.
3.1 Totalkulbrinter
Metoden kan anvendes i forbindelse med kvantificering af olieforurening i jordprøver. Olieforurening omfatter produkter som benzin, gasolie, motorolie og lignende. Olieprodukter består af organiske kulbrinter.
Totalkulbrinter defineres som summen af organiske stoffer, der ekstrahereres fra en given jordprøve ved ekstraktion med acetone/pentan (1:1), samt detekteres med GC-FID. Totalkulbrinterne fra benzen til og med C40 har kogepunkt imellem 80 – 525 °C.
Meget flygtige forbindelser med kogepunkt under 80 °C medregnes således ikke, da de vil skjules under solventtoppen.
Totalkulbrinter beregnes i fire fraktioner som dog opgives i tre fraktioner samt en sum af totalkulbrinter. Fraktionerne opdeles udfra visse n-alkaners kogepunkt.
Resultaterne angives i analyserapporten som følgende fraktioner, jf. tabel 3-1.
| Fraktion | Indhold | Detektionsgrænse [mg/kg TS] |
| 1. fraktion | Benzen – C10 | <2,5 |
| 2. fraktion | > C10 – C25 | <5,0 |
| 3. fraktion | >C25–C35 + >C35–C40 = > C25-C40 | <25 |
| Totalkulbrinter | Benzen–C10 + >C10–C25 + >C25–C40 = Benzen – C40 |
Tabel 3-1. Kulbrinte fraktioner.
Metodens liniære måleområde er dokumenteret op til 5000 mg/kg TS. Ved prøver med indhold højere end dette skal ekstraktet fortyndes til en koncentration indenfor måleområdet. Detektionsgrænsen for metoden er bestemt i de enkelte fraktioner, se tabel 3-1.
3.2 BTEX
Enkeltkomponenterne benzen, toluen, ethylbenzen og meta- para- og orto-xylen omtales samlet som BTEX.
BTEX i jord stammer hovedsagelig fra benzinforurening. TEX forekommer i op til 18% pr. enkeltkomponent i benzin /2/. Desuden forefindes BTEX i lavere koncentration i andre olieprodukter og kan f.eks. frigives fra tjære.
BTEX er organiske opløsningsmidler og kan forekomme som enkeltstoffer.
BTEX angives i analyserapporten som enkeltstoffer jf. tabel 3-2.
| BTEX | Detektionsgrænse MS [mg/kg TS] |
| Benzen | <0,01 |
| Toluen | <0,01 |
| Ethylbenzen | <0,01 |
| m- p-xylen | <0,01 |
| o-xylen | <0,01 |
Tabel 3-2. BTEX.
Metodens liniære måleområde er dokumenteret op til 3,33 mg/kg TS for hver enkeltkomponent. Metodens detektionsgrænse er bestemt på GC- MS og ses i tabel 3-2.
3.3 PAH
Metoden kan benyttes til bestemmelse af Poly Aromatiske Hydrocarboner (PAH). PAH-forbindelser er komponenter i tjære og i mindre mængder i de tungere olieprodukter.
Metoden kan benyttes til bestemmelse af alle 16 US-EPA PAH’er.
De 7 forbindelser som kontrolleres i forbindelse med de danske jordkvalitetskrav der er fastlagt i Vejledning fra Miljøstyrelsen /3/ er et udsnit af disse EPA PAH’er.
| 16 US-EPA PAH | 7 MST PAH |
| Naphthalen | |
| Acenaphthylen | |
| Acenaphthen | |
| Flouren | |
| Phenanthren | |
| Anthracen | |
| Fluoranthen | Fluoranthen |
| Pyren | |
| Benz(a)anthracen | |
| Chrysen/triphenylen | |
| Benzo(b+j+k)fluoranthener | Benzo(b+j+k)fluoranthener |
| Benzo(a)pyren | Benzo(a)pyren |
| Indeno(1,2,3-cd)pyren | Indeno(1,2,3-cd)pyren |
| Dibenz(a,h)anthracen | Dibenz(a,h)anthracen |
| Benzo(ghi)perylen |
Tabel 3-3. PAH-forbindelser.
Reelt er det 18 PAH-forbindelser, da benzo(j)fluoranthen ikke hører til EPA-PAH forbindelserne, men kun meget vanskeligt kan adskilles fra henholdsvis benzo(b)fluoranthen og benzo(k)fluoranthen. Desuden kan chrysen ikke under normale omstændigheder adskilles fra triphenylen, se kapitel 4.
Metodens liniære måleområde er dokumenteret op til 3,33 mg/kg TS for hver PAH-komponent. Metodens detektionsgrænse er bestemt på GC- MS til <0,005 mg/kg TS for hver af de enkelte PAH-forbindelser.
3.4 Kvantificering af petrogene kulbrinter
Metoden kan benyttes til bestemmelse af udvalgte petrogene kulbrinter /7/.
Petrogene kulbrinter er komponenter, der indgår i mineralolie, og som er karakterisktisk for mineralolie. Følgende petrogene kulbrinter er omfattet af denne analyseforskrift.
| Komponent |
| Naphthalen |
| Methylnaphthalener [1] |
| Dimethylnaphthalener [2] |
| Trimethylnaphthalener [3] |
| Phenanthren |
| Methylphenanthrener [4] |
| Dimethylphenanthrener [5] |
| n-Alkaner [6] |
| Acykliske isoprenoider [7] |
| Hopaner [8] |
Tabel 3-4. Petrogene kulbrinter.
3.5 kvantificering af naturlige kulbrinter
Metoden kan benyttes til kvantificering af bidrag af naturlige kulbrinter til totalkulbrinteindholdet jf. /7/.
Metoden omfatter phytosteroler og ulige n-alkaner, der skønnes at stamme fra naturligt forekomne kulbrinter, og som interfererer med totalkulbrinteindholdet.
4 Interferencer
4.1 Totalkulbrinter
Apolære og svagt polære forbindelser f.eks. stammende fra plantemateriale, mosejord eller gammel havbund vil give bidrag til indholdet af totalkulbrinter, og vil interferere på identifikationen af olieproduktet /4/.
Polære fedtstoffer, vegetabilske- og animalske olier der medekstaheres fra en given jordprøve kan give interferens ved kromatografering, men vil være let genkendelig for en erfaren kemiker.
En del af denne interferens kan identificeres og fratrækkes efter analyse ved GC-MS scan /7/.
Chlorerede opløsningsmidler samt andre forbindelse, der ikke er rene kulbrinter, vil blive medbestemt i totalkulbrinteindholdet, men detekteres med lavere respons pga. FI-detektoren svagere respons overfor chlor, ilt mm.
4.2 PAH
Mange PAH-forbindelser har identisk molekylvægt samt en struktur og fysisk-kemiske egenskaber, der ligner hinanden meget. Derfor er det vanskeligt fuldstændigt at undgå interferens i bestemmelsen af PAH. Det er derfor af stor vigtighed, at laboratoriet ved GC-MS-bestemmelsen benytter optimale kromatografiske betingelser for at opnå størst mulig separation mellem muligt interfererende forbindelser /5/.
Da forskellige kromatografiske kolonner har forskellig separation af individuelle PAH-forbindelser, og da separationen desuden er stærkt afhængig af det brugte ovntemperaturprogram, er det op til det enkelte laboratorium at sikre mindst mulig interferens i bestemmelserne. I de tilfælde, hvor interferens ikke kan undgås, bør der angives en sum af de individuelle forbindelser samt klart angives, hvilke forbindelser det drejer sig om. De bedst kendte interferenser på normal GC-kolonner er chrysen og triphenylen samt benzo(b)fluoranthen, benzo(j)fluoranthen og benzo(k)fluoranthen /5/. Disse forbindelser angives normalt som en sum af benzo(b+j+k)fluoranthen og chrysen/triphenylen.
4.3 BTEX
4.3.1 FID
Andre enkeltkomponenter med samme retentionstid kan fejlagtigt identificeres som en af BTEX'erne. Feks. har cyclohexan og benzen stort set samme retentidstid ved den her anvendte kolonne.
4.3.2 MS
Der er ikke konstateret problemer med interferens på BTEX ved GC-MS.
5 Princip
En prøve af ca. 60 gram jord ekstraheres ved mekanisk rystning med acetone og pentan i 12 – 16 timer. Herefter tilsættes vand og det organiske lag separeres. En passende mængde af dette ekstrakt analyseres til bestemmelse af totalkulbrinter ved anvendelse af kapillar gaskromatografi med flammeionisationsdetektion. En anden del af ekstraktet kan analyseres for BTEX eller PAH ved kapillar gaskromatografi med massespektrometrisk detektion i SIM-mode. Ekstraktet kan yderligere analyseres for petrogene og naturlige kulbrinter ved kapillar gaskromatografi med massespektrometrisk detektion i Scan-mode.
6 Apparatur
6.1 Prøveglas
Der skal anvendes membranglas [9]. Et glas volumen på 100 mL er passende til både jord, opløsningsmidler samt plads til omrystning. Det anbefales at anvende 100 mL Duranglas.
Glasudstyr rengøres [10] ved normal opvask efterfulgt af varmebehandling ved 400-500 °C i min. 2 timer. Renheden af glasvarene skal være dokumenteret.
Der skal en ny membran i glassene imellem hver anvendelse. Membranen skal være teflonbelagt på indersiden.
Det er en fordel at kende glassets vægt, før disse sendes ud til kunden. Således kan denne glasvægt substraheres fra vægten af det fyldte glas.
6.2 Rysteapparat
Et apparat der anvendes til at ryste prøverne i membranglassene. Det anbefales, at placere glassene i vandret liggende position, mens de rystes, således at de rystes i samme retning som centeraksen af glassene, altså på langs. Det er vigtigt, at anvende kraftig rystning for at opnå ligevægt inden for den angivne ekstraktionstid. Der anbefales en amplitude på 7 cm og en rystefrekvens på 200 slag pr. minut. Andre specifikationer kan være tilfredsstillende, men dette skal valideres af det enkelte laboratorium inden brug.
6.3 Centrifuge
En centrifuge til separation af faserne efter ekstraktion. Det er hensigtsmæssigt, hvis centrifugen har mulighed for at centrifugere membranglassene for at undgå overførsel af prøverne til nyt glasudstyr.
6.4 GC-kolonne
Følgende kolonnedimensioner anbefales: 30 m, 0,25 mm ID og 0,25 m filmtykkelse, 5 % phenylmethylsiloxan. Kolonner med lav blødning og mulighed for høje temperaturer anbefales.
Under metodeudviklingen er anvendt HP-5MS 19091S-433. Andre kolonner kan benyttes, hvis de lever op til kravene om kromatografisk ydeevne.
6.5 Gaskromatografer
6.5.1 GC-FID
Gaskromatografisk system med kapillarkolonne og flammeionisationsdetektor (FID). FID er karakteristisk ved, at alle kulbrinter stort set giver samme respons, hvilket er essentielt for metodens anvendelighed. GC-system med temperatur- og trykstyring, kapillarkolonne og splitless injektion anbefales. Et datasystem til beregning af kromatografiske arealer ved brug af tvungen basislinieprojektion, og som er i stand til at gemme og reintegrere kromatografiske data bør benyttes.
| Instrument | GC | Program |
| GC-FID | HP6890 SERIE PLUS | Chemstation A.07.01 (682) |
Tabel 6-1. Det anvendte apparatur under metodeudvikling.
6.5.2 GC-MS
Et gaskromatografisk system med kapillarkolonne og massespektrometrisk detektion (MS). MS er karakteristisk ved at detektere ioner i forhold til deres masse/ladningsforhold (m/z), hvilket gør detektionen mere specifik i forhold til enkeltstoffer.
GC-system bør have temperaturstyring, splitless injektion og anvende kapillarkolonne. Trykstyring af bæregassen anbefales for bedre reproducerbarhed.
Massespektrometret skal have mulighed for anvendelse af SIM (single ion monitoring) og Scan (scaning over alle ioner) samt være tilkoblet et datasystem, der tillader dataopsamling og lagring af alle data, der optages i det kromatografiske forløb. Datasystemet skal kunne søge datafilerne for ioner med specifikke masser og skal kunne udskrive ionresponset i forhold til tiden eller scan-nummer. Datasystemet skal kunne integrere såvel som reintegrere signalet for ethvert udtrukket ionspor.
| Instrument | GC | MS | Program |
| GC-MS | HP 6890 N | MS Detektor 5973 Network | MSD software C.00.01 |
Tabel 6-2. Det anvendte apparatur under metodeudvikling.
7 Standarder og kontrol
7.1 Kemikalier
De anvendte kemikalier skal være af analytisk renhedsgrad og velegnede til formålet.
- Acetone
- n-Pentan
- Vand
- Monobrombenzen
- O-terphenyl
- Squalan
- Phenanthren-d10
- Fluoranthen-d10
- Benzo(a)pyren-d12
Der anbefales brug af færdigkøbte ampuller [11] som kalibreringsstandarder og kontrolopløsninger. De nævnte ampuller har været anvendt under metodeudviklingen, men tilsvarende ampuller kan sandsynligvis anvendes uden yderligere afprøvning.
- Florida TRPH standard, Restek Cat.#31266
- BTEX/Naphthalen Standard, Restek Cat.#58361
- Mineral oil Standard, BAM KS 5004
- Custom BTEX/Naphtalene standard, Restek Cat.#57366
- SV Calibration Mix, Restek Cat.#31011
- PAH mixture, Agilent Technologies Cat.: 8500-6035
7.2 Ekstraktionsopløsninger
7.2.1 Afprøvning af opløsningsmidler
Der anvendes pentan og acetone af analysekvalitet. Når pentanen og acetonen modtages på laboratoriet afprøves deres renhed med hensyn til BTEX og PAH. Vandet, der anvendes, afprøves sammen med acetonen.
7.2.1.1 Pentan
Der fyldes en vial med pentan, som analyseres på GC-FID og GC-MS efter metoderne beskrevet i afsnit 8 og 10. Pentanen kan godkendes til analyse, når den ikke indeholder BTEX eller PAH i koncentrationer over detektionsgrænsen for hver enkelt komponent.
7.2.1.2 Acetone og vand
20 mL acetone håndrystes med 20 mL afprøvet pentan i ca. 1 min. Der tilsættes 30 mL vand, og der rystes 1 min. Der tappes en vial fra pentanfasen, som analyseres på GC-FID og GC-MS efter metoderne beskrevet i afsnit 8 og 10. Acetonen og vandet kan godkendes til analyse, når den pentan, det er rystet med, ikke indeholder BTEX eller PAH i koncentrationer over detektionsgrænsen for hver enkelt komponent. Hvis der opstår problemer i forhold til dette, afprøves acetonen og vandet hver for sig.
7.2.2 Stamopløsning af intern standard
De interne standarder, der anvendes til bestemmelse på GC-FID er monobrombenzen, o-terphenyl og squalan.
De interne standarder der anvendes til bestemmelse af PAH på GC-MS er phenanthren-d10, fluoranthen-d10 og benzo(a)pyren-d12.
7.2.2.1 IS Stamopløsning GC-FID (IS Stam 1, 2 og 3)
Der afvejes 1 gram af henholdsvis monobrombenzen, o-terphenyl og squalan til hver sin 100 mL målekolbe. De nøjagtige afvejede mængder noteres. Målekolberne fyldes til mærket med afprøvet pentan (7.2.1.1). De tre stamopløsninger kaldes henholdsvis IS Stam 1, 2 og 3.
Koncentrationen i hver opløsning er 10000 mg/L.
7.2.2.2 IS Stamopløsning GC-MS (IS-MS)
Der afvejes 0,1 gram af henholdvis phenanthren-d10, fluoranthen-d10 og benzo(a)pyren-d12 i samme 100 mL målekolbe. Den nøjagtige afvejede mængde noteres. Målekolben fyldes til mærket med afprøvet pentan (7.2.1.1).
Koncentrationen i opløsningen IS-MS er 1000 mg/L.
7.2.2.3 Intern standard opløsning (IS-opl.)
En 100 mL målekolbe fyldes halvt med afprøvet pentan (7.2.1.1). Der udtages med fuldpipette 1 mL af hver af IS Stam 1, 2 og 3 (7.2.2.1) samt 1 mL af IS-MS (7.2.2.2) som tilsættes ned i pentanen i en100 mL målekolbe. Målekolben fyldes op til mærket med afprøvet pentan og mærkes IS-opl.
Koncentrationen i opløsningen er 100 mg/L.
7.2.3 Pentan med intern standard (Pentan IS)
En 2000 mL målekolbe fyldes halvt op med afprøvet pentan (7.2.1.1). Der tilsættes 1 mL af hver af IS Stam 1, 2 og 3 (7.2.2.1) samt 1 mL af IS-MS (7.2.2.2) ned i pentanen i 2000 mL målekolbe. Målekolben fyldes op til mærket med afprøvet pentan.
Koncentration i opløsningen er 5 mg/L for de interne standarder til FID og 0,5 mg/L for de interne standarder til MS [12].
Det er vigtigt at Pentan IS (7.2.3) og IS opl. (7.2.2.3) er fremstillet ud fra samme Stam (7.2.2.1) og IS-MS (7.2.2.2).
7.2.4 Blindprøver
7.2.4.1 Sekvensblind
Der fyldes en række vials af den til prøverne anvendte pentan IS (7.2.3). Disse opbevares på køl indtil analysetidspunktet.
7.2.4.2 Metodeblind
Der udrystes mindst et prøveglas om dagen, præcis som om det var en prøve (8.2). Pentanen aftappes og analyseres som en prøve. Denne må ikke give indhold over detektionsgrænsen for de enkelte parametre.
7.3 Totalkulbrinter og BTEX
7.3.1 Standard totalkulbrinter og BTEX (STD-opl.)
En ampul af Florida TRPH standard, Restek Cat.#31266 konc. 500 mg/L og en ampul BTEX/Naphthalene Standard, Restek Cat.#58361 konc. 2000 mg/L tempereres til stuetemperatur.
7.3.1.1 STD-opl.
En 100 mL målekolbe fyldes halvt med afprøvet pentan (7.2.1.1). Der afpipetteres med fuldpipette 1 mL af Florida-ampullen og 250 µL af BTEX-ampullen over i 100 mL målekolbe. Der afpipetteres 5 mL af IS-opl. (7.2.2.3) over i 100 mL målekolbe. Der fyldes op med afprøvet pentan til mærket. Koncentration af STD-opl. er 5 mg/L. Denne hældes på vials uden headspace over væsken. Opbevares på køl indtil analyse.
7.3.2 Kontrol totalkulbrinter (KNT-opl.)
7.3.2.1 Stamopløsning knt.:
En ampul af Mineral oil Standard, BAM KS 5004 tempereres til stuetemperatur. Der afvejes ca. 0,8 g Mineral oil Standard i en 100 mL målekolbe. Den nøjagtige vægt noteres. Målekolben fyldes til mærket med afprøvet pentan (7.2.1.1). Kolben rystes omhyggeligt. Koncentrationen af olien i Stamopløsning knt. er ca. 8000 mg/L.
7.3.2.2 KNT-opl:
En 100 mL målekolbe fyldes halvt med afprøvet pentan (7.2.1.1). Der udtages med fuldpipette 10 mL af tempereret stamopløsning knt. til 100 mL målekolben. Der afpipetteres 5 mL af IS-opl. (7.2.2.3) over i 100 mL målekolben.
Der fyldes op med afprøvet pentan til mærket. Koncentrationen af KNT-opl er ca. 800 mg/L, koncentrationen skal beregnes nøjagtigt, før indsættelse i kontrolkort. KNT-opl. hældes på vials uden headspace over væsken. Opbevares på køl indtil analyse.
7.3.3 Kontrol BTEX (BTEX-knt.)
En ampul af Custom BTEX/Naphthalen standard, Restek Cat.#57366 konc. 200 mg/L tempereres til stuetemperatur.
7.3.3.1 BTEX-knt.
En 100 mL målekolbe fyldes halvt op med afprøvet pentan (7.2.1.1). Der udtages 500 µL af BTEX ampullen til 100 mL målekolbe. Der afpipetteres 5 mL af IS-opl. (7.2.2.3) over i 100 mL målekolbe. Der fyldes op med afprøvet pentan til mærket. Koncentrationen af BTEX-knt er 1,0 mg/L pr. enkeltkomponent. BTEX-knt. hældes på vials uden headspace over væsken. Opbevares på køl indtil analyse.
7.4 PAH på GC-MS
7.4.1 Standard og kontrol PAH-forbindelser (PAH-STD)
En ampul af (PAH'er) SV Calibration Mix, Restek Cat.#31011 med konc. 2000 mg/L tempereres til stuetemperatur.
7.4.1.1 PAH-std.
En 200 mL målekolbe fyldes halvt op med afprøvet pentan (7.2.1.1). Der udtages 500 µL af PAH ampullen til 200 mL målekolbe. Der tilsættes 1 mL af IS-MS (7.2.2.2) over i 200 mL målekolbe. Der fyldes op med afprøvet pentan til mærket. Koncentrationen af PAH-std er 5,0 mg/L pr. enkeltkomponent. PAH-std. hældes på vials uden headspace over væsken. Opbevares på køl indtil analyse.
7.4.1.2 PAH-knt.
En ampul af PAH mixture, Agilent Technologies Cat.: 8500-6035 med konc. 501 mg/L tempereres til stuetemperatur.
En 100 mL målekolbe fyldes halvt op med afprøvet pentan (7.2.1.1). Der udtages 200 µL af PAH mixture til 100 mL målekolbe. Der tilsættes 500 µL af IS-MS (7.2.2.2) over i 100 mL målekolbe Der fyldes op med afprøvet pentan til mærket. Koncentrationen af PAH-knt er 1,0 mg/L pr. enkeltkomponent. PAH-knt. hældes på vials uden headspace over væsken. Opbevares på køl indtil analyse.
7.5 Petrogene kulbrinter
7.5.1 Standarder
Ampuller kan erhverves ved. f. eks. Chiron, Restek eller tilsvarende.
| Komponent | Ampul |
| Naphthalen | Se PAH standard 7.4.1.1 |
| Methylnaphthalener (MN) | 1- og 2-Methylnaphthalen. Ampuller af 1,0 mg/ml fra Chiron i isooctan (0811,15 og 0812,15) |
| Dimethylnaphthalener (DMN) | 2,6-Dimethylnaphthalen. Ampul 1,0 mg/ml fra Chiron i isooctan (0729,12) |
| Trimethylnaphthalener (TMN) | 2,3,5-Trimethylnaphthalen. Ampul 1,0 mg/ml fra Chiron i isooctan (0706,13) |
| Phenanthren | Se PAH standard 7.4.1.1 |
| Methylphenanthrener (MP) | 2-Methylphenanthren. Ampul 1,0 mg/ml fra Chiron i isooctan (0812,15) |
| Dimethylphenanthrener (DMP) | 3,6-Dimethylphenanthren. Ampul 0,5 mg/ml fra Chiron i isooctan (0768,16) |
| n-Alkaner | S-4066 Hydrocarbon mixture C14-C32 fra Chiron 1,00 mg af hver i 1 ml isooctan og Florida TRPH standard, Restek Cat.#31266 konc. 500 mg/L |
| Acykliske isoprenoider | indgår i S-4066 Hydrocarbon mixture |
| Hopaner | 17a(H),21b(H)-Hopan (30ab) ampul fra Chiron 1 ml i isooctan, 0,1 mg/ml |
Tabel 7-1. Petrogene kulbrinter.
7.5.1.1 Petrogen kulbrintestandard
En 50 mL målekolbe fyldes halvt op med pentan IS (7.2.3). Der udtages 500 µL af MN, DMN, TMN, MP og DMP-ampullerne samt S-4066 (tabel 7-1) i 50 mL målekolbe. Der fyldes op med afprøvet pentan til mærket. Koncentrationen af Petrogen kulbrintestandard fremgår af tabel 7-2.
| Komponent | Koncentration i Petrogen kulbrintestandard |
| 1- og 2-Methylnaphthalen | 10 mg/L |
| 2,6-Dimethylnaphthalen | 10 mg/L |
| 2,3,5-Trimethylnaphthalen | 10 mg/L |
| 2-Methylphenanthren | 10 mg/L |
| 3,6-Dimethylphenanthren | 5 mg/L |
| n-Alkaner | 10 mg/L, Florida TRHP standard se 7.3.1.1 (5 mg/L) |
| Acykliske isoprenoider | 10 mg/L |
| 17a(H),21b(H)-Hopan (30ab) | 1,0 mg/L |
Tabel 7-2. Koncentration af petrogene kulbrinter.
7.5.2 Kontroller
7.5.2.1 Fuelolieopløsning
Fremstil opløsning med fuelolie til kontrol af retentionstider. Denne opløsning kan også anvendes til kontrol ved gentagne målinger.
Afvej 100 mg fuelolie i en 100 ml målekolbe. Fyld op til mærket med pentan IS. Koncentration 1000 mg/L.
8 Ekstraktionsprocedure
8.1 Prøven
8.1.1 Prøveudtagning
Der henvises til Miljøstyrelsen: Vejledning om prøveudtagning og analyse af jord /3/.
Efter udtagning af en jordprøve overføres den hurtigt til en membranglas, der lukkes umiddelbart herefter. Det er vigtigt at sikre, at glaskanten og gevindet er fri for jordpartikler, for at sikre en tæt lukning af membranglasset. Prøveudtagningskeen skal have en diameter, som er mindre end åbningen af membranglasset for at forhindre partikler på glaskant og gevind.
Det anbefales at anvende 100 mL membranglas f.eks. af typen redcap-glas. Der tilsættes jord så glasset omtrent halvfuldt, hvilket svarer til en prøvemængde på ca. 60 g. Tørstofindholdet heri vil ca. svare til 50 g.
Membranglasset må maksimalt fyldes halvt op for at tillade tilsætning af opløsningsmiddel og vand, og for at sikre tilfredsstillende bevægelser i forbindelse med ekstraktionen.
Der skal udtages en parallel prøve til bestemmelse af tørstof. Hertil kan enhver tætsluttende emballage benyttes. Tørstof bestemmes i henhold til DS 204, idet der tages en større mængde jord i anvendelse 10-20 g.
Endvidere kan det anbefales at udtage en eller flere parallelle prøver til yderligere undersøgelser eller som ekstra prøve i tilfælde af uforudsete problemer.
8.1.2 Forholdsregler
Jordprøven skal ankomme til laboratoriet i 100 mL membranglas f.eks. af typen redcap-glas. Kunden skal have fyldt glasset omtrent halvfuldt, svarende til en prøvemængde på ca. 60 g. Tørstofindholdet heri vil ca. svare til 50 g.
Der skal desuden medfølge en delprøve til tørstofbestemmelse.
Hvis prøver modtages i anden emballage end membranglas, skal dette opgives på analysecertifikatet, idet tab af flygtige forbindelser kan forventes i forbindelse med prøvehåndteringen.
Hvis mængden af jordprøve er for lille, under 30 g, skal det kommenteres på analysecertifikatet. Eventuelt skal detektionsgrænsen hæves.
Hvis prøven er på mere end 100 g, skal beholderen åbnes og overskydende jord fjernes, således at der gøres plads til opløsningsmidler samt sikres plads til optimal omrystning. Dette skal rapporteres sammen med resultaterne.
8.1.3 Opbevaring
Prøven skal opbevares ved ca. 4 °C i mørke. Prøven skal transporteres til laboratoriet så hurtigt som muligt og inden for 1 døgn efter udtagning.
8.2 Ekstraktionsproceduren
Ekstraktion af prøverne skal påbegyndes indenfor et døgn efter modtagelse på laboratoriet.
8.2.1 Afvejning
Jordprøverne foreligger i 100 mL membranglas f.eks. af typen Duranglas, hvor kunden har fyldt glasset omtrent halvfuldt, hvilket svarer til en prøvemængde på ca. 60 g.
Det halvt fyldte membranglas vejes og mængden af jord bestemmes ved at subtrahere glasvægten. Hvis glasvægten ikke kendes, vejes den tomme flaske efter ekstraktion, rengøring og tørring. Det er en fordel, at kunden anvender forvejede glas.
Hvis prøver modtages i en anden type beholder, afvejes der 60 g prøve i et 100 mL redcap-glas. Det skal sikres ved homogenisering at prøven repræsenterer den samlede prøve, og der udtages delprøver jævnt fordelt over den totale prøve.
8.2.2 Tilsætning af acetone/pentan
Der injiceres 20 mL af den afprøvede acetone (7.2.1.2) til prøven gennem membranen i låget. Ryst glasset kort i hånden. Injicer 20 mL pentan med intern standard (7.2.3) til prøven gennem membranen [13]. Skru låget af og skift membranen. Dette sikrer, at glasset er helt tæt under udrystning. Ryst glasset kraftigt i hånden, indtil en ensartet suspension er nået [14].
8.2.3 Rystning
Glassene ligges ned på langs på rysteapparatet og spændes fast. Rysteapparatet indstilles på en rystefrekvens 200 slag pr. min. med en amplitude på 7 cm. Prøvene rystes i 12 til 16 timer. Derefter tages de af rysteapparatet og tappes umiddelbart herefter.
8.2.4 Aftapning
Når prøverne er taget af rysteapparatet tilsættes 30 mL vand (7.2.1.2). Prøverne rystes 1 min. og henstilles et kort øjeblik. Ved mange prøver dannes en fri klar pentanfase øverst i glasset, denne kan umiddelbart tappes i vials. I andre tilfælde kan det være nødvendigt at centrifugere glasset for at få faserne til at adskille.
Der aftappes det antal vials, der skal benyttes til analysen [15] samt en ekstra til opbevaring. Vials opbevares på køl indtil analysetidspunktet.
9 Fremgangsmåde for totalkulbrinter og BTEX ved GC-FID
9.1 Klargøring og kalibrering af GC-FID
9.1.1 Klargøring/opstart af GC
Gaskromatografen skal være klargjort og vedligeholdt før opstart af prøvesekvens. Injektionsnålen skal være ren og skylleskålene fyldt op. Det anbefales at skifte liner dagligt samt skære 1-2 cm af kolonnen dagligt. Guldpladen skal være ren og uden ridser. Kolonnen skal opretholde tilfredsstillende adskillelse af enkeltkomponenter [16], samt have minimal kolonne blødning.
9.1.2 Metode/betingelser
Der skal vælges betingelser, så der opnås en tilfredsstillende kromatografering af hele området fra benzen til n-alkan C40.
Kravene til ydeevne er:
- Der skal kunne opnås ensartet respons for de enkelte n-alkaner i hele det kromatografiske område. Enkelttoppene skal have en pæn kromatograffisk form. Bedømmes visuelt.
- 2. fraktion kvantificeres overfor summen af C12, C16, C20 og C24.
3. fraktion kvantificeres overfor C28, C30, C32 og C34.
4. fraktion kvantificeres overfor C36 og C38. Det stillede krav er, at det laveste respons indenfor et interval skal udgøre mindst 2/3 af det største. - Der må ikke være uhensigtsmæssig diskriminering af tungere forbindelser. Arealet af C40 skal udgøre mindst 60% af arealet for C20.
I tabel 9-1 ses GC-programmet, anvendt under metodeudviklingen.
| Betingelser | GC-FID |
| Injektionsbetingelser | 350 °C Tryk 11,7 psi. Splitless-tid 1,0 min. Helium som bæregas. |
| Ovnprogram | Starttemp 35 °C. Starttemp holdes 3 min, herefter 15 °C/min til 315 °C. Sluttemp holdes 18 min. |
| Kolonne | HP5 MS 30.0 meter 250 µm ID 0,25 µm filmtykkelse. Flow 1,0 ml/min |
| Detektor | Temperatur 315 °C Hydrogen 30 ml/min. Luft 400 ml/min. Makeup 30 ml/min. |
Tabel 9-1. GC-metoden, der anbefales.
9.1.3 Sekvens/prøveserie
Det anbefales at analysere en sekvensblind (7.2.4.1), en STD-opl. (7.3.1.1), en KNT-opl. (7.3.2.2) og en BTEX-knt [17]. (7.3.3.1) først og sidst i hver prøveserie. Desuden en sekvensblind efter 10 prøver. Der er erfaring for, at en sekvens ikke bør være længere end 20 prøver af hensyn til GC'ens performance. Der skal hver dag analyseres en metodeblind (7.2.4.2).
9.1.4 Kalibrering og kontrol
9.1.4.1 Totalkulbrinter
Det kontrolleres, at:
- der ikke er megen kolonneblødning eller overslæb på blindprøverne.
- forholdet af C40/C20 i standarden (STD-opl. (7.3.1.1)) er mindst 60%.
- indholdet i kontrolprøven (KNT-opl. (7.3.2.2)) giver 80-120% af nominel værdi [18].
9.1.4.2 BTEX på GC-FID
Det kontrolleres, at:
- der ikke er indhold af BTEX i blindprøven højere end detektionsgrænsen.
- arealet af de enkelte BTEX'er i standarden (STD-opl. (7.3.1)) er fornuftige i forhold til hinanden. Arealet af toluen ethylbenzen ½*m-p-xylen o-xylen, benzen ned til 80% af de andre.
- indholdet i kontrolprøven BTEX-knt.(7.3.3.1) giver 80-120% af nominel værdi.
9.2 Resultater
9.2.1 Beregning
9.2.1.1 Total kulbrinter
Retentionstiden for benzen, C10, C25, C35 og C40 i standarden (STD-opl. (7.3.1)) findes. Disse tider plus 0,2 min [19] benyttes til at opdele kromatogrammet af prøver og blindprøver i fraktioner.
Responset af en given prøve fratrækkes blind og arealerne af de interne standarder inden koncentrationen beregnes (sekvensblind 7.2.4.1). Dette gøres for de enkelte fraktioner.
Indholdet i prøverne beregnes efter følgende formel.
![]()
Hvor,
| Afrak. | : | Arealet for den pågældende fraktion. |
| AIS frak. | : | Arealet for den interne standard i pågældende fraktion. |
| Astd. | : | Summen af arealet for de n-alkaner, der hører til pågældende fraktion, se tabel 9-2. |
| AIS std. | : | Arealet for den tilsvarende interne standard i standarden. |
| Cstd. | : | Koncentrationen af standarden i mg/L |
| Vext | : | Volumen af ekstraktionsmiddel i mL |
| m | : | Prøvens vægt i gram |
| TS % | : | Procent tørstof |
| Beregning af fraktioner | ||||
| Beregnes som totalarealet |
Beregnes overfor sumarealet af |
Beregnes i forhold til intern standard |
Opgives som | Totalindholdet [mg/kg TS]: |
| Benzen – C10 | Toluen | monobrombenzen | 1. fraktion benzen – C10 |
fra benzen til og med C10 |
| > C10 – C25 | C12 + C16 +C20 +C24 | o-terphenyl | 2. fraktion > C10 – C25 |
fra C10 til og med C25 |
| > C25–C35 | C28 + C30 + C32 +C34 | o-terphenyl | 3. fraktion >C25–C35 + >C35–C40 |
fra C25 til og med C35 summeret med fra C35 til og med C40 |
| > C35–C40 | C36 + C38 | o-terphenyl | ||
| Totalkulbrinter benzen–C10 + >C10–C25 + >C25–C40 |
Fra og med benzen til og med C10 summeret med fra C10 til og med C25 summeret med fra C25 til og med C40 |
|||
Tabel 9-2. Beregning af de enkelte fraktioner.
Hvis der forekommer interferens på o-terphenyl, således at dens areal afviger mere end 20% i forhold til det gennemsnitlige areal for o-terphenyl i sekvensblinderne beregnes i forhold til squalan.
9.2.1.2 BTEX på GC-FID
BTEX'ernes retentionstider identificeres i standardkromatogrammet (STD-opl. (7.3.1)). BTEX i prøverne integreres udfra retentionstiden af standarden +/- 0,2 min. Arealet for den enkelte BTEX fratrækkes eventuelt blindindhold (7.2.4.1).
Indholdet af enkeltkomponenter beregnes efter følgende formel:
![]()
Hvor,
| Akomp. | : | arealet for den enkelte komponent. |
| AIS pr. | : | arealet for interne standard (monobrombenzen) i prøven. |
| Astd. | : | arealet for samme komponent i standarden. |
| AIS std. | : | arealet for intern standard (monobrombenzen) i standarden. |
| Cstd. | : | koncentrationen af standarden i mg/L |
| Vext | : | volumen af ekstraktionsmiddel i mL |
| m | : | prøvens vægt i gram |
| TS % | : | procent tørstof |
Resultater opgives med to betydende cifre.
10 Fremgangsmåde for PAH og BTEX-MS
10.1 Klargøring og kalibrering af GC-MS
10.1.1 Klargøring/opstart af GC
GC vedligeholdes og klargøres som i afsnit 9.1.1. Kolonnen skal opretholde tilfredsstillende adskillelse af enkeltkomponenter [20]. MS skal være kontrolleret for utætheder samt tunet.
10.1.2 Metode/betingelser
I tabel 10-1 ses GC-MS metoden som anbefales til bestemmelse af PAH og BTEX ved MS.
| Betingelser | GC-MS |
| Injektionsbetingelser | 300 °C Tryk 6,77 psi. Splitless-tid 0,30 min. Helium som bæregas. |
| Ovnprogram | Starttemp 35 °C. Starttemp holdes 3 min, herefter 15 °C/min til 315 °C. Sluttemp holdes 8 min |
| Kolonne | HP5 MS 30.0 meter 250 µm ID 0,25 µm filmtykkelse. Flow 1,0 ml/min |
| Detektor | HP standard |
Tabel 10-1. GC-MS metoden, der har været anvendt under validering.
10.1.3 Sekvensopbygning
10.1.3.1 BTEX på MS
Det anbefales at analysere en blindprøve, en STD-opl. (7.3.1) og en BTEX-knt. (7.3.2.2) først og sidst i hver prøveserie. Desuden en blindprøve (7.2.4.1) og en BTEX-knt. (7.3.2.2) efter hver 10 prøver.
10.1.3.2 PAH på MS
Det anbefales at analysere en blindprøve, en PAH-std. (7.4.1.1) og en PAH-knt. (7.4.1.2) først og sidst i hver prøveserie. Desuden en blindprøve (7.2.4.1) og en PAH-knt. (7.4.1.2) efter hver 10 prøver.
10.1.4 Kalibrering og kontrol
Kvantificeringsdelen i MS skal opdateres i forhold til standard (std-opl.7.3.1.1 /PAH-std. 7.4.1.1), således at retentionstider og forholdet mellem target og qualifierion passer i forhold til den aktuelle standard.
Det kontrolleres, at:
- der ikke er indhold af BTEX/PAH i blindprøven højere end detektionsgrænsen (sekvensblind 7.2.4.1).
- arealet af enkeltkomponenterne i standarden (std-opl. (7.3.1.1)/PAH-std.(7.4.1.1)) er fornuftige i forhold til hinanden.
- indholdet i kontrolprøven (BTEX-knt.(7.3.3.1)/PAH-knt. (7.4.1.2) giver 80-120% af nominel værdi.
10.2 Resultater
10.2.1 Beregning
PAH/BTEX i prøverne integreres udfra retentionstiden af standarden. Det kontrolleres at target/qualifierion forholdet er tilsvarende forholdet i standarden, normalt 80-120%. Dette kan dog fraviges specielt i nærheden af detektionsgrænsen, hvor en visuel vurdering kan godtages.
Arealet for den enkelte PAH/BTEX fratrækkes eventuelt blindindhold (7.2.4.1).
Indholdet af enkeltkomponenter beregnes efter følgende formel:
![]()
Hvor;
| Akomp. | : | arealet for den enkelte komponent. |
| AIS pr . | : | arealet forden interne standard i prøven. |
| Astd. | : | arealet for samme komponent i standarden. |
| AIS std. | : | arealet for den interne standard i standarden. |
| Cstd. | : | koncentrationen af standarden i mg/L |
| Vext | : | volumen af ekstraktionsmiddel i mL |
| m | : | prøvens vægt i gram |
| TS % | : | procent tørstof |
Alle BTEX beregnes overfor monobrombenzen som intern standard. I tabel 10-2 kan det ses hvilken intern standard de enkelte PAH'er beregnes overfor.
| Intern standard | phenanthren-d10 | flouranthen-d10 | benzo(a)pyren-d12 |
| PAH-forbindelser | Naphtalen | Anthracen | Benzo(bjk)fluoranthen |
| Acenaptylen | Fluoranthen | Benzo(a)pyren | |
| Acenaphten | Pyren | Indeno(1,2,3-cd)pyren | |
| Fluoren | Benz(a)anthracen | Dibenz(ah)anthracen | |
| Phenanthren | Chrysen | Benzo(ghi)perylen |
Tabel 10-2. Interne standarder til PAH-MS.
Resultater opgives med to betydende cifre.
11 Fremgangsmåde GC-MS-Scan
11.1 Klargøring og kalibrering af GC-MS
11.1.1 Klargøring/opstart af GC
GC vedligeholdes og klargøres som i afsnit 9.1.1. Kolonnen skal opretholde tilfredsstillende adskillelse af enkeltkomponenter [21]. MS skal være kontrolleret for utætheder samt tunet.
11.1.2 Metode/betingelser
I tabel 11-1 ses GC-MS metoden som anbefales til bestemmelse af petrogene kulbrinter.
| Betingelser | GC-MS |
| Injektionsbetingelser | 300 °C Tryk 6,77 psi. Splitless-tid 0,30 min. Helium som bæregas. |
| Ovnprogram | Starttemp 35 °C. Starttemp holdes 3 min, herefter 15 °C/min til 315 °C. Sluttemp holdes 8 min |
| Kolonne | HP5 MS 30.0 meter 250 µm ID 0,25 µm filmtykkelse. Flow 1,0 ml/min |
| Detektor | HP standard, scan ion 35 m/z – 500 m/z |
Tabel 11-1. GC-MS metoden, der har været anvendt under validering.
11.1.3 Sekvensopbygning
11.1.3.1 Petrogen kulbrintestandard
Det anbefales at analysere en blindprøve, en std. opl. (7.3.1.1.) en petrogen kulbrintestandard (7.5.1.1) og fuelolieopløsning (7.5.2.1) først og sidst i hver prøveserie. Desuden en blindprøve (7.2.4.1) og en fuelolieopløsning (7.5.2.1) efter hver 10 prøver.
11.1.4 Kalibrering og kontrol
Kvantificeringsdelen i MS skal opdateres i forhold til standard (std-opl.7.3.1.1 /Petrogen kulbrintestandard. 7.5.1.1), således at retentionstider passer i forhold til den aktuelle standard.
Der foretages kontrol jf. afsnit 10.1.4.
11.2 Resultater
11.2.1 Petrogene kulbrinter. Beregning
Kulbrinterne i prøverne integreres udfra retentionstiden af standarden og de homologe rækker af naphthalener og phenanthrener integreres udfra retentionstider fundet i fuelolien. Tilsvarende integreres hopaner udfra retentionstider opnået fra fuelolien.
Følgende ionspor anbefales:
| Komponent | MS ion (m/z) | |
| Naphthalen | Naphthalen | 128 |
| 1- og 2-Methylnaphthalen | 1- og 2-Methylnaphthalen | 142 |
| Dimethylnaphthalener | 2,6-Dimethylnaphthalen | 156 |
| Trimethylnaphthalener | 2,3,5-Trimethylnaphthalen | 170 |
| Phenanthren | Phenanthren | 178 |
| Methylphenanthren | 2-Methylphenanthren | 192 |
| Dimethylphenanthren | 3,6-Dimethylphenanthren | 206 |
| Trimethylphenanthren | 3,6-Dimethylphenanthren (indholdet overestimeres evt. *0,01) | 220 |
| Hopanerne | 17a(H),21b(H)-Hopan (30ab) evt. n-alkan C30, areal std*0,23 |
191 |
| Phytosteroler | Indhold beregnes ved GC-FID |
Spor undersøges ved 93 m/z. Integration kan foretages i TIC. |
| n-alkaner* | Lige; overfor n-alkaner Ulige; overfor nærmeste foregående lige n-alkan |
113 |
| Pristan* | Pristan | 113 |
| Phytan* | Phytan | 113 |
Tabel 11-2. Valg af kalibrant og ionspor
Indholdet af enkeltkomponenter beregnes efter følgende formel:
![]()
Hvor;
| Akomp. | : | arealet for den enkelte komponent. |
| AIS pr . | : | arealet forden interne standard i prøven. |
| Astd. | : | arealet for samme komponent i standarden. |
| AIS std. | : | arealet for den interne standard i standarden. |
| Cstd. | : | koncentrationen af standarden i mg/L |
| Vext | : | volumen af ekstraktionsmiddel i mL |
| m | : | prøvens vægt i gram |
| TS % | : | procent tørstof |
Alle kulbrinter kan beregnes overfor o-terphenyl som intern standard.
Resultater opgives med to betydende cifre.
Følgende forhold i tabel 11-3 beregnes jf /7/.
| Parameter |
| Isoprenoidforhold |
| n-alkan C17/pristan |
| n-alkan C18/phytan |
| pristan/phytan |
| CPI-indeks |
| CPI(15 – 21) |
| CPI(25 – 33) |
Tabel 11-3: Indeks
Hvor CPI [22] beregnes som følger;
![]()
![]()
11.2.2 Naturlige kulbrinter. Beregning
Sitosterol lokaliseres udfra ionspor af ion 93 m/z, samt udfra massespektrum. Phytosterolerne lokaliseres ved ionspor 93 m/z, og integreres og beregnes ved GC-FID.
Fedtsyrer lokaliseres ved hjælp af ionspor 73 m/z, integreres og beregnes ved GC-FID.
Fordelingen af ulige hhv. lige n-alkaner samt summen af de ulige n-alkaner fremgår af beregningen af enkeltkomponenterne.
De ulige n-alkaner, der kan fratrækkes totalkulbrinteindholdet fundet ved GC-FID, beregnes som differencen mellem den ulige n-alkan fratrukket indholdet af den lige n-alkan umiddelbart før.
Ulige n-alkaner fra naturlige kulbrinter =
(C25–C24) + (C27–C26) + (C29–C28) + (C31–C30) + (C33–C32)
11.3 Vejledning
11.3.1 Petrogene kulbrinter
De fundne indhold anvendes til vurdering af, hvorvidt der er indhold af petrogene kulbrinter i jordprøven /6/.
11.3.2 Naturlige kulbrinter
Indhold fundet ved GC-MS/GC-FID fratrækkes totalkulbrinteindholdet fundet ved GC-FID.
12 Metodeoplysninger
12.1.1 Detektionsgrænse, præcision og nøjagtighed
Der er under metodeudviklingen valideret detektionsgrænser svarende til dem angivet i tabel 12-1. Præcision og nøjagtighed er også opgivet i tabel 12-1 som middelværdier for BTEX og PAH. Værdierne fundet for de enkelte stoffer kan forefindes i valideringsrapporten for analysemetoden /8/. Disse emner bør eftervises regelmæssigt på det enkelte laboratorium samt når metoden tages i brug.
| Prøve | DL [mg/kg TS] |
Præcision | |||||
| Sb % | Sw % | RSD % | |||||
| Lav høj |
Lav høj |
Lav høj |
|||||
| Kulbrinter: | |||||||
| Benzen - C10 | <2,5 | ||||||
| C10 - C25 | <5 | ||||||
| C25 – C40 | <25 | ||||||
| Totalkulbrinter sum | <32,5 | 15 | 10 | 12 | 8 | 19 | 12 |
| BTEX – FID: | <0,1 | ||||||
| BTEX – MS: | <0,01 | 14 | 8 | 5 | 5 | 14 | 9 |
| PAH – MS: | <0,005 | 15 | 13 | 12 | 10 | 19 | 17 |
Tabel 12-1. Detektionsgrænse, Præcision og nøjagtighed.
12.1.2 Linearitet
Der er under valideringen vist koncentrationsområder, hvori der er en liniær sammenhæng mellem respons og koncentration. I disse områder kan metoden anvendes med enkeltpunktskalibrering (som i denne forskrift). Liniariteten skal eftervises regelmæssigt af de enkelte laboratorier, der anvender denne metode.
13 Analyserapport
Analyserapporten skal som minimum indeholde følgende:
- Henvisning til denne metode.
- Nøjagtig identifikation og prøvemærkning af den enkelte prøve.
- Angivelse af tidspunktet for prøvernes modtagelse på laboratoriet, og hvornår ekstraktionen er indledt.
- Koncentrationen i mg/kg TS for hver komponent, der er bestemt. Samt evt. sum af totalkulbrinter/PAH eller BTEX, der er bestemt.
- Afvigelser fra analyseforskriften skal angives.
- Hvis BTEX er bestemt ved GC-FID skal det anføres at identificationen alene er udført udfra retentionstid.
- Detektionsgrænse og usikkerhed for den enkelte analyseparameter.
- Kvalitativ information udfra GC-FID kromatogrammet som angivelse af olietype, nedbrydningsgrad, tegn på PAH eller chlorerede opløsningsmidler.
- Udspecificering ved hjælp af vejledning /6/ af indhold af kulbrinter, der ikke umiddelbart kan henføres til mineralolieprodukter.
14 Litteratur
/1/ ”ISO/DIS 16703 Soil Quality – Determination of Mineral Oil Content by Gas Chromatography – 2001 February”, Draft International Standard.
/2/ Kemiske stoffers opførsel i jord og grundvand: Bind 2, Projekt om jord og grundvand fra Miljøstyrelsen nr. 20 1996.
/3/ Vejledning fra miljøstyrelsen: Prøveudtagning og analyse af jord, nr. 13. 1998.
/4/ Bestemmelse af olie i jord. Gaskromatografisk metode, november 1998, Miljøstyrelsens Referencelaboratorium. VKI
/5/ Bestemmelse af PAH i jord, Februar 2000, Miljøstyrelsens Jordforureningskontor, VKI.
/6/ DS 204:1980. Vandundersøgelser. Tørstof og glødetest.
/7/ Videreudvikling af metoder til analyse af olie i jord. Delrapport 2. Miljørapport nr. xxx 2003.
/8/ Videreudvikling af metoder til analyse af olie i jord. Delrapport 1. Miljørapport nr. xxx 2003.
Fodnoter
[1] 2-mehylnaphthalen og 1-methylnaphthalen
[2] 2,6-; 2,7-; 1,3-; 1,7-; 1,6-; 2,3-; 1,4-; 1,5- og 1,2-Dimethylnaphthalen
[3] 1,3,7-; 1,3,6-; 1,3,5-; 1,4,6-; 2,3,6-; 1,6,7- (=2,3,8-); 1,2,7-; 1,2,6-; 1,2,4- og 1,2,5-trimethylnaphthalen
[4] 3-; 2-; 9- og 1-Methylphenanthren
[5] 3,6-; 2,6-; 2,7-; 3,5-; 1,3-; 2,10-; 3,9-; 3,10-; 1,6-; 2,5-; 2,9-; 1,7-; 2,3-; 1,9-; 4,9-; 4,10- og 1,8-Dimethylphenanthren
[6] N-Alkanerne C14; C15; C16; C17; C18; C19; C20; C21; C22; C24; C25; C26; C27; C28; C29; C30; C31; C32; C33 og C34.
[7] Farnesan, nor-pristan, pristan og phytan
[8] Ts, Tm, 29ab, 30ab, 31abS og 31abR
[9] Forsynet med et låg, hvori der sidder et septum, som tillader at opløsningsmidler injiceres igennem membranen.
[10] Medmindre der anvendes engangsglasvarer.
[11] Med certifikater.
[12] Dette gør sig også gældende for alle standarder og kontroller.
[13] Det er muligt, at låget kan tages af et kort øjeblik, når pentanen tilsættes. Dette er ikke afprøvet under metodeudviklingen.
[14] For visse jordtyper samt referencematerialer kan det være nødvendigt at tilsætte ekstra acetone eller vand for at opnå suspension.
[15] Afhængigt af antal analyseparametre.
[16] Kontrolleres let ved adskillelse af C17-pristan og C18-phytan.
[17] Kun nødvendigt hvis der skal kvantificeres BTEX på prøverne.
[18] Beregnet udfra den nøjagtige afvejede mængde.
[19] Benzen minus 0,2 min.
[20] Kontrolleres let ved adskillelse af indeno(1,2,3-cd)pyren og dibenz(ah)anthracen
[21] Kontrolleres let ved adskillelse af indeno(1,2,3-cd)pyren og dibenz(ah)anthracen
[22] ”Carbon preference indeks”, giver som navnet antyder et udtryk for foretrukne kulstofforbindelser. Jf. afsnit 12.8 i hovedrapporten for yderligere forklaring.
Bilag 1
Dichlormethan ekstraktion
1.1 Beskrivelse af metoden
Metoden er en gammel metode der er på vej væk fra de danske laboratorier, pga. de miljømæssige konsekvenser ved anvendelse af dichlormethan.
Jordprøven opslemmes i 20 ml vand og der tilsættes 20 ml ekstraktionsopløsning/dichlormethan med IS. Prøven rystes 2 timer på rysteapparat.
Hvis dichlormethan og jord efter henstand stadig er emulgeret, faseadskilles disse ved centrifugering. Vandfasen suges bort.
Dichlormethan dekanteres til 2 ml skruelågsglas med teflonindlæg og komponenterne bestemmes ved gaschromatografi på kapillarkolonne med "Flame Ionisation Detektor" GC/FID eller på GC-MS.
Brombenzen og o-terphenyl benyttes som intern standard(IS).
1.2 Kemikalier
- Dichlormethan
- Brombenzen
- o-terphenyl
- Methanol
- Vand (frisk hanevand)
1.3 Fremstilling af opløsninger
1.3.1 STAM1 og STAM2:
Der fremstilles stamopløsninger med hver af de to interne standarder med en koncentration på 10 g/L (Brombenzen = STAM1, o-terphenyl = STAM2). (1 g af hver intern standard tilsættes 100 mL methanol i en målekolbe → 10 g/L.)
1.3.2 FORT1
Af STAM1 og STAM2 udtages 1 mL til en 100 mL kolbe, og der fyldes op med Dichlor → 100 mg/L af de to interne standarder.
1.3.3 IS Dichlor (5 mg/L)
Fra STAM1 og STAM 2 udtages 1 mL til 2 L målkolbe og der fyldes op med Dichlor → 5 mg/L
1.3.4 STANDARDOPLØSNING 1 (5 mg/L C10-C40)
Af ampul nr. 31266 fra Restek (500 mg/L) tilsættes 1 mL til en 100 mL kolbe og der fyldes op med IS Dichlor → 5 mg/L.
1.3.5 STANDARDOPLØSNING 2 (5 mg/L af 16 PAH'er)
Der udtages 1 mL af ampul PAH Mix fra Agilent med nummer 8500-6035 (konc. 500 mg/L) + 5 mL af FORT 1 til en 100 mL målekolbe som fyldes op med Dichlor (uden IS) → 5 mg/L (PAH) og 5 mg/L (IS).
1.4 Forbehandling af prøven
- 50 g prøve udtages til et 100 mL redcap glas med membranlåg
- Udtag prøve til tørstofbestemmelse.
1.5 Ekstraktion
- Prøveglasset tilsættes gennem membranen 20 mL IS Dichlor, samt 20 mL H2O med dispensette.
- Prøveglasset rystes på rystebord ved den aktuelle rystetid og hastighed (se forsøgsplan).
- Der udtages ekstrakt fra dichlormethanfasen.
2 ISO/DIS 16703
2.1 Beskrivelse af metoden
ISO/DIS 16703 metoden /1/ er en ny officiel ISO standard der er til høring (25.10.01 – 25.03.02). Metoden kan anvendes til kvantificering af totalkulbrinter fra olieholdige prøver i intervallet fra n-alkan C10 til og med C40.
Jordprøven ekstraheres med acetone/n-heptan i 30 minutter. Ekstraktet vaskes med vand og de polære komponenter fjernes ved adsorption til florisil, og det rensede ekstrakt analyseres ved GC-FID.
2.2 Kemikalier
- Acetone
- n-Heptan
- Brombenzen
- o-terphenyl
- Methanol
2.3 Fremstilling af opløsninger
2.3.1 STAM1 og STAM2:
Der fremstilles stamopløsninger med hver af de to interne standarder med en koncentration på 10 g/L (Brombenzen = STAM1, o-terphenyl = STAM2). (1 g af hver intern standard tilsættes 100 mL methanol i en målekolbe → 10 g/L.)
2.3.2 FORT1
Af STAM1 og STAM2 udtages 1 mL til en 100 mL kolbe, og der fyldes op med n-heptan → 100 mg/L af de to interne standarder.
2.3.3 IS Heptan (5 mg/L)
Af hver af de to stamopløsninger (STAM1 og STAM2) udtages 1 mL til 2 L n-Heptan → 5 mg/L pr. intern standard.
2.3.4 STANDARDOPLØSNING 1 (5 mg/L C10-C40)
Af ampul nr. 31266 fra Restek (500 mg/L) tilsættes 1 mL til en 100 mL kolbe og der fyldes op med IS Heptan 5 mg/L.
2.3.5 STANDARDOPLØSNING 2 (5 mg/L 16 PAH'er)
Der udtages 1 mL af ampul PAH Mix fra Agilent med nummer 8500-6035 (konc. 500 mg/L) + 5 mL af FORT 1 til en 100 mL målekolbe som fyldes op med n-Heptan(uden IS) → 5 mg/L (PAH) og 5 mg/L (IS).
2.4 Ekstraktion
- 20 g homogeniseret jordprøve afvejes til et 100 mL redcap-glas
- Udtag prøve til tørstof
- Tilsæt 20 mL acetone med dispensette.
- Ryst et kort øjeblik.
- Tilsæt 10 mL IS Heptan
- Prøven placeres på rystebord med den aktuelle rystehastighed og ekstraktionstid (se forsøgsplan).
2.5 Ekstrakt
- Tilsæt 30 mL destilleret H2O og ryst ca. 1 min. Centrifugér indtil passende faseseparation.
- Der udtages ekstrakt fra heptanfasen.
3 PAH-metode, VKI sag 11670
3.1 Beskrivelse af metoden
PAH-metoden stammer fra en metode Miljøstyrelsens Jordforureningskontor offentliggjorde i februar 2000 /28/. Metoden er meget tidskrævende pga. mange trin både før og efter selve ekstraktionen.
Metoden omfatter bestemmelse af polycyklisk aromatisk hydrocarbon (PAH) i jord ved hjælp af gaskromatografi – massespektrometri (GC-MS) efter ekstraktion.
En homogeniseret jordprøve opslemmes i vand, og der tilsættes en blanding af pentan:acetone(1:1), hvorefter prøven kort gives ultralyd og ekstraheres på rystebord. Ekstraktet centrifugeres, dekanteres og tørres efterfulgt af inddampning til passende volumen.
3.2 Kemikalier
- Na2SO4 (vandfrit, aktiveret ved opvarmning til 450 C i 16 timer)
- Acetone
- n-Pentan
- Brombenzen
- o-terphenyl
- Methanol
- H2O (frisk hanevand)
- 4 M NaOH-opløsning i H2O
3.3 Fremstilling af opløsninger
3.3.1 STAM1 og STAM2:
Der fremstilles stamopløsninger med hver af de to interne standarder med en koncentration på 10 g/L (Brombenzen = STAM1, o-terphenyl = STAM2). (1 g af hver intern standard tilsættes 100 mL methanol i en målekolbe →10 g/L.)
3.3.2 FORT1
Af STAM1 og STAM2 udtages 1 mL til en 100 mL kolbe, og der fyldes op med Pentan → 100 mg/L af de to interne standarder.
3.3.3 IS Pentan (0,75 mg/L)
Fra FORT1 udtages 15 mL til 2 L pentan som fremstilles i 2 L kolbe.
3.3.4 ACETONE:IS-PENTAN-OPLØSNING
En 1 L kolbe fyldes med IS Pentan og en 1 L kolbe fyldes med acetone. Kolbernes indhold blandes sammen i en tom pentanflaske.
3.3.5 STANDARDOPLØSNING 1 (5 mg/L C10-C40)
Af ampul nr. 31266 fra Restek (500 mg/L) tilsættes 1 mL til en 100 mL kolbe og der fyldes op med IS Pentan → 5 mg/L.
3.3.6 STANDARDOPLØSNING 2 (5 mg/L af 16 PAH'er)
Der udtages 1 mL af ampul PAH Mix fra Agilent med nummer 8500-6035 (konc. 500 mg/L) + 5 mL af FORT 1 til en 100 mL målekolbe som fyldes op med Pentan(uden IS) → 5 mg/L (PAH) og 5 mg/L (IS).
3.3.7 4 M NaOH-opløsning
Der afvejes 80 g NaOH, som tilsættes en 500 mL kolbe, som er mindst halv fuld med H2O før tilsætning (frisk koldt hanevand). Der fyldes op med H2O.
3.4 Forbehandling af prøven
- Jordprøven findeles og lufttørres i stinkskab i ca. 18-24 timer.
- Prøverne homogeniseres grundigt og sigtes gennem en 2 mm si for at fjerne træstykker, sten m.m. (tjæreklumper må dog ikke fjernes, men skal homogeniseres med prøven).
- Udtag prøve til tørstofbestemmelse.
3.5 Ekstraktion
- Der afvejes 10 g prøve i en 500 mL red cap flaske (Så præcist som muligt).
- Der tilsættes 40 mL H2O (frisk hanevand).
- pH justeres med NaOH-opløsningen til pH=10-12.
- Der tilsættes 150 mL Acetone:IS-Pentan-opløsning med dispensette.
- Prøven sættes 5 min. i ultralydsbad.
- Prøven sættes på rystebord i den aktuelle tid og med den aktuelle rystehastighed (se forsøgsplan).
- Prøven stilles i ro natten over indtil jordprøven er sedimenteret.
- Væskefasen overføres til skilletragt med stangpipette, og den organiske fase filtreres gennem et faseseparationsfilter med 25-30 g glødet Na2SO4 til en rundbundet kolbe.
- Ekstraktet inddampes til ca. 5 mL på rotationsfordamper uden varme.
- Det inddampede ekstrakt overføres til en 10 mL målekolbe, som fyldes efter til mærket med n-pentan.
- Der udtages ekstrakt fra pentan-opløsningen.
4 PAH-modificeret
4.1 Beskrivelse af metoden
PAH-metoden stammer fra en metode Miljøstyrelsens Jordforureningskontor offentliggjorde i februar 2000 /28/. Metoden er modificeret i forhold til metode 3 (PAH-metoden) således at den er mere simpel pga. at de indledende trin er fjernet.
4.2 Kemikalier
- Acetone
- n-Pentan
- Brombenzen
- o-terphenyl
- Methanol
- 0,05 M pyrofosfat-opløsning
4.3 Fremstilling af opløsninger
4.3.1 STAM1 og STAM2:
Der fremstilles stamopløsninger med hver af de to interne standarder med en koncentration på 10 g/L (Brombenzen = STAM1, o-terphenyl = STAM2). (1 g af hver intern standard tilsættes 100 mL methanol i en målekolbe → 10 g/L.)
4.3.2 FORT1
Af STAM1 og STAM2 udtages 1 mL til en 100 mL kolbe, og der fyldes op med n-pentan &rarr 100 mg/L af de to interne standarder.
4.3.3 IS Pentan (5 mg/L)
Fra STAM1 og STAM 2 udtages 1 mL til 2 L målkolbe og der fyldes op med n-Pentan &rarr 5 mg/L
4.3.4 STANDARDOPLØSNING 1 (5 mg/L C10-C40)
Af ampul nr. 31266 fra Restek (500 mg/L) tilsættes 1 mL til en 100 mL kolbe og der fyldes op med IS Pentan &rarr 5 mg/L.
4.3.5 STANDARDOPLØSNING 2 (5 mg/L af 16 PAH'er)
Der udtages 1 mL af ampul PAH Mix fra Agilent med nummer 8500-6035 (konc. 500 mg/L) + 5 mL af FORT 1 til en 100 mL målekolbe som fyldes op med n-pentan(uden IS)&rarr 5 mg/L (PAH) og 5 mg/L (IS).
4.4 Forbehandling af prøven
- 50 g prøve udtages til et 100 mL redcap glas med membranlåg.
- Udtag prøve til tørstofbestemmelse.
4.5 Ekstraktion
- Prøveglasset tilsættes gennem membranen 20 mL IS-Pentan, samt 20 mL pyrophosfat og 20 mL Acetone fra dispensette.
- Prøveglasset rystes på rystebord ved den aktuelle rystetid og hastighed (se forsøgsplan).
- Prøveglassset centrifugeres 2 min. ved 2000 rpm.
- Der udtages ekstrakt fra pentanfasen.
5 Pentan ekstraktion
5.1 Beskrivelse af metoden
Gaskromatografisk metode til bestemmelse af olie i jord, der har været anvendt siden 1998 /2/.
50 g jord tilsættes pentan med IS og pyrofosfatopløsning (0,05 M). Efter udrystning i 16 timer, udtages ekstrakt fra pentanfasen som analyseres ved GC-FID.
5.2 Kemikalier
- n-Pentan
- 0,05 M pyrofosfat-opløsning
- Brombenzen
- o-terphenyl
- Methanol
5.3 Fremstilling af opløsninger
5.3.1 STAM1 og STAM2:
Der fremstilles stamopløsninger med hver af de to interne standarder med en koncentration på 10 g/L (Brombenzen = STAM1, o-terphenyl = STAM2). (1 g af hver intern standard tilsættes 100 mL methanol i en målekolbe → 10 g/L.)
5.3.2 FORT1
Af STAM1 og STAM2 udtages 1 mL til en 100 mL kolbe, og der fyldes op med n-Pentan → 100 mg/L af de to interne standarder.
5.3.3 IS Pentan (5 mg/L)
Fra STAM1 og STAM 2 udtages 1 mL til 2 L målkolbe og der fyldes op med n-Pentan → 5 mg/L
5.3.4 STANDARDOPLØSNING 1 (5 mg/L C10-C40)
Af ampul nr. 31266 fra Restek (500 mg/L) tilsættes 1 mL til en 100 mL kolbe og der fyldes op med IS Pentan → 5 mg/L.
5.3.5 STANDARDOPLØSNING 2 (5 mg/L af 16 PAH'er)
Der udtages 1 mL af ampul PAH Mix fra Agilent med nummer 8500-6035 (konc. 500 mg/L) + 5 mL af FORT 1 til en 100 mL målekolbe som fyldes op med n-pentan(uden IS) → 5 mg/L (PAH) og 5 mg/L (IS).
5.4 Forbehandling af prøven
- 50 g prøve udtages til et 100 mL redcap glas med membranlåg
- Udtag prøve til tørstofbestemmelse.
5.5 Ekstraktion
- Prøveglasset tilsættes gennem membranen 20 mL IS-Pentan, samt 20 mL pyrophosfat med dispensette.
- Prøveglasset rystes på rystebord ved den aktuelle rystetid og hastighed (se forsøgsplan).
- Der udtages ekstrakt fra pentanfasen.
Bilag 2
Forsøgsplan for ekstraktioner
| Prøvenr. | Analysenr. | Gentagelse | Behandling | Ekstraktionsmetode | Tid [timer] |
| 1 | 1 | A | 11 | Dichlormethan | 8 |
| 2 | 2 | B | 16 | Dichlormethan | 16 |
| 3 | 3 | A | 6 | Dichlormethan | 4 |
| 4 | 4 | B | 11 | Dichlormethan | 8 |
| 5 | 5 | B | 1 | Dichlormethan | 0,5 |
| 6 | 6 | A | 1 | Dichlormethan | 0,5 |
| 7 | 7 | B | 6 | Dichlormethan | 4 |
| 8 | 8 | A | 16 | Dichlormethan | 16 |
| 9 | 1 | A | 12 | ISO/DIS 16703 | 8 |
| 10 | 2 | B | 17 | ISO/DIS 16703 | 16 |
| 11 | 3 | A | 7 | ISO/DIS 16703 | 4 |
| 12 | 4 | B | 12 | ISO/DIS 16703 | 8 |
| 13 | 5 | B | 2 | ISO/DIS 16703 | 0,5 |
| 14 | 6 | A | 2 | ISO/DIS 16703 | 0,5 |
| 15 | 7 | B | 7 | ISO/DIS 16703 | 4 |
| 16 | 8 | A | 17 | ISO/DIS 16703 | 16 |
| 17 | 1 | A | 13 | PAH (VKI) | 8 |
| 18 | 2 | A | 8 | PAH (VKI) | 4 |
| 19 | 3 | A | 18 | PAH (VKI) | 16 |
| 20 | 4 | B | 18 | PAH (VKI) | 16 |
| 21 | 5 | B | 8 | PAH (VKI) | 4 |
| 22 | 6 | A | 3 | PAH (VKI) | 0,5 |
| 23 | 7 | B | 13 | PAH (VKI) | 8 |
| 24 | 8 | B | 3 | PAH (VKI) | 0,5 |
| 25 | 1 | A | 19 | PAH-modificeret | 16 |
| 26 | 2 | B | 9 | PAH-modificeret | 4 |
| 27 | 3 | A | 4 | PAH-modificeret | 0,5 |
| 28 | 4 | A | 9 | PAH-modificeret | 4 |
| 29 | 5 | B | 14 | PAH-modificeret | 8 |
| 30 | 6 | A | 14 | PAH-modificeret | 8 |
| 31 | 7 | B | 19 | PAH-modificeret | 16 |
| 32 | 8 | B | 4 | PAH-modificeret | 0,5 |
| 33 | 1 | B | 10 | n-Pentan | 4 |
| 34 | 2 | A | 15 | n-Pentan | 8 |
| 35 | 3 | B | 15 | n-Pentan | 8 |
| 36 | 4 | A | 5 | n-Pentan | 0,5 |
| 37 | 5 | A | 10 | n-Pentan | 4 |
| 38 | 6 | A | 20 | n-Pentan | 16 |
| 39 | 7 | B | 5 | n-Pentan | 0,5 |
| 40 | 8 | B | 20 | n-Pentan | 16 |
Bilag 3
KULBRINTER (C10-C40)
Tjæreforurent jord (F1)
Bilag 4
Summering af de benyttede olieprodukter
| Olietype | Betegnelse | Kogepunkts-interval* | Kommentarer |
| Gasolie | Fuelolie #4, (Restek ampul catalog # 31244) | C10-C26 | Ampul konc. 50,000 μg/mL |
| Gasolie | Dieselfuel #2, (Restek ampul (catalog # 31258) | C10-C26 | Ampul konc. 50,000 μg/mL |
| Motorolie | motor oil Composite standard, (Restek ampul (catalog # 31464) | C18-C36 | Ampul konc. 50,000 μg/mL |
| Motorolie | Shell X-100 Motor oil | C18-C40 | Afvejet fra rent produkt |
| Gearolie | Shell Donax TA | C13-C34 | Afvejet fra rent produkt |
| Motorolie | Shell Helix plus 10W-40 | C18-C38 | Afvejet fra rent produkt |
| Bionedbrydelig motorolie | Shell Nautilus Bio. Outboard Oil TC-W3 |
C9-C16 og C22-C40 |
Afvejet fra rent produkt |
| Kædeolie, til kædesav | Shell Betula 100 | C16-C39 | Afvejet fra rent produkt |
| Bitumen | Bitumen B, Dansk Shell | C20-? | Afvejet fra rent produkt |
| Stenkulstjære | Stenkulstjære/H-tjære, Koppers Denmark A/S | C10-C34 | Afvejet fra rent produkt |
| Gasolie | Dieselolie fra Shell | C8-C30 | Afvejet fra rent produkt |
| Fuelolie | Shell Fuelolie 77 | C8-C38 | Afvejet fra rent produkt |
* Kogepunktsintervallet er vurderet udfra GC-FID kromatogrammer.
Bilag 5
Valideringsplan
| Valideringsplan | ||||||||
| Total kulbrinter Niveau |
Jordtype | Koncentration [mg/kg TS] |
Dag 1 | Dag 2 | Dag 3 | Dag 4 | Dag 5 | Dag 6 |
| N1 | Alm. lerjord | 110 | N1-1a N1-1b |
N1-2a N1-2b |
N1-3a N1-3b |
N1-4a N1-4b |
N1-5a N1-5b |
N1-6a N1-6b |
| N2 | Spiket sand | 255 | N2-1a N2-1b |
|||||
| N3 | Jord med motor olieforurening |
268 | N3-1a N3-1b |
N3-2a N3-2b |
N3-3a N3-3b |
N3-4a N3-4b |
N3-5a N3-5b |
N3-6a N3-6b |
| N4 | Spiket jord med motorolieforurening | 818 | N4-1a N4-1b |
N4-2a N4-2b |
N4-3a N4-3b |
N4-4a N4-4b |
N4-5a N4-5b |
N4-6a N4-6b |
| N5 | Spiket sand | 998 | N5-1a N5-1b |
N5-2a N5-2b |
N5-3a N5-3b |
N5-4a N5-4b |
N5-5a N5-5b |
N5-6a N5-6b |
| N6 | Spiket sand | 4995 | N6-1a N6-1b |
|||||
| BTEX/PAH | Jordtype | BTEX/PAH [mg/Kg TS] |
Dag 1 | Dag 2 | Dag 3 | Dag 4 | Dag 5 | Dag 6 |
| N1 | Alm. lerjord spiket med BTEX |
0,033 / 0,047 | PN1-1a PN1-1b |
PN1-2a PN1-2b |
PN1-3a PN1-3b |
PN1-4a PN1-4b |
PN1-5a PN1-5b |
PN1-6a PN1-6b |
| N2 | Sand spiket med BTEX og PAH |
0,066 / 0,066 | PN2-1a PN2-1b |
|||||
| N3 | Jord med tjæreforurening spilet med BTEX |
0,33 / 596 | PN3-1a PN3-1b |
PN3-2a PN3-2b |
PN3-3a PN3-3b |
PN3-4a PN3-4b |
PN3-5a PN3-5b |
PN3-6a PN3-6b |
| N4 | Jord med tjæreforurening spiket med BTEX og PAH | 0,67 /0,926 | PN1-4a PN1-4b | PN4-2a PN4-2b | PN4-3a PN4-3b | PN4-4a PN4-4b | PN4-5a PN4-5b | PN4-6a PN4-6b |
| N5 | Sand spiket med BTEX og PAH | 1,67 / 1,67 | PN1-5a PN1-5b | PN5-2a PN5-2b | PN5-3a PN5-3b | PN5-4a PN5-4b | PN5-5a PN5-5b | PN5-6a PN5-6b |
| N6 | Sand spiket med BTEX og PAH | 3,33 / 0,33 | PN6-1a PN6-1b | |||||
Alle prøver er analyseret i randomiseret rækkefølge på GC-apparaterne inden for hver dag.
Bilag 6
Target-og kvalificeringsioner.
| Forbindelse | Retentionstid [min.] |
Target ion (m/z) | Qualifierioner (m/z) 1 2 | |
| Naphtalen | 10,11 | 128,0 | 102,0 | - |
| Acenaphtylen | 12,63 | 152,0 | 151,0 | 153,0 |
| Acenaphten | 12,93 | 153,0 | 154,0 | 152,0 |
| Fluoren | 13,74 | 166,0 | 165,0 | - |
| Phenanthren-d10 | 15,23 | 188,0 | 178,0 | - |
| Phenanthren | 15,26 | 178,0 | 176,0 | 188,0 |
| Anthracen | 15,34 | 178,0 | 176,0 | - |
| Fluoranthen-d10 | 17,15 | 212,0 | 202,0 | - |
| Fluoranthen | 17,18 | 202,0 | 101,0 | 212,0 |
| Pyren | 17,53 | 202,0 | 101,0 | - |
| Benz(a)anthracen | 19,48 | 228,0 | 114,0 | - |
| Chrysen | 19,54 | 228,0 | 114,0 | - |
| Benzo(bjk)fluoranthener | 21,13 | 252,0 | 250,0 | - |
| Benzo(a)pyren-d12 | 21,52 | 264,0 | 252,0 | 250,0 |
| Benzo(a)pyren | 21,56 | 252,0 | 264,0 | - |
| Indeno(1,2,3-cd)pyren | 23,24 | 276,0 | 278,0 | 274,0 |
| Dibenz(ah)anthracen | 23,27 | 278,0 | 276,0 | 274,0 |
| Benzo(ghi)perylen | 23,66 | 276,0 | 274,0 | 278,0 |
Bilag 7
Hopanmønster
Hopaner, anvendte forkortelser, og ionspor 191 m/z /18/.
Bilag 8
Hopaner, responsfaktorer
| Ret. tid Min. |
Rt. IS | RRT | Areal | Areal IS | Korr. Areal IS |
A/Ais | Konc. (mg/l) |
Amount /area |
|
| n-alkan C-18 | 15,34 | 16,16 | 0,95 | 4513595 | 3899698 | 1,16 | 5,00 | 0,231 | |
| 17?(H), 22.29.30-trisnorhopan | 22,06 | 16,16 | 1,37 | 1361969 | 178168 | 356336 | 3,82 | 49,50 | 0,077 |
| 17?(H),21?(H)-30-norhopan | 22,64 | 16,07 | 1,41 | 450468 | 3758439 | 3854809 | 0,12 | 2,48 | 0,047 |
| 17?(H),21?(H)-30-norhopan | 23,07 | 16,16 | 1,43 | 1481930 | 262268 | 524536 | 0,50 | 49,00 | 0,010 |
| 17?(H),21?(H)-30-norhopan | 23,07 | 16,16 | 1,43 | 670427 | 165430 | 330860 | 2,03 | 24,80 | 0,082 |
| 17?(H),21?(H)-hopan | 23,28 | 16,16 | 1,44 | 8080488 | 1371108 | 274220 | 2,947 | 49,50 | 0,060 |
| 17?(H),21?(H)-30-hopan | 23,54 | 16,16 | 1,46 | 10452381 | 1621113 | 3242226 | 3,22 | 49,50 | 0,065 |
| 17?(H),21?(H)-22S-homohopan | 23,94 | 16,16 | 1,48 | 2515244 | 1487164 | 2974328 | 0,85 | 48,00 | 0,018 |
| 17?(H),21?(H)-22R-homohopan | 23,88 | 16,07 | 1,49 | 487851 | 4246405 | 4355287 | 0,11 | 2,28 | 0,049 |
| 17?(H),21?(H)-hopan | 24,31 | 16,16 | 1,50 | 12230475 | 1588458 | 3176916 | 3,85 | 47,50 | 0,081 |
Responsfaktorer (amount/area) for udvalgte hopaner. Rene opløsninger anskaffet fra Chiron AS (se phenanthrenerne i bilag 7), og analyseret i ved GC-MS SIM.
Det er ikke alle hopaner, der kan købes som rene opløsninger, og beregning af et eksaktindhold af hopaner er næsten umuligt. Der må derfor i højere grad beregnes på forhold mellem enkeltkomponenterne. Gennemsnitligt responsfaktor er 0,054. Korrektionsfaktorfaktor i forhold til n-alkan C18 er her 0,23.
Beregnes n-alkan C18 i forhold til n-alkan C30 ses ingen nævneværdig forskel i amount/area (responsforholdet):
(MSTSIM9 Stoffer 130103A, std2)
| Areal | Areal IS | Korr. Areal IS |
A/Ais | Konc. (mg/l) |
Amount /area |
|
| n-alkan C-18 | 4513595 | 5,00 | 1,108*10-6 | |||
| n-alkan C-30 | 4800216 | 5,00 | 1,041*10-6 |
Indholdet af hopanerne kan tilnærmelsesvis kvantificeres overfor n-alkan C30, med en korrektionsfaktor på 0,23 ganget på reponset for standarden.
Bilag 9
Naphthalener, responsfaktorer
Indledningsvis er alle naphthalenerne undersøgt enkeltvis, men der er desværre ikke udført analyse af n-alkan C18 samt fluoranthen, således at de relative forhold har kunnet beregnes.
(MFSPAH; Andrea 090500b)
| Forbindelse | Konc. (amount) [mg/L] |
Rt. [min] |
Respons | Intern standard Respons Phenanthren – d10 |
A Respons/IS |
Amount/A |
| Phenanthren-d10 | 0,5 | 13,2 | - | - | - | - |
| 2,6-Dimethylnaphthalen | 10,0 | 8,8 | 45.245.001 | 170.443 | 265,5 | 0,03767 |
| 2,7-Dimethylnaphthalen | 1,0 | 8,8 | 4.070.904 | 175.798 | 23,2 | 0,04318 |
| 1,7-Dimethylnaphthalen | 1,0 | 9,0 | 3.996.696 | 182.693 | 21,9 | 0,04571 |
| 1,3-Dimethylnaphthalen | 11,2 | 9,0 | 41.366.896 | 186.992 | 221,2 | 0,05045 |
| 1,6-Dimethylnaphthalen | 1,0 | 9,1 | 4.017.457 | 184.066 | 21,8 | 0,04582 |
| 2,3-Dimethylnaphthalen | 10,0 | 9,3 | 43.305.244 | 174.873 | 247,6 | 0,04038 |
| 1,4-Dimethylnaphthalen | 11,1 | 9,3 | 40.775.627 | 187.634 | 217,3 | 0,05126 |
| 1,5-Dimethylnaphthalen | 1,0 | 9,3 | 3.900.903 | 190.416 | 20,5 | 0,04881 |
| 1,2-Dimethylnaphthalen | 16,2 | 9,5 | 51.170.609 | 188.554 | 271,4 | 0,05969 |
| 1,8-Dimethylnaphthalen | 10,0 | 9,7 | 13.055.027 | 181.954 | 71,7 | 0,13937 |
| Gennemsnit =0,05624 | ||||||
| 1,3,7-Trimethylnaphthalen | 1,0 | 10,3 | 4.664.412 | 191.844 | 24,3 | 0,04113 |
| 1,4,6-Trimethylnaphthalen | 1,0 | 10,6 | 9.025.251 | 193.877 | 46,6 | 0,02148 |
| 2,3,6-Trimethylnaphthalen | 1,0 | 10,6 | 3.940.586 | 198.134 | 19,9 | 0,05028 |
| 2,3,5-Trimethylnaphthalen | 1,0 | 10,8 | 4.142.629 | 196.402 | 21,1 | 0,04741 |
| 1,2,6-Trimethylnaphthalen | 1,0 | 10,8 | 202.323 | 210.208 | 1,0 | * |
| 1,2,4-Trimethylnaphthalen | 1,0 | 11,0 | 2.599.625 | 200.566 | 13,0 | 0,07715 |
| 2,4,5-Trimethylnaphthalen | 1,0 | 11,0 | 3.616.838 | 189.210 | 19,1 | 0,05231 |
| 1,2,3-Trimethylnaphthalen | 1,0 | 11,3 | 2.072.236 | 193.176 | 10,7 | 0,09322 |
| 1,4,5-Trimethylnaphthalen | 1,0 | 11,3 | 3.676.876 | 192.074 | 19,1 | 0,05224 |
| Gennemsnit =0,05440 | ||||||
| * ikke medregnet i gennemsnit. | ||||||
Det har derfor været nødvendigt at udføre en fornyet undersøgelse af responsfaktorer, da naphthalen ikke var undersøgt i 1. omgang
(NOVAPAH kaj, 27.09.02);
| Areal | Areal IS | A/Ais | Konc. (mg/l) |
Responsfaktor | Indbyrdes forhold | |
| Phenanthren-d10 | 5567325 | 5,0 | ||||
| naphthalen | 9759740 | 5567325 | 1,753 | 1,6 | 0,9127 | 1,0 |
| 2-methylnaphthalen | 4111929 | 5567325 | 0,7386 | 0,8 | 1,083 | 1,2 |
| 1-methylnaphthalen | 3829361 | 5567325 | 0,6878 | 0,8 | 1,163 | 1,3 |
| 2,6-dimethylnaphthalen | 3655457 | 5567325 | 0,6566 | 0,8 | 1,218 | 1,3 |
| 2,3,5-trimethylnaphthalen | 3142516 | 5567325 | 0,5645 | 0,8 | 1,417 | 1,6 |
Der ses nogen forskel i reponsfaktorer indenfor gruppen af naphthalener, men i nærværende undersøgelse er der ikke udført korrektion.
Forskellen mellem naphthalen og fluoranthen
(NOVAPAH kaj, 27.09.02);
| Areal | Areal IS | A/Ais | Konc. (mg/l) |
Responsfaktor | Indbyrdes forhold | |
| fluoranthen | 15752473 | 5567325 | 1,614 | 1,6 | 0,9913 | 1,1 |
| naphthalen | 9759740 | 5567325 | 1,753 | 1,6 | 0,9127 | 1,0 |
Forskellen mellem fluoranthen og n-alkan C18
(MST stoffer,130103a);
| Areal | Areal IS | A/Ais | Konc. (mg/l) |
Responsfaktor | Indbyrdes forhold | |
| fluoranthen | 19055849 | 5,0 | 2,623*10-7 | 1,0 | ||
| n-alkan C18 | 9055134 | 5,0 | 5,522*10-7 | 0,475 |
Ved at anvende n-alkan C18 som kalibreringsstandard for naphthalenerne får man en fejl i forbindelse med kvantificering. Denne fejl er i størrelsesordenen en faktor 2. Det er valgt at kvantificere udfra n-alkan C18, og ikke at udføre korrektion i alle beregninger. I forbindelse med fastsættelse af kvalitetskriterier, er der vurderet på indhold, og her er der taget højde for den kvantificeringsfejl.
Bilag 10
Phenanthrener, responsfaktorer
Korrigeret areal IS er det opnåede areal korrigeret med en evt. fortyndingsfaktor pga. ampullernes lave koncentration.
Der er ikke helt samme kromatografiske respons for de forskellige dimethylphenanthrener og trimethylphenanthrener.
I denne rapport er der fortaget en approksimation med henblik på kvantificering af phenanthrenerne, da der ikke i alle de analyserede serier med prøver har været kørt referencemateriale.
Følgende omregningsfaktorer er anvendt:
| Komponent | Responsfaktor | Korrektionsfaktor |
| n-alkan C18 | 0,231 | 1,000 |
| phenanthren | 0,419 | 1,814 |
| Methylphenanthrener (MP) | 0,219 | 0,948 |
| Dimethylphenanthrener (DMP) | 0,144 | 0,623 |
| Trimethylphenanthrener (TMP) | Som ovenstående | samme |
Der anvendes n-alkan C18 som kalibreringsstandard, og denne multipliceres med en aktuelle korrektionsfaktor.
Det anbefales, at der kvantificeres overfor udvalgte standarder. Dels er det nemmere at håndtere beregningsmæssigt, og der er betydelig større præcision i kvantificering.
Alle analyserede enkeltkomponenter er referencestandarder fra
Chiron AS
Stiklestadveien 1
N-7041 Tronheim
Norway
e-mail: chiron@chiron.no
web: www.chiron.no
Bilag 11
Scanmetode
Method Information For: C:\MSDCHEM\1\METHODS\MSTSCAN.M
Method Comments:
Metode til GC-MS TIC scan af total kulbrinter
=============================================
HP6890 GC METHOD
=============================================
OVEN
Initial temp: 36 'C (On) Maximum temp: 325 'C
Initial time: 3.00 min Equilibration time: 0.00 min
Ramps:
# Rate Final temp Final time
1 15.00 315 18.00
2 0.0(Off)
Post temp: 0 'C
Post time: 0.00 min
Run time: 39.60 min
FRONT INLET (UNKNOWN)
Mode: Splitless
Initial temp: 350 'C (On)
Pressure: 7.86 psi (On)
Purge flow: 40.0 mL/min
Purge time: 0.40 min
Total flow: 43.6 mL/min
Gas saver: On
Saver flow: 20.0 mL/min
Saver time: 3.00 min
Gas type: Helium
COLUMN
Capillary Column
Model Number: HP 19091J-433
HP-5 5% Phenyl Methyl Siloxane
Max temperature: 325 'C
Nominal length: 30.0 m
Nominal diameter: 250.00 um
Nominal film thickness: 0.25 um
Mode: constant flow
Initial flow: 1.1 mL/min
Nominal init pressure: 7.87 psi
Average velocity: 38 cm/sec
Inlet: Front Inlet
Outlet: MSD
Outlet pressure: vacuumMS Information
-- -----------
| Solvent Delay : |
3.00 min |
| EM Absolute : | False |
| EM Offset : | 400 |
| Resulting EM Voltage : |
2164.7 |
| [Scan Parameters] |
|
| Low Mass : | 35.0 |
| High Mass : | 500.0 |
| Threshold : | 0 |
| Sample # : | 3 A/D Samples 8 |
| Plot 2 low mass : | 271.5 |
| Plot 2 high mass : |
272.5 |
| [MSZones] |
|
| MS Quad : | 150 C maximum 200 C |
| MS Source : | 230 C maximum 250 C |
Bilag 12
Boreprofiler af de rene lokaliteter H1 – H9
Klik her for at se boreprofiler
Bilag 13
GC-MS Scan af prøver fra 0 – 10 cm's boreprofilerne
H1: Agerland, Sand
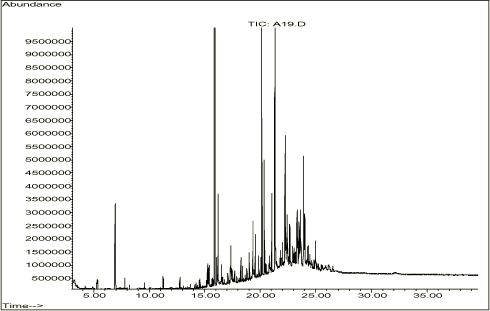
H2: Agerland, Ler
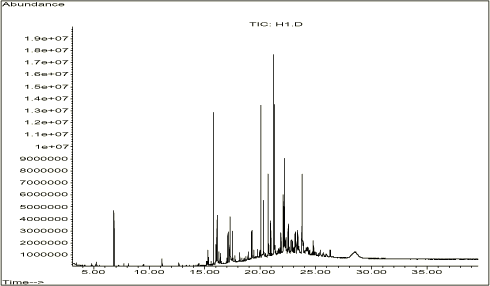
H3: Ferskvandseng
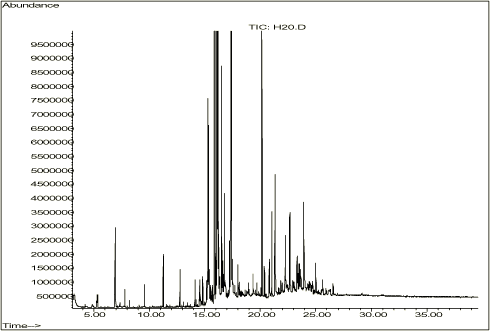
H4: Nåleskov
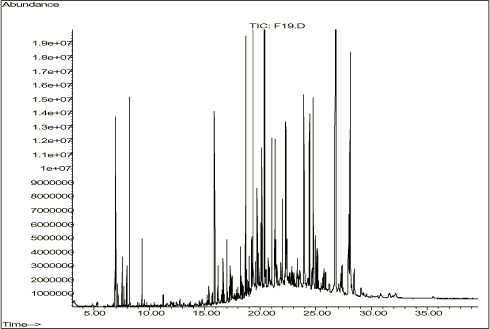
H5: Tørvemose
H6: Bøgeskov
H7: Frederiksdal Inddæmningen
H8: Strandeng
H9: Botanisk Have
Bilag 14
Tabeller over resultater for rene lokaliteter
Bilag 15
SPIMFAB kalibreringsoplĝsninger


Bilag 16
Svenske kvalitetskrav til olie i jord
Uddrag fra SNV; Förslag til riktvärden för förorenade bensinstationer, 1998
Bilag 17
Tabeller over resultater for 50 prøver
Bilag 18
Resultater af metodeafprøvning
Klik her for at se figurer og tabeller til Bilag 18
Version 1.0 Marts 2004 • © Miljøstyrelsen.