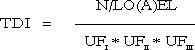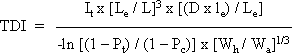|
Miljøprojekt nr. 974, 2004 Principper for sundhedsmæssig vurdering af kemiske stoffer med henblik på fastsættelse af kvalitetskriterier for luft, jord og vandIndholdsfortegnelse
2 Datagrundlaget for vurdering af farlighed
3 Farlighedsvurdering og karakterisering
4 Fastlæggelse af TDI for stoffer med en tærskelværdi for effekter
5 Fastlæggelse af TDI for genotoksiske carcinogener
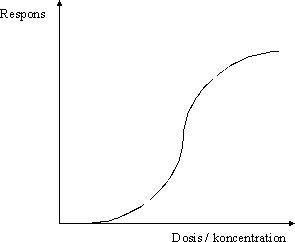
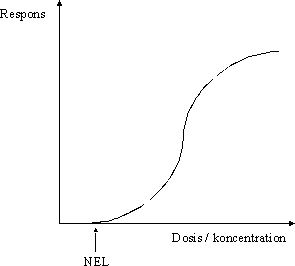
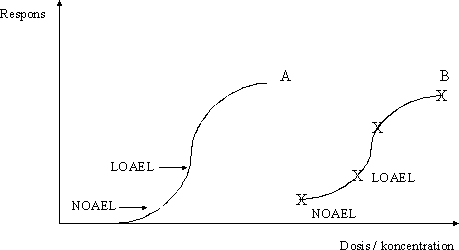
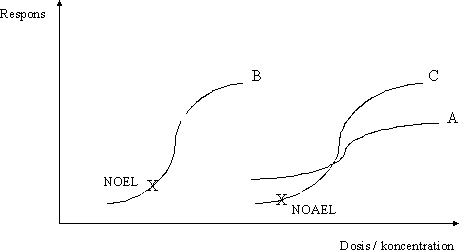
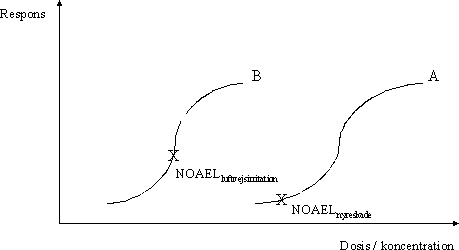
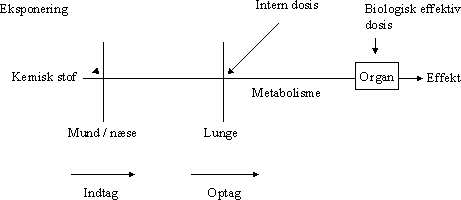
ForordMiljøstyrelsen har i Vejledning nr. 1 om Sundhedsmæssig vurdering af kemiske stoffer i drikkevand fra 1992, og i bilag til Vejledning nr. 6 fra 1990, Begrænsning af luftforurening fra virksomheder beskrevet principper for hvorledes en sundhedsmæssig vurdering bør foretages i forbindelse med fastsættelse af sundhedsmæssigt baserede kvalitetskriterier. Beskrivelse af disse principper og metoder trænger nu til en opdatering set i lyset af den udvikling, der er foregået inden for risikovurderingsområdet i de seneste ti år og i lyset af den praksis, der er fremkommet ved anvendelse af de hidtidige principper. Ikke mindst beskyttelse af børn og gravide samt hensyntagen til eventuelle hormonforstyrrende effekter har påkaldt sig opmærksomhed, og i forbindelse med udarbejdelse af en redegørelse for beskyttelse af børn og gravide mod farlige stoffer (september 1999), har man fra Miljøstyrelsens side anført, at der er behov for at indarbejde (tydeliggøre) sådanne aspekter nøjere i fremtidige, opdaterede retningslinier for fastsættelse af sundhedsmæssigt baserede kvalitetskriterier. Derudover har det været et generelt ønske, at rationalet bag anvendelse af sikkerhedsfaktorer beskrives i et mere detaljeret omfang end tidligere. Formålet med dette projekt er derfor at gennemgå nyere paradigmer for risikovurdering og give forslag til, hvorledes elementer herfra kan inkluderes i Miljøstyrelsens arbejde fremover. Endvidere er formålet, at den praksis, der har udmøntet sig på områderne i de år, hvor de hidtil gældende principper har været anvendt i forbindelse med fastsættelse af jord-/ luft- og drikkevandskvalitetskriterier og B-værdier, i størst muligt omfang indarbejdes ved beskrivelse af procedurerne. Hensigten med projektet er således, at det beskriver den fagligt/tekniske baggrund for, at Miljøstyrelsen kan udarbejde en ny vejledning på området. Miljøstyrelsen har bedt Institut for Fødevaresikkerhed og Ernæring (IFSE) (tidligere: Institut for Fødevaresikkerhed og Toksikologi, IFT), Fødevaredirektoratet om at udarbejde denne opdaterede beskrivelse, idet Instituttet dels har medvirket til udformningen af til de tidligere vejledninger og dels har fungeret som konsulent i de forløbne ti år i forbindelse med udarbejdelse af kvalitetskriterier og B-værdier. For projektet har været nedsat en styregruppe med følgende medlemmer: Poul Bo Larsen, Miljøstyrelsen (formand) Rapporten reflekterer forfatternes såvel som styregruppemedlemmernes holdninger og synspunkter, og der er ikke nødvendigvis opnået konsensus vedrørende alle aspekter i rapporten. Rapporten kan således ikke betragtes som dækkende for de involverede institutioners, Fødevaredirektoratets og Miljøstyrelsens, holdninger og synspunkter. SammenfatningI henhold til Miljøbeskyttelsesloven kan ministeren fastsætte krav til udledning af miljøfaktorer til miljøet og til kvaliteten af de omgivende medier, jord, luft og vand. I administrationen af miljølovgivningen fastsætter Miljøstyrelsen sundhedsmæssigt baserede kvalitetskriterier for en lang række kemiske stoffer i jord, luft og drikkevand. Principperne for fastsættelse af disse kvalitetskriterier er beskrevet i Miljøstyrelsens Vejledning nr. 1 om Sundhedsmæssig vurdering af kemiske stoffer i drikkevand fra 1992, og i bilag til Miljøstyrelsens Vejledning nr. 6 fra 1990, Begrænsning af luftforurening fra virksomheder. Disse principper er beskrevet i rapportens kapitel 1, hvor der endvidere er en meget kort gennemgang af de eksisterende principper for risikovurdering af kemiske stoffer inden for EU samt en kort omtale af forsigtighedsprincippet. Set i lyset af den udvikling, der er foregået inden for området i de seneste ti år, er der opstået et behov for en opdatering af principperne for fastsættelse af kvalitetskriterierne. Formålet med denne rapport er således at gennemgå den nyere udvikling inden for risikovurdering af kemiske stoffer og give forslag til, hvorledes elementer herfra kan inkluderes i Miljøstyrelsens arbejde fremover. Endvidere er formålet, at den praksis, der er fremkommet ved anvendelse af de hidtidige principper for fastsættelse af kvalitetskriterier for luft, jord og drikkevand, i størst muligt omfang inkluderes i de opdaterede principper. Hensigten med rapporten er således at beskrive den fagligt/tekniske baggrund for, at Miljøstyrelsen kan udarbejde en ny vejledning på området. Det videnskabelige grundlag for fastsættelse af sundhedsmæssigt baserede kvalitetskriterier for kemiske stoffer i luft, jord og drikkevand udgøres af en farlighedsvurdering, en dosis-respons(effekt) vurdering (farlighedskarakterisering), samt en eksponeringsvurdering. Farlighedsvurderingen og farlighedskarakteriseringen tager udgangspunkt i undersøgelser af det pågældende stofs toksikologiske effekter i mennesker og i dyr. Datagrundlaget herfor er beskrevet i rapportens kapitel 2. Data hentes primært fra internationale og nationale kriteriedokumenter, via litteratursøgning i internationale databaser, samt fra originalartikler. Endvidere anvendes også upublicerede data fra risikovurderingsrapporterne i EU's risikovurderingsprogram samt i visse tilfælde upublicerede data stillet til rådighed af industrien. Da data således oftest er samlet sammen fra sekundære kilder, foretages der som sådan ikke en egentlig toksikologisk bedømmelse af de enkelte studier. Dog søges originalartiklen altid fremskaffet for de(t) studie(r), der ligger til grund for udpegning af de(n) kritiske effekt(er). Humane data kan stamme fra case reports (f.eks. forgiftningssager), kliniske undersøgelser, undersøgelser af frivillige forsøgspersoner, arbejdspladsundersøgelser, samt epidemiologiske undersøgelser (befolkningsundersøgelser). For langt de fleste kemiske stoffer foreligger der imidlertid ikke velegnede humane data, hvorfor kvalitetskriterierne hyppigst baseres på dyreeksperimentelle undersøgelser. Ideelt set ønskes der ved fastsættelsen af kvalitetskriterier et fuldt datasæt bestående af dyreeksperimentelle undersøgelser til vurdering af følgende toksikologiske egenskaber: toksikokinetik, akut toksicitet, irritation, sensibilisering, toksicitet ved gentagen administration af stoffet, mutagenicitet og genotoksicitet, carcinogenicitet, samt effekter på reproduktion og fosterudvikling. Det skal her understreges, at der ved fastsættelse af kvalitetskriterier altid tages udgangspunkt i den eksisterende viden. Det vil sige, at der ikke igangsættes nye undersøgelser med henblik på at belyse toksikologiske effektområder, hvorom den eksisterende viden er mangelfuld. Det skal også bemærkes, at ikke nødvendigvis alle tilgængelige oplysninger om et givent kemisk stof er relevante i farlighedsvurderingen af det pågældende stof med henblik på fastsættelsen af kvalitetskriterier. Eksponering for et givent stof kan medføre forskellige effekter varierende fra lette gener til dødeligt forløbende forgiftninger. Forløbet er ofte relateret til koncentrationen eller dosis af stoffet. Et enkelt studie (epidemiologisk såvel som dyreeksperimentelt) kan således ofte afsløre forskellige typer af effekter afhængigt af de forskellige koncentrationer eller doser, som mennesker eller dyr har været udsat for i det pågældende studie. Som et led i farlighedsvurderingen foretages der således for hvert enkelt studie en vurdering af dosis-effektsammenhænge, det vil sige en karakterisering af sammenhængen mellem dosis (eller koncentration) af stoffet og de observerede effekter. Derudover foretages også en vurdering af dosis-responssammenhænge, det vil sige en karakterisering af sammenhængen mellem dosis (eller koncentration) af stoffet og forekomst/hyppighed og alvorlighed af de observerede effekter. Principperne for selve farlighedsvurderingen og farlighedskarakteriseringen er beskrevet i rapportens kapitel 3. I dette kapitel er der endvidere inkluderet en omtale af sensibilisering for kemiske stoffer (3.5), kvalitativ vurdering af kræftfremkaldende stoffer (3.6), samt specifikke effekter hos forsøgsdyr hvor relevansen for mennesker er omdiskuteret (3.7). Ved vurdering af alvorligheden af de observerede effekter skelnes der mellem effekter som sådan og skadelige effekter. Generelt betragtes effekter som følge af eksponering for et givent stof som skadelige, når der er tale om væsentlige forandringer i morfologi, fysiologi, funktion, vækst, udvikling, og/eller levetid hos eksponerede individer, og når forekomsten hos denne gruppe er signifikant højere end i kontrolgruppen. Ved farlighedsvurderingen skelnes der endvidere mellem systemiske og lokale effekter. Systemiske effekter optræder i organismen efter optagelse af stoffet via luftvejene, mave-tarmkanalen, og/eller huden. Under lokale effekter henregnes effekter, der optræder lokalt i luftvejene eller i mave-tarmkanalen samt direkte effekter på hud og øjne. I vurderingen af det enkelte studie indgår også en udpegning af nul-effektniveau og det laveste effektniveau for en given effekttype. Farlighedsvurderingen og farlighedskarakteriseringen munder ud i en udpegning af den kritiske effekt, det vil sige den effekt, der anses for at være den væsentligste for fastsættelse af et sundhedsmæssigt baseret kvalitetskriterium. Ved identifikation af den kritiske effekt sammenholdes og vurderes alle de informationer, der er samlet sammen under vurderingerne af alle de enkelte studier. Det vurderes, hvor alvorlige de pågældende effekter er og ved hvilke doser (koncentrationer), de pågældende effekter optræder, ligesom der sondres mellem effekter og skadelige effekter, mellem systemiske og lokale effekter, samt hvorvidt effekter observeret hos forsøgsdyr er relevante for mennesker. Endelig fastlægges der et nul-effektniveau for den kritiske effekt hvis muligt. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand vurderes effekter i reglen som værende mere kritiske desto lavere koncentrationer, de optræder ved, således at nul-effektniveauet for stoffet ofte, men ikke nødvendigvis, vil være identisk med det laveste af de fastlagte nul-effektniveauer fra de enkelte studier. Det næste skridt i fastsættelse af kvalitetskriterierne er fastlæggelse af en tolerabel daglig indtagelse (TDI). TDI er en beregnet størrelse (koncentration eller dosis), som mennesker vurderes at kunne udsættes for (tolerere) gennem et helt livsforløb, uden at der optræder sundhedsskadelige effekter. For stoffer, hvor den akutte toksicitet vurderes som værende den kritiske effekt med henblik på fastsættelse af kvalitetskriterier i luft, jord og drikkevand, giver det ikke mening at fastsætte en TDI og i stedet fastlægges den maksimale tolerable dosis/koncentration (MTD/K) som udgangspunkt for fastsættelse af kvalitetskriterierne. Ved beregningen af TDI skelnes der mellem, hvorvidt der antages at findes en tærskel for stoffets kritiske effekt eller ej. For langt de fleste typer af effekter findes der en tærskel, det vil sige en grænse (koncentration eller dosis), hvorunder der ikke ses effekt. Tærskelværdiens størrelse afhænger af det enkelte stofs potens såvel som af det eksponerede individs følsomhed. For eksempel vil allergikere reagere på udsættelse for allergifremkaldende stoffer ved langt lavere koncentrationer end ikke-allergikere, det vil sige, at allergikere har en lavere tærskelværdi for sådanne stoffer end ikke-allergikere. For stoffer, hvor der er en tærskelværdi for den kritiske effekt, beregnes TDI med udgangspunkt i det observerede nul-effektniveau (NO(A)EL) eller det laveste observerede effektniveau (LO(A)EL) for den kritiske effekt under anvendelse af en usikkerhedsfaktor. Principperne herfor er beskrevet i rapportens kapitel 4. Formålet med usikkerhedsfaktoren (som i andre sammenhænge er benævnt sikkerhedsfaktor, korrektionsfaktor, ekstrapolationsfaktor) er at tage højde for eventuelle mangler i den eksisterende viden og i datagrundlaget. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand opdeles usikkerhedsfaktoren (UF) i tre hovedkategorier (UFI, UFII, UFIII). Usikkerhedsfaktor I (UFI) anvendes med henblik på at tage højde for, at mennesker kan være mere følsomme over for et givent stof end forsøgsdyr. Denne faktor har historisk været sat til 10. Ekstrapolation af data fra dyr til mennesker kan opfattes som omhandlende to forskellige aspekter: 1) korrektion af dosis for forskelle i kropsstørrelse mellem forsøgsdyr og mennesker, såkaldt allometrisk skalering, og 2) andre former for forskelle mellem forsøgsdyr og mennesker, som ikke nødvendigvis afspejles i forskellene i kropsstørrelse. Almindeligvis anvendes legemsvægten (W) som et udtryk for kropsstørrelsen, og korrektion for forskelle i kropsstørrelse mellem dyr og mennesker foretages sædvanligvis på basis af legemsvægt. Denne form for dosiskorrektion er imidlertid blevet kritiseret som værende utilstrækkelig, og internationalt hælder man efterhånden mest til dosiskorrektion for forskelle i kropsstørrelse på basis af stofskiftet (W0,67). En gennemgang af den eksisterende viden understøtter, at der i relation til fastsættelse af kvalitetskriterier for luft, jord og vand fortsat anvendes en standardværdi på 10 for UFI, når korrektion af dosis for forskelle i kropsstørrelse mellem dyr og mennesker foretages på basis af legemsvægt. Det skal dog understreges, at det for visse stoffer og effekter samt for visse forsøgsdyr vurderes, at der sandsynligvis vil være større forskelle i følsomheden mellem forsøgsdyr og mennesker end en 10-faktor. Omvendt vurderes det også, at der i visse tilfælde kan være mindre forskelle i følsomheden mellem forsøgsdyr og mennesker end en 10-faktor. Usikkerhedsfaktor II (UFII) anvendes med henblik på at tage højde for, at nogle individer i befolkningen kan være mere følsomme over for et givent stof end den generelle befolkning (for eksempel børn, gravide, ældre, svækkede, kronisk syge). Denne faktor har oftest været sat til 10. Forskellene i følsomhed skyldes den biologiske variation, der findes mellem mennesker. Faktorer som alder, køn, graviditet, genotype, helbred, og livsstil kan være medvirkende til en øget biologisk følsomhed, som afspejler dels forskelle i toksikokinetik og dels i toksikodynamik. Langt de fleste data vedrørende et stofs toksiske effekter stammer som bekendt fra dyreforsøg. Datagrundlaget for et givent stof giver således sjældent kendskab til, hvorvidt det enkelte individ er mere eller mindre følsomt for det pågældende stof end den gennemsnitlige befolkning. Der er publiceret en række analyser med henblik på at belyse den interindividuelle variation mellem mennesker og dermed en vurdering af størrelsesordenen af usikkerhedsfaktor II. Sammenfattende understøtter disse analyser en standardværdi på 10 for interindividuel variation, idet denne 10-faktor for mange stoffer vurderes at tage højde for den interindividuelle variation inden for størstedelen (mere end 99%) af den humane population. Det skal dog understreges, at det for visse stoffer og effekter samt for visse undergrupper eller enkeltindivider i populationen vurderes, at der vil være større forskelle i den interindividuelle variation end denne 10-faktor. Usikkerhedsfaktor III (UFIII) anvendes med henblik på at tage højde for kvalitet og relevans af de tilgængelige data, det vil sige usikkerheder ved fastlæggelse af nul-effektniveau eller laveste effektniveau for den kritiske effekt. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand har denne faktor typisk varieret fra 1 til 100 afhængigt af datagrundlaget for det pågældende stof. I UFIII indgår elementer som for eksempel kvaliteten af datasættet, route-to-route” ekstrapolation (for eksempel omregning fra oral dosis til inhalationsdosis), “LOAEL-to-NOAEL” ekstrapolation , “duration of exposure” (for eksempel ekstrapolation fra subkronisk nul-effektniveau til kronisk nul-effektniveau), og “nature and severity of toxicity” (for eksempel kræftfremkaldende effekt eller skader på reproduktionssystemet). På baggrund af den eksisterende viden om de enkelte delelementer af UFIII er det ikke muligt at pege på en specifik størrelsesorden for en standardværdi, hverken for de enkelte delelementer af UFIII eller for UFIII som sådan. I relation til fastsættelse af kvalitetskriterier for luft, jord og vand anbefales det derfor fortsat at fastlægge størrelsen af UFIII på baggrund af en ekspertvurdering og case-by-case under hensyntagen til det givne stofs samlede datasæt. Baggrunden for vurderingen af de enkelt delelementer i UFIII bør være så transparent som overhovedet mulig. Som beskrevet ovenfor, vurderes de enkelte elementer (UFI, UFII og UFIII) i den samlede UF hver for sig. Derefter beregnes UF som produktet af hver af de enkelte UF. Ved denne fremgangsmåde kan der imidlertid i visse tilfælde opnås en meget høj samlet UF. Især kan de enkelte delelementer i UFIII bidrage til en høj samlet UF. Derfor skal der foretages endnu en vurdering af den samlede UF med henblik på rimeligheden af denne faktor i forhold til det givne datasæt. Hvis den samlede UF har en meget høj værdi (f.eks. over 10.000), bør datagrundlaget vurderes med hensyn til, om det er meningsfyldt at estimere TDI og beregne kvalitetskriterier på det foreliggende grundlag. For nogle typer af effekter findes der muligvis ikke en tærskel. Dette antager man for eksempel for genotoksiske carcinogener, hvor den tilgrundliggende mekanisme for udvikling af tumorer er en beskadigelse af arvematerialet. Dette betyder i teorien, at en hvilken som helst eksponering, hvor lav den end måtte være, vil medføre en risiko for udvikling af tumorer, der er større end nul. Det skal dog understreges, at man internationalt i de senere år har diskuteret og fortsat diskuterer, hvorvidt der foreligger er tærskel for genotoksiske effekter eller ej. Under antagelse af at der for genotoksiske carcinogener ikke findes en tærskel, fastlægges TDI på baggrund af en kvantitativ risikovurdering. Principperne herfor er beskrevet i rapportens kapitel 5. Der er udviklet en række modeller med henblik på at foretage den kvantitative risikovurdering. I relation til fastsættelse af kvalitetskriterier i luft, jord og drikkevand har Miljøstyrelsen siden 1990 ved estimering af en TDI for genotoksiske carcinogener anvendt den såkaldte one-hit model. Den lineære multistadiemodel (engelsk: Linearised MultiStage (LMS) model) har gennem mange år været US-EPAs foretrukne model til kvantitativ risikovurdering af genotoksiske carcinogener, og denne har ligeledes været anvendt af WHO i relation til guidelines for drikkevandskvalitet. Andre metoder/modeller til kvantitativ risikovurdering af genotoksiske carcinogener er blevet foreslået i de seneste år. For eksempel har US-EPA fremsat nye retningslinier og anbefaler nu som standard at anvende lineær ekstrapolation med udgangspunkt i en dosisdeskriptor kaldet LED10 (den nedre 95% konfidensgrænse for den dosis, der er associeret med 10% ekstra risiko). Inden for EU har der været fremsat forslag om at anvende en mere simplificeret metode baseret på en dosisdeskriptor kaldet T25 (den kroniske dosis (enhed: mg/kg legemsvægt per dag), som vil give 25% af forsøgsdyrene tumorer i et specifikt væv, efter korrektion for den spontane hyppighed, indenfor den standardiserede levetid for det pågældende species). T25-metoden er blevet evalueret ved at sammenligne resultater opnået ved denne metode med resultater opnået ved anvendelse af LMS og LED10. Sammenligningen med både LMS (for 33 stoffer) og med LED10 (for 68 stoffer) viste en god korrelation mellem de 2 metoder indbyrdes. Dog har T25-metoden en tendens til at give et højere resultat end LMS og LED10, det vil sige, at T25-metoden ikke er ligeså konservativ som LMS og LED10. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand anbefales det enten at anvende one-hit modellen i den kvantitative risikovurdering til beregning af TDI eller alternativt T25-metoden. En af disse to metoder anbefales frem for LED10-metoden, idet begge metoder vurderes at give resultater, der er sammenlignelige med LED10-metoden, men beregningerne er mere simple og gennemskuelige og kan udføres uden anvendelse af computerbaserede beregningsprogrammer. Et forhold, der taler for anvendelse af T25-metoden frem for one-hit modellen, er, at denne metode er accepteret i relation til kvantitativ risikovurdering af genotoksiske carcinogener inden for EU's risikovurderingsprogram for eksisterende stoffer. Som led i den kvantitative risikoberegning skal der også tages stilling til, hvilken livstidsrisiko der kan tolereres eller accepteres. Der findes hverken nationalt eller internationalt faste regler herfor, da dette i høj grad er et politisk/administrativt spørgsmål. Den tolerable livstidsrisiko kan derfor være forskellig hos forskellige myndigheder. En livstidsrisiko for at udvikle tumorer på 10-6 betyder, at livslang eksponering for den pågældende dosis eller koncentration kan medføre, at én ud af en million eksponerede individer i princippet kan udvikle én tumor, men ikke at det nødvendigvis sker. Miljøstyrelsens hidtidige administrative praksis i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand har været en livstidsrisiko på 10-6. Den generelle befolkning kan eksponeres for kemiske stoffer ved inhalation af dampe, aerosoler og støv i luften (i indeklima såvel som udendørs), indtagelse af levnedsmidler, drikkevand og jord (jord- og støvpartikler), samt ved hudkontakt med drikkevand, jord og forbrugerprodukter. Ved en eksponeringsvurdering foretages normalt en kvantitativ vurdering af den koncentration (eller dosis) af et kemisk stof i et givent medie (luft, jord, drikkevand, og levnedsmiddel/foder), som et individ eller en population udsættes for i en given situation. I relation til fastsættelse af sundhedsmæssigt baserede kvalitetskriterier for luft, jord og drikkevand foretages der ikke en regelret eksponeringsvurdering som sådan. I stedet for tages der udgangspunkt i nogle standardværdier for indtagelse af jord og drikkevand, for hudkontakt med jord samt for inhalation af luft. Ved omregning af eksponeringskoncentrationer i de respektive medier til en gennemsnitlig daglig dosis anvendes disse standardværdier ligeledes. Dette er beskrevet i rapportens kapitel 6. Endvidere er i dette kapitel for de mest anvendte forsøgsdyr angivet de standardeksponeringsestimater for foder, drikkevand og luft, der ligger til grund for omregning af koncentrationer af kemiske stoffer i disse medier til indtagen dosis (enhed: mg/kg legemsvægt per dag). Ved eksponering for kemiske stoffer via inhalation af luft kan der foretages omregninger mellem eksponeringskoncentrationen i luften (enhed: mg/m³) og en gennemsnitlig daglig dosis (enhed: mg/kg legemsvægt per dag) ved at inddrage kropsvægten og den daglige gennemsnitlige ventilation (volumet af luft indåndet per dag, enhed: m³/dag). Ventilationen (VR) varierer afhængigt af alder, køn, vægt, helbred og fysisk aktivitetsniveau (arbejde, løb, gang, hvile). I relation til fastsættelse af kvalitetskriterier for luft har MST som udgangspunkt hidtil anvendt en standardværdi for VR på 20 m³/dag (gennemsnitsværdi for voksne). De fleste nyere undersøgelser af den gennemsnitlige daglige VR over kortere eller længere tid, herunder variationer i relation til alder, køn, vægt, helbred og fysisk aktivitetsniveau, er foretaget i USA. På baggrund af disse undersøgelser har US-EPA anbefalet for eksponering gennem længere tid at anvende en standardværdi for den gennemsnitlige daglige VR på 11,3 m³/dag for kvinder og 15,2 m³/dag for mænd (alder voksne: 19 år og opefter). For gruppen børn anbefales en række aldersspecifikke standardværdier. For eksponering i kortere tid er der på basis af forskellige aktivitetsmønstre anbefalet en række specifikke standardværdier. Standardværdien på 20 m³/dag, som MST hidtil har anvendt i relation til fastsættelse af kvalitetskriterier for udeluft, vurderes i lyset af de nyere undersøgelser som værende en værdi, der ligger over gennemsnittet for befolkningen som helhed. Ved eksponering for kemiske stoffer via indtagelse af jord kan der foretages omregninger mellem eksponeringskoncentrationen i jorden (enhed: mg/kg jord) og en gennemsnitlig daglig dosis (enhed: mg/kg legemsvægt per dag) ved at inddrage kropsvægten og den daglige gennemsnitlige indtagelse af jord (enhed: mg/dag). Denne eksponeringsvej er især relevant for mindre børn, idet børn af natur er nysgerrige og ofte undersøger ukendte ting ved at putte disse i munden. Der kan således været tale om decideret jordspisning (geophagia), men også indtagelse af jord- eller støvpartikler ved, at børnene sutter på deres hænder, legetøj eller andre genstande. Der kan være en ulige fordeling børn imellem, idet de fleste børn kun indtager relativt små mængder, mens nogle få børn spiser større mængder jord. Det sidste fænomen kaldes pica. MST har i relation til fastsættelse af kvalitetskriterier for jord som udgangspunkt hidtil anvendt en standardværdi for daglig gennemsnitlig indtagelse af jord på 200 mg/dag. I relation til meget kortvarig eksponering har der som udgangspunkt været anvendt en standardværdi på 10 g/dag for at tage højde for eventuelle pica-børn. I løbet af 1990-erne er publiceret en række studier, der alle peger på, at den gennemsnitlige indtagelse af jord for mindre børn ligger under eller omkring 100 mg/dag med en 95 percentil på omkring 200 mg/dag. De fleste estimater er baseret på undersøgelser foretaget i USA. På baggrund af de nyere undersøgelser har US-EPA i 1997 for børn anbefalet en standardværdi for gennemsnitlig indtagelse af jord på 100 mg/dag (børn i alderen op til 6 år) med en 95 percentil på 400 mg/dag og for voksne en gennemsnitsværdi på 50 mg/dag. For pica børn i relation til meget kortvarige (akutte) eksponeringer er anbefalet en standardværdi på 10 g/dag. Standardværdien på 200 mg/dag, som MST hidtil har anvendt som en daglig gennemsnitsværdi for et barns indtagelse af jord i relation til fastsættelse af kvalitetskriterier for jord, vurderes i lyset af de nyere undersøgelser som værende en værdi, der ligger væsentligt over gennemsnitsværdien for børn generelt og vurderes (forsigtigt skøn) som svarende til omkring 95 percentilen for børns daglige indtagelse af jord i de nyere studier. Den generelle befolkning kan også eksponeres for kemiske stoffer i jorden ved direkte hudkontakt. Denne eksponeringsvej er især relevant for mindre børn, idet børn oftest leger på jorden og også mange gange med jorden. Voksne kan også eksponeres for kemiske stoffer i jorden ved direkte hudkontakt, for eksempel ved havearbejde. MST har i relation til fastsættelse af kvalitetskriterier for jord som udgangspunkt hidtil anvendt en standardværdi for hudkontakt med jord på 1000 mg/dag (som en daglig gennemsnitsværdi for børn). Der er ikke fundet nogle studier vedrørende den gennemsnitlige daglige hudkontakt, hverken for børn eller voksne. Ved eksponering for kemiske stoffer via indtagelse af drikkevand kan der foretages omregninger mellem eksponeringskoncentrationen i vandet (enhed: mg/liter) og en gennemsnitlig daglig dosis (enhed: mg/kg legemsvægt per dag) ved at inddrage kropsvægten og den daglige gennemsnitlige indtagelse af drikkevand (enhed: liter/dag). Indtagelsen af drikkevand varierer afhængigt af alder, fysisk aktivitetsniveau (arbejde, løb, gang, hvile), samt omgivelsernes temperatur. MST har i relation til fastsættelse af kvalitetskriterier for drikkevand som udgangspunkt hidtil anvendt en standardværdi for daglig indtagelse af drikkevand på 2 liter/dag (gennemsnitsværdi for voksne). De fleste nyere undersøgelser af den daglige indtagelse af drikkevand er foretaget i USA. På baggrund af udvalgte undersøgelser har US-EPA for voksne anbefalet at anvende en standardværdi for den gennemsnitlige daglige indtagelse af drikkevand på 1,4 liter/dag og en 90 percentil på 2,3 liter/dag (alder: 19 år og opefter). For gruppen børn som sådan anbefales aldersspecifikke standardværdier. Derudover er der anbefalet specifikke standardværdier for gravide og ammende kvinder, og for aktivitet under forskellige temperaturforhold. Standardværdien på 2 liter/dag, som MST hidtil har anvendt som en gennemsnitsværdi i relation til fastsættelse af kvalitetskriterier for drikkevand, vurderes i lyset af de nyere undersøgelser som værende en værdi, der ligger noget over den gennemsnitlige indtagelse for befolkningen som helhed. Standardværdien på 2 liter/dag kan betragtes som en repræsentativ værdi for en 95 percentil for børn i alderen op til 19 år, men ikke for voksne. Selve kvalitetskriteriet for luft, jord og drikkevand beregnes ud fra den beregnede tolerable daglige indtagelse (TDI) ved at dividere TDI (eller en procentdel af denne) med den gennemsnitlige daglige standardeksponering for det relevante medie. Det vil sige, at selve fastsættelsen af det enkelte kvalitetskriterie ud fra TDI er ens for stoffer, hvor der findes en tærskelværdi for de(n) kritiske effekt)er) og for genotoksiske carcinogener. Beregning af kvalitetskriterierne for luft, jord og drikkevand er beskrevet i rapportens kapitel 7. I dette kapitel er endvidere inkluderet en kort gennemgang af betydningen af overskridelse af kvalitetskriterier eller TDI samt af principperne for allokering, det vil sige tildeling af kun en vis procentdel af TDI til eksponering fra et givent medie. English SummaryIn Denmark, health based quality criteria are set for chemical substances in soil, drinking water and ambient air according to the principles laid down in the Guideline [1] for health based evaluation of chemical substances in drinking water and in Appendix A of the Industrial Air Pollution Control Guidelines [2]. These principles are currently undergoing a revision based on the experiences gained during the last ten years within this area as well as on the improved knowledge internationally regarding the various steps in the risk assessment process. The aim of this report is to update the current knowledge and experiences in order to provide the basis for the revision of the principles for the setting of health based quality criteria for chemical substances in soil, drinking water and ambient air. The most essential features of the principles are summarised below. The report has been prepared for the Danish Environmental Protection Agency (DEPA) by the Institute of Food Safety and Nutrition, Danish Veterinary and Food Administration. The scientific basis for the assessment of health based quality criteria for chemical substances in soil, drinking water and ambient air, below referred to as `quality criteria', consists of a hazard identification, a dose (concentration) – response (effects) assessment (hazard characterisation), and an exposure assessment. The hazard identification and characterisation are based on data elucidating the toxicological effects in humans and experimental animals of a given chemical substance. Data are collected from national and international criteria documents and monographs, by searching in international databases, and from original scientific literature. Furthermore, unpublished data from the risk assessment reports within the EU programme on the evaluation and control of existing substances are also included in the hazard assessment and occasionally unpublished data from industry or other sources. Human data include information from case reports (e.g., poisonings), clinical examinations, studies on volunteers, experiences from the working environment, and epidemiological studies. For most chemical substances however, human data are not adequate or available. Therefore, the quality criteria for chemical substances in soil, drinking water and ambient air are primarily based upon data from studies in experimental animals. Ideally, a complete database including information on toxicokinetics, acute toxicity, irritation, sensitisation, repeated dose toxicity, mutagenicity and genotoxicity, carcinogenicity, and toxicity to reproduction should be available. In relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air, this requirement is most often not fulfilled. According to the current administrative practice of the DEPA, the quality criteria are based on the existing database and to compensate for the inadequacy of the database, an uncertainty factor is applied. Exposure to a chemical substance can result in a broad spectrum of effects varying from mild effects as e.g., irritation to fatal poisonings. The type and severity of the effects observed is most often correlated with the exposure concentration. The first step in the hazard assessment is the hazard identification, i.e., an identification of the toxicological effects, which a substance has an inherent capacity to cause. The next step is the hazard characterisation, i.e., estimation of the relationship between dose or exposure concentration to a substance, and the incidence and severity of an effect. Regarding the severity of a given effect, it is evaluated whether the effect can be considered as being adverse or not. Generally, an effect is considered as being adverse when there is a change in morphology, physiology, functional capacity, development, and/or life span in the exposed individuals, and when the incidence of the effect is statistically significantly different from that in the control group. The hazard assessment also includes an evaluation of the `no observed adverse effect level' (NOAEL) and `the lowest observed adverse effect level' (LOAEL) for the various effects observed. When all the relevant toxicological data have been evaluated, the hazard(s) considered most important, “the critical effect(s)”, is identified, i.e., the effect(s), which is considered as being the essential one for the setting of the quality criteria. The critical effect(s) can be considered to be of two types: those considered to have a threshold and those for which there is considered to be some risk at any level (non-threshold, e.g. genotoxic carcinogens). For threshold effects, a NOAEL (or LOAEL) is identified for the critical effect. In relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air, effects are generally considered to be of more concern the lower the concentration (or dose) at which they occur, and the effect observed at the lowest concentration (or dose level) often forms the basis for the quality criteria. The next step in the setting of quality criteria for chemical substances in soil, drinking water and ambient air is the derivation of the tolerable daily intake (TDI), which is an estimate of the intake of a substance, which can occur over a lifetime without appreciable health risk. For acutely toxic substances, the maximal tolerable dose or concentration (MTD/K) forms the basis for the quality criteria. The derivation of the TDI depends on the type of the critical effect: threshold or non-threshold. For threshold effects, the TDI is calculated by dividing the NOAEL (or LOAEL) for the critical effect(s) with an uncertainty factor (UF). The current practice according to the DEPA in relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air is to divide the UF into three categories (UFI, UFII, UFIII). UFI accounts for the interspecies variation in susceptibility, i.e., accounts for that humans may be more susceptible to a given effect than experimental animals. According to the current practice of the DEPA in relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air, a default value of 10 is used for UFI. The interspecies differences can be divided into differences in metabolic size and remaining species-specific differences. To account for differences in metabolic size, three methods are used in practice: extrapolation based on 1) body weight, 2) surface area, and 3) caloric demand. Scaling on the basis of caloric demand to adjust oral NOAELs for metabolic size is internationally considered as being more appropriate compared to extrapolation based on body weight. The data on the remaining (i.e., after metabolic scaling) uncertainty in the extrapolation from animals to humans are too limited in order to suggest a default value. A number of analyses [3] have been performed in order to propose a default value to account for interspecies differences (metabolic as well as the remaining species-specific differences). These analyses support to continue to use a default value of 10 for UFI in relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air when correction for metabolic size is based on body weight. UFII accounts for the differences in interindividual susceptibility, i.e., accounts for subgroups in the population such as children and the unborn child, pregnant women, elderly, or sick people may be more susceptible than the general population. According to the current practice of the DEPA in relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air, a default value of 10 is used for UFII. The responses of humans to xenobiotics may vary because of a number of biological factors, such as age, sex, genetic composition, health status, and lifestyle, and reflect both differences in toxicokinetics and in toxicodynamics. A number of analyses [4] have been performed in order to propose a default value to account for the interindividual differences. These analyses support to continue to use a default value of 10 for UFII in relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air. UFIII accounts for the quality and relevance of the data, i.e., accounts for the uncertainties in the establishment of a NOAEL for the critical effect. According to the current practice of the DEPA in relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air, a value from 1 to 100 is used for UFIII. The UFIII includes elements such as the 1) quality of the database, e.g., data on specific toxic endpoints are lacking or inadequate; 2) route-to-route extrapolation, e.g., no studies using the appropriate exposure route are available; 3) LOAEL-to-NOAEL extrapolation, e.g., a NOAEL cannot be established for the critical effect; 4) subchronic-to-chronic extrapolation, e.g., no chronic studies on which to establish the NOAEL are available; and 5) nature and severity of toxicity, e.g., the critical effect is toxicity to reproduction, carcinogenicity or sensitisation. A number of analyses4 have been performed in order to propose a default value to account for the various elements of UFIII. Based on these analyses, a default value for UFIII as well as for the various elements of UFIII cannot be suggested. In relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air it is therefore recommended to evaluate the value of the UFIII case-by-case based on expert judgement. Finally, the total UF has to be reviewed. If the magnitude of the total UF is very high (e.g., above 10000), the database might be considered as too limited in order to set a health based quality criteria for a given chemical substances in soil, drinking water and ambient air. For non-threshold effects (e.g., genotoxic carcinogens), there is no clear consensus on appropriate methodology for the calculation of the TDI. A number of approaches based largely on characterisation of dose response analyses and low-dose extrapolation have been adopted for assessment of such effects, which all require administrative/political judgements of acceptable health risk. A number of mathematical models have been developed for extrapolation from responses at the high experimental doses generally used in animal carcinogenicity tests to those of the substantially lower exposure levels encountered in human situations. The most extensively used mathematical model to date is the Linearised Multi-Stage (LMS) model, which has been used by US-EPA and by WHO in relation to guidelines for drinking water quality. Another model is the One-hit model, which according to the current practice of the DEPA is used in relation to the setting of quality criteria for genotoxic carcinogens in soil, drinking water and ambient air. US-EPA has recently published (1996, 1999) [5] proposed guidelines for carcinogen risk assessment and one of the key issues is to use linear extrapolation downwards from a benchmark dose (LED10, the 95% lower confidence limit on a dose associated with 10% extra tumour risk adjusted for background) in cases where there is no evidence for non-linearity in the dose response. Within the EU, it has recently been proposed to use the dose descriptor T25 from animal studies as a basis for the quantitative risk characterisation for carcinogens [6]. T25 is defined as the chronic dose rate (in mg/kg b.w./day), which will give 25% of the animals tumours at a specific tissue site, after correction for spontaneous incidence, within the standard lifetime of that species. The results obtained with the T25-approach, which can be calculated without computer programmes, are in agreement with results from computer-based extrapolation methods such as the LMS-model and the benchmark method using LED10. Based on the current knowledge and experiences, it is recommended in relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air either to continue to use the One-hit model or alternatively, to implement the T25-approach. There are no regulations on acceptable health risk, but there is an administrative practice followed by various authorities. Generally, a lifetime risk between 10-6 and 10-7 is considered a tolerable level. A lifetime risk of 10-6 for developing tumours means that exposure for life time to a specific dose or concentration may result in that one individual of a million of exposed individuals develops one tumour. The current administrative practice according to the DEPA in relation to the setting of health based quality criteria for genotoxic carcinogens in soil, drinking water and ambient air is to use a lifetime risk of 10-6. The general population is exposed to chemical substances via inhalation of vapours, aerosols and dust in the air (indoor air as well as ambient air), intake of food, drinking water and soil, and dermal contact to water, soil and consumer products. The exposure assessment generally estimates the concentrations/doses to which human populations are or may be exposed. According to the current practice of the DEPA in relation to the setting of quality criteria for chemical substances in soil, drinking water and ambient air, no such straightforward exposure assessment is performed, but standard exposure rates for soil, drinking water, and air are used. The exposure concentration in the air (expressed in mg/m³) can be converted to an average daily dose (expressed as mg/kg b.w./day) by using the body weight and the daily average inhalation rate (ventilation rate VR, expressed as m³/day). Breathing rates are affected by numerous individual characteristics, including age, gender, body weight, health status, and levels of activity. According to the current practice of the DEPA in relation to the setting of health based quality criteria for chemical substances in ambient air, a standard inhalation rate of 20 m³/day (daily average for adults) is used. A number of analyses on inhalation rates have been performed during the last 10 years, predominantly in the USA. Based on selected key inhalation rate studies, US-EPA [7] has recommended a daily average inhalation rate (VR) for long-term exposure of 11.3 m³/day for women and of 15.2 m³/day for men (Table 6.1.1B); an upper percentile has not been recommended. The recommended inhalation rates for men and women are different than the 20 m³/day, which has commonly been assumed in the past US-EPA risk assessments. For children, US-EPA7 has recommended various age-specific inhalation rates (Table 6.1.1B). Recommended inhalation rates have also been given children and adults for short-term exposures in which distribution of activity patterns are specified (Table 6.1.1B). The exposure concentration in soil (expressed in mg/kg soil) can be converted to an average daily dose (expressed as mg/kg b.w./day) by using the body weight and the average daily soil intake (expressed as mg/day). The potential for exposure to chemical substances via this source is greater for children because they are more likely to ingest more soil than adults as a result of behavioural patterns present during childhood such as the mouthing of objects or hands. Deliberate soil ingestion is defined as pica. According to the current practice of the DEPA in relation to the setting of health based quality criteria for chemical substances in soil, a standard estimate for soil intake of 200 mg/day (daily average for an infant) is used with an intake of 10 g for acutely toxic substances in relation to pica. A number of analyses on soil intake among children have been performed during the last 10 years, predominantly in the USA. Based on selected key studies on soil intake among children, US-EPA [8] has recommended a daily average of 100 mg/day for soil intake among children under 6 years of age with an upper percentile (95 percentile) of 400 mg/day (Table 6.1.2.1C). For children who deliberately ingest soil, an intake of 10 g/day has been recommended by US-EPA8 as a reasonable value for use in acute exposure assessments. Adults may also ingest soil or dust particles that adhere to food, cigarettes, or their hands. According to US-EPA8, only three studies have attempted to estimate adult soil ingestion; a daily average of 50 mg/day for adult soil ingestion has been recommended (Table 6.1.2.1C). The general population can also be exposed to chemical substances in soil by dermal contact, e.g., via outdoor recreation, gardening, construction, and playing. No data are available regarding a daily average for dermal contact to soil. According to the current practice of the DEPA in relation to the setting of health based quality criteria for chemical substances in soil, a standard estimate for dermal contact to soil of 1 g/day is used for children (daily average) and of 0.1 g/day for adults (daily average). The exposure concentration in drinking water (expressed in mg/litre) can be converted to an average daily dose (expressed as mg/kg b.w./day) by using the body weight and the average daily drinking water intake (expressed as litre/day). The daily consumption of drinking water varies with age, levels of physical activity, and fluctuations in temperature and humidity. According to the current practice of the DEPA in relation to the setting of health based quality criteria for chemical substances in drinking water, a standard estimate for drinking water intake of 2 litres/day (daily average for an adult) is used. A number of analyses on drinking water intake have been performed during the last 10 years, predominantly in the USA. Based on selected key studies on drinking water intake, US-EPA8 has recommended a daily average of 1.4 litre/day for drinking water intake among adults (age: above 19 years) with an upper percentile (90 percentile) of 2.3 litres/day (Table 6.1.3D). These recommended values are different than the 2 litres/day commonly assumed in US-EPA risk assessments. For children, US-EPA8 has recommended various age-specific drinking water intake rates (Table 6.1.3C). Finally, the health based quality criteria for chemical substances in soil, drinking water and ambient air are derived for the relevant media by dividing the TDI with the standard exposure rate of this media. To ensure that the total daily intake of a given chemical substance from the various media does not exceed the TDI, a certain percentage of the TDI can be assigned to the relevant media (allocation) and in this way account for other important exposure routes such as exposure through the food. The health based quality criteria derived as described above are used as the basis for the setting of quality criteria for chemical substances in soil and drinking water, and of C-values [9] in ambient air. In this step, other than health based viewpoints may be taken into account, including aesthetical factors such as odour (all media), discoloration (soil, drinking water), taste (drinking water), and microbial growth (drinking water). The C-value is fixed as a mean hourly value that must not be exceeded by more than about seven hours a month, i.e., 1% of the time. When implementing health based quality criteria in air to C-values, the nature of the toxicological effects and the period during which the substances remain active are of crucial importance. Four different categories have been established, for further details are referred to the Appendix A of the Industrial Air Pollution Control Guidelines [10]. Furthermore, economic or political administrative factors may also be taken into account in relation to the setting of quality criteria for chemical substances in soil, drinking water, and ambient air. Fodnoter[1] Sundhedsmæssig vurdering af kemiske stoffer i drikkevand. Vejledning fra Miljøstyrelsen Nr. 1 1992, Miljøministeriet, Miljøstyrelsen. [2] Industrial Air Pollution Control Guidelines. Vejledning fra Miljøstyrelsen Nr. 9 1992, Ministry of the Environment, Denmark, Danish Environmental Protection Agency. [3] For references, see section 4.6. [4] For references, see section 4.6. [5] For references, see section 5.5. [6] For references, see section 5.5. [7] US-EPA (1997). Exposure Factors Handbook EPA/600/P-95/002Fa. Update to Exposure Factors Handbook EPA/600/8-89/043 - May 1989. http://www.epa.gov/nceawww1/pdfs/efh/front.pdf [8] US-EPA (1997). Exposure Factors Handbook EPA/600/P-95/002Fa. Update to Exposure Factors Handbook EPA/600/8-89/043 - May 1989. http://www.epa.gov/nceawww1/pdfs/efh/front.pdf [9] Contribution value. Defined as the maximum amount of any pollutant a company is allowed to emit in the air as immission. The C-value is used in the calculations for all chimneys that emit pollutants into the atmosphere. [10] Industrial Air Pollution Control Guidelines. Vejledning fra Miljøstyrelsen Nr. 9 1992, Ministry of the Environment, Denmark, Danish Environmental Protection Agency. 1 Indledning1.1 Hidtil anvendt praksis for fastsættelse af sundhedsmæssigt baserede kvalitetskriterier Ifølge Miljøbeskyttelsesloven kan ministeren for at beskytte befolkningens sundhed og miljøet fastsætte krav til udledningen af miljøfaktorer til miljøet og til kvaliteten af de omgivende medier, jord, luft og vand. I administrationen af miljølovgivningen har Miljøstyrelsen i den forbindelse således fastsat sundhedsmæssigt baserede grænseværdier (= kvalitetskriterier) for en lang række kemiske stoffer i jord, luft og drikkevand (samt i et mindre omfang i grundvand). De sundhedsmæssigt baserede kvalitetskriterier, som fastsættes for konkrete stoffer ud fra et vurderet behov, er nationale værdier, der anvendes som vejledende værdier som et værktøj ved centrale og decentrale myndigheders håndtering af forureningssager. Hvad angår industriens udledning af luftbåren forurening har dette gennem Luftvejledningen (MST 1990a, MST 2001) medført en række krav. Et vigtigt og styrende element i denne sammenhæng er fastsættelse af bidragsværdier, B-værdier, for en række konkrete udledninger (kemiske stoffer), hvor udgangspunktet for fastsættelsen af disse er sundhedsmæssige vurderinger samt beregning af et sundhedsmæssigt baseret luftkvalitetskriterium (tidligere ofte benævnt som sundhedsmæssig baseret grænseværdi). Tilsvarende er der i forbindelse med jordforureninger opstået en praksis, hvor sundhedsmæssigt baserede jordkvalitetskriterier er et centralt værkstøj i forbindelse med registrering af forurenet jord og i forbindelse med risikovurdering og anvendelse af forurenet jord. Inden for drikkevandsområdet gælder EU-harmoniserede retningslinier for kvalitetskrav. Direktivet for vandkvalitet er dog et minimumsdirektiv og opstiller for en række miljøfaktorer, herunder en række kemiske stoffer, konkrete kravværdier. Ved dansk implementering af dette direktiv kan særlige nationale vurderinger og hensyn medføre afvigelse fra disse kravværdier. For forureninger, der ikke er omfattet af direktivet, kan der udarbejdes nationale kvalitetskriterier for drikkevand. I forbindelse med udarbejdelse af disse nationale værdier anvendes typisk et sundhedsmæssigt baseret drikkevandskvalitetskriterium som udgangspunkt. Drikkevandskvalitetskriterier anvendes desuden som vejledende værdier i konkrete situationer, hvor der ikke er opstillet drikkevandskrav. 1.1 Hidtil anvendt praksis for fastsættelse af sundhedsmæssigt baserede kvalitetskriterierDer findes i dag internationalt anerkendte principper for sundhedsmæssige vurderinger af kemiske stoffer. Miljøstyrelsen tager udgangspunkt i disse principper, når der skal fastsættes sundhedsmæssigt baserede kvalitetskriterier for kemiske stoffer i luft, drikkevand og jord. De principper, som Miljøstyrelsen hidtil har lagt til grund for fastsættelse af sundhedsmæssigt baserede kvalitetskriterier (tidligere benævnt grænseværdier) for kemiske stoffer, er skitseret i Bilag A til Vejledning nr. 6 1990 Begrænsning af luftforurening fra virksomheder (MST 1990a), i daglig tale kaldet Luftvejledningen. I Vejledning nr. 1 1992 Sundhedsmæssig vurdering af kemiske stoffer i drikkevand (MST 1992) er skitseret de principper, som Miljøstyrelsen anvender ved sundhedsmæssige vurderinger af drikkevand. Endvidere er principperne kort sammenfattet i Projekt om jord og grundvand fra Miljøstyrelsen nr. 12 1995 (Nielsen et al. 1995) (dansksproget udgave) samt i Environmental Review no. 6 1996 (Nielsen & Larsen 1996) (engelsksproget udgave). Med udgangspunkt i formuleringerne i de eksisterende vejledninger gengives de hidtil anvendte principper gengives kort: Det videnskabelige grundlag for fastsættelse af kvalitetskriterier for et kemisk stof udgøres af undersøgelser over det pågældende stofs mulige skadevirkninger. Alle relevante data om et givent stof samles i et engelsksproget baggrundsdokument, som danner grundlag for fastsættelsen af kvalitetskriterierne. Endvidere udarbejdes der et datablad på dansk omhandlende de væsentligste data samt grundlaget for fastsættelsen af kvalitetskriteriet. Data hentes fra internationale og nationale kriteriedokumenter samt via litteratursøgning i internationale databaser. Som grundlag for kvalitetskriterier anvendes dels data for, hvordan stoffet virker på mennesker og dels data fra dyreforsøg. Humane data kan foreligge i form af epidemiologiske undersøgelser og for nogle stoffer også som data fra kliniske undersøgelser, fra eksperimentelle undersøgelser på frivillige personer eller fra forgiftningstilfælde. For langt de fleste kemiske stoffer findes imidlertid ikke tilstrækkelig viden om stoffets virkning på mennesker, hvorfor fastsættelse af kvalitetskriterier hyppigst baseres på eksperimentelle undersøgelser i forsøgsdyr. Ud fra de tilgængelige data foretages en farlighedsvurdering, og den kritiske effekt identificeres. Dernæst fastlægges et nul-effektniveau, det vil sige en tærskelværdi (koncentration eller dosis), hvorunder der ikke observeres sundhedsskadelige effekter (engelsk: No Observed Adverse Effect Level - NOAEL). I de tilfælde, hvor der ikke kan fastlægges et NOAEL, fastlægges den laveste koncentration eller dosis, hvor der er observeret sundhedsskadelige effekter (engelsk: Lowest Observed Adverse Effect Level - LOAEL). Efter fastlæggelse af NOAEL (eller LOAEL) anvendes der en række (u)sikkerhedsfaktorer for at nå frem til den koncentration (eller dosis), den tolerable daglige indtagelse (TDI), som mennesker vurderes at kunne udsættes for gennem et helt livsforløb uden, at der optræder skadelige virkninger. Traditionelt har Miljøstyrelsen anvendt tre typer af sikkerhedsfaktorer: SFI sættes traditionelt til 10 og tager højde for, at mennesker kan være mere følsomme over for stoffet end forsøgsdyr. SFII sættes traditionelt til 10 og tager højde for, at nogle individer i befolkningen (f.eks. børn, gravide, gamle, og kronisk syge) kan være mere følsomme over for stoffet end den generelle befolkning. SFIII sættes typisk i intervallet fra 1 til 100 og tager højde for kvalitet og relevans af data, det vil sige usikkerheder ved fastsættelse af NOAEL (LOAEL). For visse typer af sundhedsskadelige effekter menes det, at det ikke er muligt at fastlægge en koncentration (eller dosis), hvorunder stoffet ikke udviser en skadelig effekt. Det vil sige, at for den pågældende effekttype (for eksempel udvikling af tumorer, hvor den tilgrundliggende mekanisme skyldes en beskadigelse af generne) antages det, at der ikke findes en tærskelværdi. I disse tilfælde foretages en risikoberegning ud fra en fastlagt livstidsrisiko. Ved hjælp af en matematisk ”one-hit” model beregnes (sædvanligvis ud fra tumorfundene i dyreforsøg) den daglige livstidsdosis, der vil medføre ét ekstra tilfælde af cancer blandt en population på 1 million dyr (10-6 livstidsrisiko). Ved ekstrapolation af denne dosis til humandosis fås den daglige livstidsdosis (TDI), der anses for at være tolerabel for mennesker. Efter fastlæggelse af TDI beregnes et sundhedsmæssigt baseret kvalitetskriterium for det relevante medie (luft, jord eller drikkevand) ved at dividere med en standardeksponering for det pågældende medie. Standardeksponeringerne for luft, jord og drikkevand er angivet i Miljøprojekt nr. 123 1990 Risikovurdering af forurenede grunde (MST 1990b). Ved beregning af kvalitetskriterier tages endvidere hensyn til, at den generelle befolkning ofte bliver eksponeret for et givent kemisk stof via flere medier (luft, indeklima, drikkevand, levnedsmidler, jord) ved kun at tildele (allokere) en vis procentdel af TDI til det pågældende medie. Ved fastsættelsen af kvalitetskriteriet for et givent medie inddrager Miljøstyrelsen, udover de sundhedsmæssige aspekter, også andre aspekter så som hygiejniske og administrative betragtninger som for eksempel udseende, lugt og smag, samt i relevante tilfælde baggrundsniveauet af det givne stof i mediet. Anvendelse af ovenstående principper og den praksis, der hermed er indarbejdet, repræsenterer efter Miljøstyrelsens opfattelse en forsigtighedstilgang, der har medført, at de fastsatte kvalitetskriterier opfylder målsætningen om et højt beskyttelsesniveau for befolkningen, idet efterlevelse af de anførte værdier anses at sikre mod sundhedsskadelige effekter i befolkningen. Kvalitetskriterierne skal således opfattes som sikkerhedsgrænser og ikke faregrænser. Det vil sige, at overskridelse af et kvalitetskriterium ikke automatisk skal opfattes som ensbetydende med fare, men som et udtryk for, at det høje beskyttelsesniveau, man sædvanligvis tilstræber, er blevet formindskes. 1.2 Risikovurdering af kemiske stoffer i EUFor nye og eksisterende kemiske stoffer, biocider samt pesticider er der i EU-regi fastlagt principper for risikovurdering. De overordnede retningslinier for risikovurderingen af disse forskellige stofgrupper er fastlagt i forskellige direktiver eller forordninger. 1.2.1 Nye og eksisterende kemiske stofferDe overordnede retningslinier for risikovurdering af nye kemiske stoffer er fastlagt i direktiv 93/67/EEC (EEC 1993a) og for eksisterende stoffer i forordningerne 793/93/EEC (EEC 1993a) og 1488/94 (EEC 1994). Principperne er udførligt beskrevet i Technical Guidance Document (TGD) (EC 1996), som for øjeblikket er under revision og som, udover nye og eksisterende stoffer, også til en vis grad skal omfatte biociderne. Danmark tager del i EU's risikovurderingsprogrammer for nye og eksisterende stoffer i henhold til de gældende EU-reguleringer, men disse reguleringer er ikke implementeret i dansk lovgivning som sådan. For både sundhed og miljø omfatter risikovurderingen flere trin: eksponeringsvurdering, effektvurdering (farlighedsvurdering samt dosis-respons/effekt-vurdering), og risikokarakterisering (eksponeringsvurderingen og effektvurderingen sammenholdes). Ved risikokarakteriseringen vurderes den såkaldte Margin of Safety (MOS), det vil sige forholdet mellem nul-effektniveauet (eller det laveste effektniveau) for en given effekt og eksponeringsestimatet for et givent scenarie. Risikokarakteriseringen udarbejdes separat for tre undergrupper i den humane population: arbejdere, forbrugere, og den generelle befolkning eksponeret indirekte via miljøet. Ud fra en overordnet synsvinkel er principperne for farlighedsvurderingen af kemiske stoffer i relation til fastsættelse af sundhedsmæssigt baserede kvalitetskriterier i luft, jord eller drikkevand sammenlignelige med retningslinierne for effektvurderingen af nye og eksisterende kemiske stoffer i EU-regi. Det er således naturligt, at der drages nytte af og sker en vekselvirkning mellem arbejdet i EU og arbejdet med de nationale vurderinger med hensyn til anvendelse af såvel overordnede principper som i konkrete stofvurderinger. 1.2.2 Plantebeskyttelsesmidler og biociderOverordnede retningslinier for risikovurdering af aktive stoffer i plantebeskyttelsesmidler og biocidmidler findes henholdsvis i direktiverne 91/414/EØF (EEC 1991) om markedsføring af plantebeskyttelsesmidler og 98/8/EF (EC 1998) om markedsføring af biocidholdige midler. Den mere detaljerede risikovurdering af midlerne, hvori de aktive stoffer indgår, foregår nationalt efter endnu ikke harmoniserede retningslinier. Det skal dog bemærkes, at det reviderede Technical Guidance Document (se 1.2.1) også til en vis grad omfatter biociderne. Principperne for den sundhedsmæssige risikovurdering af aktivstoffer er meget ens i de to direktiver. De bygger begge på en beskrivelse af stoffernes iboende toksikologiske egenskaber (farevurdering) og en udredning af mulige eksponeringsveje af mennesker og dyr (eksponeringsvurdering). Farevurderingen for aktive stoffer i plantebeskyttelsesmidler og biocidmidler foregår i princippet på helt samme måde som for nye og eksisterende kemikalier og munder ligeledes kvalitativt ud i en klassificering i henhold til kriterierne i direktiv 67/548/EØF om klassificering, emballering, mærkning af kemiske stoffer og produkter (EEC 1967). Kvantitativt udregnes der forskellige referenceværdier, eksempelvis AOEL (Acceptable Operator Exposure Level) og ADI (Acceptable Daily Intake) ved brug af usikkerhedsfaktorer, der følger det traditionelle mønster med tre typer af usikkerhedsfaktorer. Både plantebeskyttelsesmidler og biocidmidler er underlagt en forhåndsgodkendelsesordning, det vil sige, at der er omfattende datakrav, som skal være opfyldt. Til forskel fra proceduren ved fastsættelse af kvalitetskriterier i jord, luft og vand, som bygger på eksisterende data, kræves det således, at den, der ansøger om godkendelse af et plantebeskyttelsesmiddel eller et biocid, genererer de fornødne data inden risikovurderingen kan iværksættes. 1.3 Harmonisering af risikovurdering inden for DG SANCO's Videnskabelige KomiteerVidenskabelige risikovurderingsprocedurer inkluderer både vurdering af risikoen for menneskers helbred og risikoen for miljøet. En væsentlig del af EU's risikovurderingsarbejde blev i 1997 i forbindelse med skandalen omkring BSE (kogalskab) i Europa samlet i DG SANCO, hvor det blev organiseret i 8 komiteer under en Styringskomité (de områder som ikke behandles i DG SANCO's komiteer, er lægemidler, arbejdsmiljø og helbredspåvirkninger hidrørende fra livsstil f.eks. kost, rygning, alkohol). Det viste sig hurtigt, at hver af komiteerne begyndte at udvikle sin egen praksis for risikovurdering, som vanskeliggjorde sammenlignelighed, og Styringskomiteen nedsatte en arbejdsgruppe til at se på mulighederne for harmonisering af risikovurderingsprocedurerne mellem de forskellige komiteer. Som fordele ved en sådan harmonisering fremhævede styringskomiteen:
Den Videnskabelige Styringskomité vedtog den første rapport om harmonisering af risikovurderingsprocedurerne I oktober 2000 (SSC 2000). 1.4 ForsigtighedsprincippetBåde i Danmark og i udlandet har der været tiltagende fokus på forsigtighedsprincippet i de senere år. Men der foreligger ikke konkrete retningslinier for, hvordan princippet anvendes i det daglige arbejde. Således er forsigtighedsprincippet ikke direkte nedfældet i dansk lovgivning. Men i praksis afspejles princippet ofte i de indledende bemærkninger til diverse miljølove, som for eksempel miljøbeskyttelsesloven, lov om kemiske stoffer og produkter, havmiljøloven og genteknologiloven. I kommentarerne til §3 til Miljøbeskyttelsesloven henvises til formuleringen i stk. 2 vedrørende ”forureningens sandsynlige virkninger”, som, ifølge kommentarerne, indebærer, at lovens administration skal bygge på forsigtighedsprincippet. Videre anføres det, at Miljøministeriet ved udstedelse af regler og vejledninger kan operere med for eksempel sikkerhedsfaktorer ved fastsættelse af grænseværdier eller retningslinier for forureningsmæssige beregninger på de områder, hvor der ikke foreligger et tilstrækkeligt eksakt vidensgrundlag. (Bjerring & Møller 1998). I Lov om kemiske stoffer og produkter anføres det i § 1 Denne lov har til formål at forebygge sundhedsfare og miljøskade i forbindelse med fremstilling, opbevaring, anvendelse og bortskaffelse
af kemiske stoffer og produkter. I § 2 er det anført Loven skal sikre, at der tilvejebringes fornødne oplysninger om kemiske stoffer og produkter, der sælges her i landet, og sikre, at der kan
foretages regulering af salg og anvendelse af de kemiske stoffer og produkter, som er eller på grundlag af foreliggende undersøgelser eller erfaringer formodes at være farlige for sundheden
eller skadelige for miljøet. Forsigtighedsprincippet er et princip, hvor tvivl og usikkerheder kommer mennesker og miljø til gode, idet princippet åbner op for regulatoriske tiltag på basis af mistanke om sundhedsfare og miljøskade frem for at afvente endelige videnskabelige beviser. Også i forbindelse med fastsættelse af kvalitetskriterier for luft, jord og drikkevand afspejles dette, idet der ved valg og anvendelse af metoderne til fastsættelse af disse kriterier er inkluderet en forsigtighedstilgang, så man på grund af manglende viden ikke tager unødige chancer, men sikrer et højt beskyttelsesniveau for befolkningen. EU Kommissionen har for nyligt publiceret en meddelelse (engelsk: communication) om forsigtighedsprincippet. Formålet med denne meddelelse er 1) at beskrive Kommissionens indledende fremgangsmåde at anvende forsigtighedsprincippet på; 2) at fastlægge Kommissionens retningslinier for anvendelse af forsigtighedsprincippet; 3) at opbygge en almen forståelse i relation til fastlæggelse, vurdering, håndtering og kommunikation af risici, som videnskaben endnu ikke fuldt ud er i stand til at evaluere; samt 4) at undgå uberettigede forbehold overfor forsigtighedsprincippet som en forklædt form for protektionisme. (EU 2000). 1.5 ReferencerBjerring J og Møller G (1998). Miljøbeskyttelsesloven af 1991 med kommentarer. Jurist- og Økonomforbundets forlag. EC (1998). Directive 98/8/EC of the European Parliament and of the Council of 16 February 1998 concerning the placing of biocidal products on the market. EC (1996). Technical Guidance Document in support of Commission Directive 93/67/EEC on risk assessment for new notified substances and Commission Regulation (EC) No 1488/94 on risk assessment for existing substances. EEC (1994). Commission Regulation (EC) No 1488/94 of 28 June 1994 on risk assessment for existing substances. EEC (1993a). Commission Directive 93/67/EEC of 20 July 1993, lying down the principles for the assessment of risks to man and the environment of substances notified in accordance with Council Directive 67/548/67. EEC (1993b). Council Regulation 793/93/EEC of 23 March 1993 on the evaluation and control of the risks of existing substances. EEC (1991). Council Directive 91/414/EEC of 15 July 1991 concerning the placing of plant protection products on the market. EEC (1967). Council Directive 67/548/EEC of 27 June 1967 on the approximation of the laws, regulations and administrative provisions relating to the classification, packaging and labelling of dangerous substances. EU (2000). Communication from the Commission of the precautionary principle. Commission of the European Communities, Brussels, 2.2.2000 COM(2000) 1 final. MST (2001). Luftvejledningen. Vejledning Nr. 2 2001. Miljøstyrelsen, Miljø- og Energiministeriet. MST (1998). Forsigtighedsprincippet. Udskrift og resumé fra Miljøstyrelsens konference om forsigtighedsprincippet. Eigtveds Pakhus, København 29. maj 1998. Miljø- og Energiministeriet, Miljøstyrelsen. MST (1992). Sundhedsmæssig vurdering af kemiske stoffer i drikkevand. Vejledning fra Miljøstyrelsen Nr. 1 1992. Miljøministeriet, Miljøstyrelsen. MST (1990a). Begrænsning af luftforurening fra virksomheder. Vejledning fra Miljøstyrelsen Nr. 6 1990. Miljøministeriet, Miljøstyrelsen. MST (1990b). Risikovurdering af forurenede grunde. Miljøprojekt nr. 123 1990. Miljøministeriet, Miljøstyrelsen. Nielsen E og Larsen PB(1996). Toxicological evaluation and limit values for DEHP and phthalates, other than DEHP. Environmental Review No. 6 1996. Miljø- og energiministeriet, Miljøstyrelsen. Nielsen E, Larsen PB, Hansen E, Ladefoged O, Mortensen I, Strube M og Poulsen M (1995). Toksikologiske kvalitetskriterier for jord og drikkevand. Projekt om jord og grundvand fra Miljøstyrelsen Nr. 12 1995. Miljøministeriet, Miljøstyrelsen. SSC (2000). Opinion of the Scientific Steering Committee on harmonisation of risk assessment procedures (adopted on 26-27 October 2000). First report published on the internet 18.12.2000). http://europa.eu.int/comm/food/fs/sc/ssc/out82_en.html 2 Datagrundlaget for vurdering af farlighed2.1 Humane data Det videnskabelige grundlag for fastsættelse af sundhedsmæssigt baserede kvalitetskriterier for kemiske stoffer i luft, jord og drikkevand udgøres af en farlighedsvurdering, en dosis-respons(effekt) vurdering (farlighedskarakterisering), samt en eksponeringsvurdering. Som udgangspunkt for fastsættelse af kvalitetskriterier vil viden om, hvordan stoffet påvirker mennesker i princippet udgøre det mest ideelle grundlag. For de fleste kemiske stoffer foreligger der imidlertid ikke velegnede humane data, hvorfor kvalitetskriterier hyppigst baseres på dyreeksperimentelle undersøgelser. Det skal her understreges, at der ved fastsættelse af kvalitetskriterier altid tages udgangspunkt i den eksisterende viden. Det vil sige, at der ikke igangsættes nye toksikologiske undersøgelser med henblik på at belyse effektområder, hvorom den eksisterende viden er mangelfuld. I tilfælde af et mangelfuldt datagrundlag kompenseres der herfor ved anvendelse af en usikkerhedsfaktor. Dette er nærmere beskrevet i 4.4.3. Farlighedsvurderingen og farlighedskarakteriseringen tager udgangspunkt i undersøgelser af det pågældende stofs toksikologiske effekter i mennesker og i dyr og munder ud i en udpegning af den kritiske effekt. Det vil sige den effekt, der anses for at være den væsentligste for fastsættelse af et sundhedsmæssigt baseret kvalitetskriterium, og er nærmere beskrevet i 3.4. Datagrundlaget for farlighedsvurderingen er nærmere beskrevet nedenfor, mens principperne for selve farlighedsvurderingen er nærmere beskrevet i kapitel 3. Ved dosis-respons(effekt)-vurderingen foretages en karakterisering af sammenhængen mellem dosis (eller koncentration) af stoffet og forekomst (engelsk: incidence) og alvorlighed (engelsk: severity) af en effekt. Principperne herfor er nærmere beskrevet i kapitel 4 og 5. Ved eksponeringsvurderingen foretages en kvalitativ eller kvantitativ vurdering af den koncentration (eller dosis) af et kemisk stof i et givent medie (luft, jord, drikkevand, og levnedsmiddel/foder), som et individ eller en population udsættes for. Ved fastsættelsen af sundhedsmæssigt baserede kvalitetskriterier for luft, jord og drikkevand foretages der som sådan ikke en regelret eksponeringsvurdering, idet der tages udgangspunkt i standardbetragtninger for indtagelse af jord og drikkevand samt for inhalation af luft. Dette er nærmere beskrevet i kapitel 6. 2.1 Humane dataSom udgangspunkt for fastsættelse af kvalitetskriterier vil viden om, hvordan stoffet påvirker mennesker i princippet udgøre det mest ideelle grundlag. Fordele ved anvendelse af humane data frem for data fra dyreeksperimentelle undersøgelser er, at disse relaterer sig til den relevante species (mennesket), således at der ikke opstår fortolkningsmæssige problemer med hensyn til ekstrapolering af data fra forsøgsdyr til mennesker. Endvidere kan visse effekter som for eksempel neurologiske effekter, der undersøges ved intellektuelle og psykologiske parametre, i dag kun afsløres ved undersøgelser på mennesker, da der ikke på nuværende tidspunkt findes velegnede forsøgsdyrmodeller til afsløring af sådanne effekter. Som en begrænsende faktor kan nævnes, at der oftest skal fortolkes fra effekter observeret hos en mindre og selekteret gruppe, som regel unge og raske personer til en hel befolkning med følsomme undergrupper som for eksempel børn, ældre, svækkede og syge personer. Når humane data anvendes som udgangspunkt for fastsættelse af sundhedsmæssigt baserede kvalitetskriterier, anvendes i reglen en usikkerhedsfaktor til at tage højde for interindividuelle forskelle i biologisk følsomhed over for kemiske stoffer. Dette er nærmere beskrevet i 4.4.2. Humane data kan stamme fra case reports (f.eks. forgiftningssager), kliniske undersøgelser, undersøgelser af frivillige forsøgspersoner, arbejdspladsundersøgelser, samt epidemiologiske undersøgelser (befolkningsundersøgelser). 2.1.1 Case reports og kliniske undersøgelserEksponering for kemiske stoffer har i en række tilfælde medført forgiftningstilfælde enten som følge af uheld eller på grund af anvendelse af et givent stof i selvmordsøjemed. I sådanne forgiftningstilfælde optræder primært akutte effekter og symptomerne herpå kan i visse tilfælde være velbeskrevne. Der kan dog også for visse stoffer optræde effekter efter en kortere eller længere latenstid, hvor effekterne på grund af tidsforskydningen ikke nødvendigvis vil blive sat i relation til selve det akutte forgiftningstilfælde. I forgiftningssager er det som regel vanskeligt at klarlægge, hvor meget af stoffet den pågældende person har været udsat for, hvilket betyder, at en mere præcis dosis-effekt sammenhæng som oftest ikke kan klarlægges. Data fra enkeltstående forgiftningstilfælde er således ikke særligt velegnede med henblik på fastsættelse af kvalitetskriterier, hvorimod flere velbeskrevne sager med klarlagt dosis-effekt sammenhæng i visse tilfælde vil kunne anvendes. For visse stoffer kan der foreligge humane data som følge af kliniske og fysiologiske undersøgelser, herunder provokationstests i relation til udredning af allergier. Sådanne informationer vil selvsagt indgå i farlighedsvurderingen af et givent stof, men vil som oftest ikke være særligt velegnede med henblik på selve fastsættelsen af kvalitetskriteriet. 2.1.2 Undersøgelser af frivillige forsøgspersonerI kontrollerede forsøg udsættes frivillige forsøgspersoner (oftest unge, raske individer) for specifikke koncentrationer eller doser af et givent stof i et veldefineret tidsrum. Undersøgelserne er i reglen af kortere varighed og har derfor størst værdi i relation til undersøgelser af akutte effekter, herunder irritative og sensibiliserende egenskaber, samt toksikokinetiske aspekter. Fordele ved denne type undersøgelser er, at de foretages under standardiserede betingelser med veldefinerede eksponeringsniveauer, at der kan afsløres lettere grader af effekter som for eksempel irritative gener, at de kan anvendes til vurdering af toksikokinetiske forhold og virkningsmekanismer, samt at der kan foretages en direkte vurdering af sammenhængen mellem eksponeringen og de observerede effekter. Som begrænsende faktorer kan nævnes, at der skal fortolkes fra effekter hos en lille gruppe, ofte unge, raske forsøgspersoner til en hel befolkning med følsomme undergrupper som for eksempel børn, ældre, svækkede og syge personer. Endvidere er det af praktiske grunde kun muligt at foretage undersøgelser af kortere varighed. Sidst men ikke mindst er de etiske aspekter af stor betydning, hvorfor undersøgelser af kemiske stoffer på frivillige forsøgspersoner kun sjældent igangsættes i Danmark og kun under velkontrollerede forhold, hvor marginale effektpåvirkninger udløses. Eksempler herpå er påvirkning af lugt og/eller smag, lette påvirkninger af lungefunktionsparametre, samt målinger af biomarkører eller lignende parametre repræsenterende meget lette grader af påvirkninger. Miljøstyrelsen tager således af etiske årsager afstand fra at anvende humantestning som standardtest i forbindelse med godkendelsesprocedurer og fastlæggelse af generelle testningsprocedurer for kemikalier. 2.1.3 ArbejdspladsundersøgelserFor nogle kemiske stoffer foretages der jævnligt målinger af koncentrationer i arbejdsmiljøet. Endvidere har medarbejderne mulighed for at rapportere sundhedsmæssige gener til læger knyttet til arbejdspladsen. Ligeledes undersøges visse medarbejdere jævnligt for en række sundhedsmæssige parametre blandt andet med henblik på at vurdere, hvorvidt de pågældende medarbejdere udsættes for kemiske stoffer. I nogle tilfælde rapporteres sådanne arbejdspladsundersøgelser i den åbne litteratur, og data herfra kan således inddrages i farlighedsvurderingen af et givent kemisk stof. En begrænsning i anvendelsen af sådanne data ligger dog i, at flertallet af arbejdere i reglen udsættes for mere end et enkelt kemisk stof, hvorfor det ofte kan være vanskeligt at klarlægge en dosis-effekt sammenhæng for et givent stof. Endvidere består arbejdere af en mere selekteret og homogen gruppe end den almene befolkning, hvor sidstnævnte også omfatter særligt følsomme/sårbare undergrupper som for eksempel børn og ældre samt særligt svækkede og syge personer. 2.1.4 Epidemiologiske undersøgelserFor kemiske stoffer, som har været i anvendelse over længere tid og i afgrænsede befolkningsgrupper, kan der foreligge epidemiologiske undersøgelser over stoffets toksikologiske effekter hos mennesker. Effektmålene i epidemiologiske undersøgelser er typisk dødelighed, sygelighed, antal lægebesøg/indlæggelser, forekomst af generende symptomer og lignende. Epidemiologiske undersøgelser er således også relevante i relation til undersøgelser af langtidseffekter, herunder cancer. Fordele ved epidemiologiske undersøgelser er, at de baseres på eksisterende data. Endvidere tages der højde for, at den udvalgte befolkningsgruppe ofte består af en heterogen population, samt at eksponeringen har fundet sted i befolkningsgruppens ”naturlige” miljø, det vil sige i tilstedeværelse af andre risikofaktorer end det givne kemiske stof. Blandt ulemperne kan nævnes, at det ofte kan være svært at foretage en korrekt eksponeringsvurdering, det vil sige at kunne vurdere hvor meget af et givent stof den udvalgte befolkningsgruppe rent faktisk har været udsat for. Endvidere har den udvalgte befolkningsgruppe sædvanligvis også været udsat for mere end et enkelt kemisk stof og/eller andre relevante påvirkninger (for eksempel støj og stress), hvilket kan vanskeliggøre en vurdering af en årsagssammenhæng. Tolkningsvanskeligheder kan også skyldes, at befolkningsgruppen i en given undersøgelse ofte vil være selekteret og for eksempel kun omfatte ansatte på en virksomhed, hvor der i nogle tilfælde stort set kun er ansat det ene køn, og hvor særligt følsomme individer aldrig har været ansat eller er holdt op på grund af helbredsmæssige problemer ved arbejdet. Man får således i disse tilfælde ikke noget indtryk af effekterne på børn, ældre, og syge. Antallet af personer, der har været udsat for et givent kemisk stof, kan ofte være for lille til, at man kan få et statistisk signifikant resultat og dermed afsløre en årsagssammenhæng. Endelig vil der ofte være tale om en ”slørende” indflydelse på undersøgelsesresultaterne af andre faktorer (confounders) som for eksempel alder, køn, socio-økonomisk status, og rygning; disse faktorer er der dog i varierende grad mulighed for at korrigere for, hvilket er gjort i velgennemførte epidemiologiske undersøgelser. Et velgennemført epidemiologisk studie forudsætter, at både udfaldet (effekten) og eksponeringen er dokumenteret, objektiviseret og om muligt kvantificeret. Der er således en række kriterier, som ofte benyttes ved vurdering af sammenhængen mellem årsag og effekt, de såkaldte kausalitetskriterier udviklet af Hill (1965):
Det skal understreges, at epidemiologiske undersøgelser, hvor der ikke er fundet en sammenhæng mellem effekter og eksponering for et kemisk stof, ikke uden videre kan frikende det pågældende stof. For eksempel kræves der for nogle typer af effekter temmelig store datamaterialer, hvis man skal være i stand til at finde beskedne påvirkninger. 2.2 Dyreeksperimentelle undersøgelserFor de fleste kemiske stoffer foreligger der ikke velegnede humane data, hvorfor kvalitetskriterier hyppigst baseres på dyreeksperimentelle undersøgelser. Resultaterne fra dyreforsøgene anvendes således til at forudsige den mulige virkning på mennesker. Det vil sige, at resultater fra dyreforsøg anvendes som model for tilsvarende effekter i mennesker. Dyreeksperimentelle data bruges også til at supplere humane data, der ikke er entydige eller til at udpege de aktive stoffer, når mennesker har været udsat for blandinger af stoffer. Ideelt set ønskes der ved fastsættelsen af kvalitetskriterier et fuldt datasæt bestående af dyreeksperimentelle undersøgelser til vurdering af følgende toksikologiske egenskaber: toksikokinetik, akut toksicitet, irritation, sensibilisering, toksicitet ved gentagen administration af stoffet, mutagenicitet og genotoksicitet, carcinogenicitet, samt effekter på reproduktion og fosterudvikling. Fordelene ved dyreforsøg er, at der er tale om standardiserede forsøgsbetingelser, at der er mulighed for at afsløre væsentligt flere effekter hos forsøgsdyr end hos mennesker, da organer og væv kan undersøges efter forsøgets afslutning, samt at der er mulighed for at undersøge virkningsmekanismer og dosis-effekt sammenhænge for enkeltstoffer. Blandt ulemperne kan nævnes, at der er visse typer af effekter som kun vanskeligt afsløres i dyreforsøg (f.eks. lettere grader af irritation af slimhinder, visse typer af nerveskader, samt visse neuropsykologiske påvirkninger), at det er vanskeligt at registrere lettere grader af effekter (der for eksemplet kommer til udtryk som subjektive gener hos mennesker), samt at resultaterne skal fortolkes fra en homogen gruppe af dyr til at omfatte alle individer i en heterogen befolkning. Når dyreeksperimentelle data anvendes som udgangspunkt for fastsættelse af sundhedsmæssigt baserede kvalitetskriterier, anvendes i reglen en (u)sikkerhedsfaktor til at tage højde for interspeciesvariation, idet mennesker kan være mere følsomme over for udsættelse for kemiske stoffer end andre pattedyr. Dette er nærmere beskrevet i 4.4.1. 2.2.1 Guidelines for dyreforsøgVed fastsættelse af sundhedsmæssigt baserede kvalitetskriterier for kemiske stoffer i luft, jord og drikkevand tages hensyn til kvalitet og relevans af de foreliggende dyreforsøg. Der foreligger ikke nogle formelle kvalitetskrav til de studier, der danner udgangspunkt for fastsættelse af kvalitetskriterier. Studier af høj kvalitet, som er udført i henhold til guidelines (OECD, EU og lignende), foretrækkes ved fastsættelse af kvalitetskriterier, såfremt disse studier er udført på en måde, så de er velegnede til at belyse den konkrete problemstilling (relevans). Desværre er det langtfra altid tilfældet, at sådanne studier foreligger for de konkrete kemiske stoffer, der fastsættes kvalitetskriterier for, hvorfor andre former for studier ofte vil danne udgangspunkt for fastsættelse af kvalitetskriterier. For eksempel er en lang række studier (udført i forskningsøjemed og publiceret i den videnskabelige litteratur) af høj kvalitet, selvom disse ikke er udført i henhold til guidelines eller i overensstemmelse med formaliseret GLP (Good Laboratory Practice). Når der ved fastsættelse af kvalitetskriterier tages udgangspunkt i studier, der ikke lever fuldt op til det niveau, som vurderes at være nødvendigt med hensyn til kvalitet og relevans, anvendes i reglen en ekstra (u)sikkerhedsfaktor til at tage højde herfor. Dette er nærmere beskrevet i 4.4.3. OECD har udarbejdet alment accepterede retningslinier for gennemførelse af dyreeksperimentelle undersøgelser af kemiske stoffer inden for de forskellige forsøgstyper og har stået bag udarbejdelsen af principper for god laboratoriemæssig praksis (GLP). Også inden for EU er der fastlagt principper for gennemførelse af undersøgelser af kemiske stoffer i dyreforsøg (Annex V til 67-direktivet); disse principper er stort set identiske med OECD's guidelines. Disse retningslinier og principper er udarbejdet for at sikre en ensartet og høj kvalitet af de undersøgelser, der udføres. Dette er en væsentlig forudsætning for, at resultaterne af de undersøgelser, der er foretaget i et land, også kan accepteres i andre lande. Selvom der er udarbejdet retningslinier for udførelse af dyreforsøg, kan der dog være vanskeligheder forbundet med at vurdere disse forsøg. De forsøg, der ved fastsættelse af kvalitetskriterier traditionelt har været lagt mest vægt på, er langtidsdyreforsøgene. Som eksempel kan nævnes forsøg, hvor dyrene opdeles i grupper, således at én gruppe ikke får det stof, der skal undersøges (kontrolgruppen), mens dyr i andre grupper (dosisgrupperne) får stoffet i forskellige daglige doser eller koncentrationer i størsteparten af deres liv. Denne forsøgstype kan være specielt tilpasset til at afsløre en mulig kræftfremkaldende virkning, eller til at give oplysninger om en lang række andre effekter som for eksempel skader på forskellige væv og organer. Der er også udviklet forsøgsmodeller, som kan afsløre specifikke effekter, som for eksempel allergi, skader på arveanlæg, fosterskader, skader på forplantningen samt nervebeskadigelser. Det er klart, at jo flere undersøgelser, der foreligger, jo mere dækkende et billede har man af stoffets toksikologiske effekter. 2.3 In-vitro metoderIsolerede celler, væv og organer kan dyrkes i kultur (reagensglasforsøg), således at deres in vivo egenskaber tilnærmelsesvis bibeholdes. Disse in vitro metoder finder især anvendelse i relation til undersøgelser af kemiske stoffers mutagene og genotoksiske egenskaber samt til undersøgelser af stoffernes virkningsmekanismer. I de seneste 10 år er der arbejdet med udvikling af in vitro metoder som alternative metoder til at afsløre effekter, som man hidtil har anvendt dyremodeller til at undersøge, for eksempel effekter som hud- og øjenirritation samt specifikke organskader. Der er igangsat valideringsprogrammer, men ingen af de ovenfor nævnte metoder er endnu tilstrækkeligt validerede med henblik på direkte anvendelse i farlighedsvurderingen af kemiske stoffer. Denne type af undersøgelser kan således ikke alene danne grundlaget for fastsættelse af kvalitetskriterier, men anvendes oftest som støtte til det øvrige datagrundlag fra humane studier og dyreeksperimentelle undersøgelser. 2.4 Struktur-aktivitets relationerFor en del af de kemiske stoffer, som anvendes i dag, findes der hverken humane eller dyreeksperimentelle data til belysning af de toksikologiske effekter. Kvantitative struktur-aktivitets relationer (engelsk: Quantitative Structure Activity Relationships – QSARs) har i en række tilfælde vist sig at kunne anvendes som et alternativ til dyreforsøg med henblik på forudsigelse af toksikologiske egenskaber (MST 2001 [11]). Princippet bag struktur-aktivitets relationer (SAR) er, at stoffer med sammenlignelige strukturer har sammenlignelige egenskaber. Ved anvendelse af (Q)SAR forsøger man således udfra et givent kemisk stofs molekylstruktur og/eller på grundlag af stoffets lighed med andre kendte kemiske stoffer at forudsige en egenskab for det givne stof, for eksempel en toksikologisk egenskab. Når resultatet udtrykkes kvalitativt, benævnes relationen SAR, og når resultatet udtrykkes kvantitativt, benævnes relationen QSAR. (Q)SARs er baseret på en sammenligning af strukturen og fysisk-kemiske egenskaber (deskriptorer – for eksempel logP, form, størrelse, ladning osv.) med målte parametre og/eller end-points for en række stoffer, kaldet træningssættet. Sammenligningen foretages ofte ved hjælp af statistiske værktøjer. Målet er at fastlægge, hvilke deskriptorer, der har afgørende betydning for forudsigelse af et givent end-point og derefter fastlægge en relation mellem disse deskriptorer og det pågældende end-point. Når der er fastlagt en relation mellem struktur og egenskaber, kan denne anvendes til forudsigelse af end-points for andre stoffer, for hvilke deskriptorerne er kendte eller kan estimeres. I reglen foregår arbejdet med udvikling og anvendelse af (Q)SARs under anvendelse af computere. Der er indtil videre udviklet flere forskellige computermodeller til forudsigelse af en række forskellige end-points. Nyere computermodeller består ofte af flere QSARs til forudsigelse af et givent end-point. De senere års validering af computermodellerne har vist, at der kan opnås korrekte forudsigelser for ca. 70 til 85% af de testede stoffer (MST 20011). Forudsigelsernes validitet kan variere for de forskellige kemiske strukturer og typer af effekter. For nærværende er der ikke tilstrækkelig erfaring med at bruge (Q)SAR modeller som erstatning for dyreforsøgsdata til at sådanne modelberegninger kan anvendes som udgangspunkt til fastsættelse af kvalitetskriterier. (Q)SARs anvendes primært med henblik på forudsigelse ved vurdering af stoffer, hvor der ikke findes valide testdata samt i prioriteringsøjemed. 2.5 Indhentning af dataData hentes primært fra internationale og nationale kriteriedokumenter, via litteratursøgning i internationale databaser, samt fra originalartikler. Endvidere anvendes også upublicerede data fra risikovurderingsrapporterne i EU's risikovurderingsprogram (se 1.2) samt i visse tilfælde upublicerede data stillet til rådighed af industrien. Da data således oftest er samlet sammen fra sekundære kilder foretages der i reglen ikke en egentlig toksikologisk bedømmelse af de enkelte studier. Dog søges originalartiklen altid fremskaffet for de(t) studie(r), der ligger til grund for udpegning af de(n) kritiske effekt(er). Det skal understreges, at ikke nødvendigvis alle tilgængelige oplysninger om et givent kemisk stof er relevante i farlighedsvurderingen af det pågældende stof med henblik på fastsættelsen af kvalitetskriterier. 2.5.1 KriteriedokumenterSom eksempler på kriteriedokumenter kan nævnes: Environmental Health Criteria (EHC), monografier udgivet af IPCS De enkelte monografier indeholder en meget detaljeret gennemgang og vurdering af det konkrete stofs toksikologi såvel som økotoksikologi. I de nyeste monografier er der endvidere en opstilling af stoffets dosis-responssammenhænge, fastlæggelse af nul-effektniveau og laveste effektniveau, beregning af tolerabel daglig indtagelse, samt identifikation af datamangler. Der er indtil videre udgivet 228 EHC dokumenter. EHC udgives af International Programme on Chemical Safety (IPCS). IPCS er et joint venture mellem FN's miljøprogram (UNEP), den Internationale Arbejderorganisation (ILO), og Verdenssundhedsorganisation (WHO). Yderligere information kan findes på http://www.who.int/pcs/pcs_pubs.html. WHO Guidelines for drinking-water quality Omfatter korte toksikologiske vurderinger af en lang række stoffer, som kan forekomme i drikkevandet, samt baggrunden for fastsættelse af vejledende grænseværdi for drikkevand. Udgives af WHO. De nyeste vurderinger er samlet i Guidelines for drinking-water quality, Second edition, Volume 2 Health criteria and other supporting information, World Health Organization, Geneva 1996 samt i Guidelines for drinking-water quality, Second edition, Addendum to Volume 2 Health criteria and other supporting information, World Health Organization, Geneva 1998. Yderligere information kan findes på http://www.who.int/water_sanitation_health/Water_quality/drinkwat.htm. WHO Air quality guidelines for Europe Omfatter kortere toksikologiske vurderinger af 35 stoffer og stofgrupper samt baggrunden for fastsættelse af vejledende grænseværdier i luft. Udgives af WHO. De nyeste vurderinger er samlet i Air quality guidelines for Europe, Second Edition, WHO Regional Publications, European Series No. 91, World Health Organization 2000. Yderligere information kan findes på http://www.who.int/peh/air/Airqualitygd.htm. Monografier fra International Agency for Research on Cancer (IARC) Omhandler primært en vurdering af det konkrete stofs kræftfremkaldende egenskaber samt baggrunden for indplacering af stoffet i en given gruppe, der relaterer til evidensen for kræftfremkaldende effekt (se også 5.1.3). Indeholder også en gennemgang af stoffets øvrige toksikologiske egenskaber. Udgives af International Agency for Recearch on Cancer. Yderligere information kan findes på http://www.iarc.fr/. Monografier fra Joint FAO/WHO Expert Committee on Food Additives (JECFA) Omfatter en detaljeret gennemgang og vurdering af det konkrete stofs toksikologiske egenskaber. Publiceres i serien Safety Evaluations of Certain Food Additives and Contaminants som udarbejdes af the Joint FAO/WHO Expert Committee on Food Additives og udgives af IPCS. Yderligere information kan findes på http://www.who.int/pcs/jecfa/jecfa.htm. Monografier fra Joint FAO/WHO Meeting on Pesticide Residues (JMPR) Omfatter en detaljeret gennemgang og vurdering af det konkrete pesticids toksikologiske egenskaber. Publiceres i serien Pesticide residues in Food som udarbejdes af the Joint FAO/WHO Meeting on Pesticide Residues og udgives af IPCS. Yderligere information kan findes på http://www.who.int/pcs/jmpr/jmpr.htm. Toxicological Profiles fra Agency for Toxic Substances and Disease Registry (ATSDR) Indeholder en meget detaljeret gennemgang og vurdering af et konkret stofs toksikologiske egenskaber. Udgives af U.S. Department of Health & Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry. Yderligere information kan findes på http://www.atsdr.cdc.gov/. American Conference on Governmental Industrial Hygienists (ACGIH) Omfatter en kort gennemgang af et konkret stofs toksikologiske egenskaber samt baggrunden for fastsættelse af en grænseværdi i arbejdsmiljøet. Udgives af American Conference on Governmental Industrial Hygienists. Yderligere information kan findes på http://www.acgih.org/. Arbete och Hälse Omfatter en detaljeret gennemgang af et konkret stofs toksikologiske egenskaber primært rettet mod fastsættelse af en grænseværdi i arbejdsmiljøet. Udarbejdes af Nordic Expert Group og udgives af Arbetarskyddsverket i Sverige. Yderligere information kan findes på http://www.niwl.se/ah/. Beatergremium für umweltrelevante Altstoffe (BUA) Omfatter en meget detaljeret gennemgang af et konkret stofs toksikologiske såvel som økotoksikologiske egenskaber. Udarbejdes og udgives af Beatergremium für umweltrelevante Altstoffe der Geschellschaft Deutscher Chemiker. Yderligere information kan findes på http://www.gdch.de/projekte/bua.htm. Tekniske rapporter fra European Centre for Ecotoxicology and Toxicology of Chemicals (ECETOC) Omfatter enten gennemgang af stoffer og stofgruppers toksikologiske egenskaber eller gennemgang af konkrete toksikologiske problemstillinger. Udarbejdes og udgives af European Centre for Ecotoxicology and Toxicology of Chemicals. Yderligere information findes på http://www.ecetoc.org/entry.htm. Risikovurderingsrapporter for eksisterende kemiske stoffer i EU Omfatter en meget detaljeret gennemgang af et konkret stofs egenskaber i relation til både sundhed og miljø, se 1.2.1. Publicerede rapporter kan findes på http://www.ecb.jrc.it/existing-chemicals/. 2.5.2 DatabaserSom eksempler på databaser kan nævnes: IUCLID International Uniform Chemical Information Database (IUCLID), som udgives af European Chemical Bureau (ECB). Indeholder toksikologiske og økotoksikologiske data for 2604 kemiske stoffer, som produceres og/eller importeres i mængder på mere end 1000 tons/år/producent eller importør. Data er indrapporteret af Industrien og ikke evalueret af myndighederne. Yderligere information kan findes på http://ecb.ei.jrc.it/Iuclid/. IRIS Integrated Risk Information System (IRIS), som udgives af US-EPA. Indeholder toksikologiske data for en lang række stoffer samt baggrund for estimering af en tolerabel daglig indtagelse eller koncentration. Yderligere information kan findes på http://www.epa.gov/iris/. HSDB Hazardous Substances Data Bank (HSDB), som udgives af the National Library of Medicine i USA. Indeholder data vedrørende toksikologi og miljøet for godt 4500 stoffer. Yderligere information kan findes på http://toxnet.nlm.nih.gov/. TOXLINE I TOXLINE (TOXicology information onLINE) stammer informationerne fra en række forskellige kilder som f.eks. National Library of Medicine, Chemical Abstracts Services, BIOSIS, MEDLINE, International Pharmaceutical Abstracts, og Kemikalieinspektionen i Sverige. Udgives af the National Library of Medicine i USA. Yderligere information kan findes på http://toxnet.nlm.nih.gov/. MEDLINE Indeholder referencer og abstracts fra mere end 4300 biomedicinske tidsskrifter. Udgives af the National Library of Medicine i USA. Yderligere information kan findes på http://toxnet.nlm.nih.gov/. Fodnoter[11] Report on the advisory list for selfclassification of dangerous substances. Environmental Project No. 636 2001. Miljøstyrelsen, Miljø- og Energiministeriet. http://www.mst.dk/udgiv/publications/2001/87-7944-694-9/html/ 3 Farlighedsvurdering og karakterisering3.1 Dosis-effekt og dosis-respons sammenhænge Formålet med en farlighedsvurdering og farlighedskarakterisering af et givent stof er, på baggrund af en kritisk vurdering af de tilgængelige data vedrørende stoffets toksikologiske effekter og virkningsmekanismer, at vurdere, hvorvidt eksponering for stoffet vil kunne udløse sundhedsskadelige effekter hos mennesker. Ved fastsættelse af kvalitetskriterier tages der altid udgangspunkt i den eksisterende viden, primært viden samlet sammen i kriteriedokumenter, monografier og reviews, hvorfor der i reglen ikke foretages en egentlig toksikologisk bedømmelse af de enkelte studier. Dog søges originallitteraturen altid fremskaffet for de(t) studie(r), der ligger til grund for udpegning af de(n) kritiske effekt(er). Hvor den eksisterende viden er mangelfuld, kompenseres der herfor ved anvendelse af en usikkerhedsfaktor, dette er nærmere beskrevet i 4.4.3. 3.1 Dosis-effekt og dosis-respons sammenhængeFarlighedsvurderingen foretages med udgangspunkt i alle de indhentede data vedrørende stoffets sundhedsskadelige effekter i mennesker og dyr, det vil sige akutte effekter, irritative/ætsende og sensibiliserende egenskaber, effekter observeret i længerevarende undersøgelser med gentagen administration af stoffet, mutagene/genotoksiske og kræftfremkaldende egenskaber, samt påvirkning af fertilitet og fosterudvikling. Endvidere lægges der vægt på de toksikokinetiske data, det vil sige stoffets skæbne i organismen (optagelse, fordeling, metabolisme og udskillelse), samt data vedrørende stoffets virkningsmekanisme(r). Eksponering for et givent stof kan medføre forskellige effekter varierende fra lette gener til dødeligt forløbende forgiftninger. Forløbet er ofte relateret til koncentrationen eller dosis af stoffet. Ved lave doser kan stoffet for eksempel give anledning til forbigående gener, mens der ved højere doser kan optræde alvorlige skader på væv og organer, i værste fald med dødelig udgang. Et enkelt studie (epidemiologisk såvel som dyreeksperimentelt) kan således ofte afsløre forskellige typer af effekter afhængigt af de forskellige koncentrationer eller doser, som mennesker eller dyr har været udsat for i det pågældende studie. Som en følge heraf kan forskellige dosis-effektsammenhænge forventes, hvis stoffet forårsager flere forskellige effekter. Som et led i farlighedsvurderingen foretages der således for hvert enkelt studie en vurdering af dosis-effektsammenhænge, det vil sige en karakterisering af sammenhængen mellem dosis (eller koncentration) af stoffet og de observerede effekter. Derudover foretages også en vurdering af dosis-responssammenhænge, det vil sige en karakterisering af sammenhængen mellem dosis (eller koncentration) af stoffet og forekomst/hyppighed (engelsk: incidence) eller graden/alvorligheden (engelsk: severity) af de observerede effekter. Ved vurdering af alvorligheden af de observerede effekter skelnes der mellem effekter som sådan og skadelige (engelsk: adverse) effekter. Generelt betragtes effekter som følge af eksponering for et givent stof som skadelige, når der er tale om væsentlige forandringer i morfologi, fysiologi, funktion, vækst, udvikling, og/eller levetid hos eksponerede individer, og når forekomsten hos denne gruppe er statistisk signifikant højere end i kontrolgruppen. Sondringen mellem effekt og skadelig effekt kan virke akademisk, men er i mange tilfælde af væsentlig betydning, blandt andet i forbindelse med vurdering af nul-effektniveau og laveste effektniveau i et givent studie (se 3.2), med udpegning af den kritiske effekt (se 3.4), og for størrelsen af usikkerhedsfaktor UFIII der anvendes ved fastsættelse af kvalitetskriterier for luft, jord og drikkevand med henblik på at tage højde for kvalitet og relevans af det tilgængelige datagrundlag (se 4.4.3). Sondringen består i en vurdering af, om de observerede effekter kan tolkes som værende af en egentlig skadelig karakter, eller om der eventuelt er tale om normale uspecifikke reaktioner, som blot er en følge af individernes reaktion på ændringer i ydre omstændigheder (homeostatiske mekanismer). Som eksempel herpå kan nævnes, at en formindsket vægtstigning hos forsøgsdyr kan være en reaktion på foderets smag eller konsistens og dermed ikke nødvendigvis en direkte reaktion på teststoffets toksikologiske effekter. Et andet eksempel er, at ændringer i en enzymaktivitet eller i en klinisk-kemisk parameter, hvilket er lig med en effekt, ikke nødvendigvis betragtes som skadelige, med mindre ændringerne i sig selv giver anledning til sundhedsmæssige påvirkninger af individerne eller er indikatorer herpå. Andre eksempler er misfarvning af et organ eller væv, hvor der ikke samtidig er observeret histologiske eller biokemiske effekter, eller induktion af enzymer involveret i metabolismen af et givent stof. Sondringen mellem effekt og skadelig effekt afhænger selvsagt af de data, der er tilgængelige for vurdering, og sondringen kan således være behæftet med en vis usikkerhed. For eksempel kan der i et givent studie ses en effekt (f.eks. morfologiske forandringer i leveren) hos nogle forsøgsdyr, uden at den pågældende effekt er observeret på et niveau, der er statistisk signifikant forskelligt fra det, der ses i kontrolgruppen. I dette tilfælde vil effekten ikke nødvendigvis tolkes som værende en skadelig effekt som følge af eksponeringen for teststoffet, men kan muligvis tilskrives tilfældige biologiske variationer. Men hvis der for eksempel havde været flere dyr i den pågældende dosisgruppe, så ville effekten måske kunne blive observeret som værende statistisk signifikant forskellig fra observationerne i kontrolgruppen, og i dette tilfælde vil effekten tolkes som værende en skadelig effekt. Der kan også være tale om atypiske effekter, som for eksempel usædvanlige fosterskader der optræder med en hyppighed, der er mindre end det, der i studiet er statistisk signifikant. I sådanne tilfælde vil effekten kunne blive vurderet som skadelig, hvis de få observationer, der ses, ikke med rimelighed kan tilskrives tilfældige biologiske variationer. Der ligger således den begrænsning i farlighedsvurderingen, at man ved gennemførelse af et studie så vidt muligt skal sikre sig at kunne være i stand til at observere en given effekt på et statistisk signifikant niveau med det anvendte antal individer ved de valgte koncentrationer eller doser. Dette kan specielt være et problem ved atypiske effekter, hvor der i givet fald skulle anvendes et uforholdsmæssigt højt antal individer per gruppe eller ved lave koncentrationer/doser, hvor den individuelle følsomhed mellem individer i en udvalgt gruppe kan 'overskygge' en eventuel effekt. Ved farlighedsvurderingen skelnes der endvidere mellem systemiske og lokale effekter. Systemiske effekter optræder i organismen efter optagelse af stoffet via luftvejene, mave-tarmkanalen, og/eller huden. Ved systemiske effekter anses det sædvanligvis at være den samlede dosis af stoffet og sjældent stoffets koncentration i de(t) pågældende medie(r), der er relevant ved fastsættelse af kvalitetskriterier i luft, jord og drikkevand. Under lokale effekter henregnes effekter, der optræder lokalt i luftvejene eller i mave-tarmkanalen samt direkte effekter på hud og øjne. I relation til lokale effekter anses sædvanligvis stoffets koncentration og sjældent den samlede dosis for relevant ved fastsættelse af kvalitetskriterier. I velgennemførte forsøg er doserne (koncentrationerne) valgt således, at der for en given (skadelig) effekt er mindst én dosis med effekt og mindst én dosis, hvor der ikke er effekt. Dosis-responssammenhængen (f.eks. antal dyr hvori den pågældende effekt er observeret eller den procentvise stigning i organvægt i forhold til kontrolgruppens organvægt) kan optegnes i et diagram med logaritmen til dosis ud ad x-aksen og respons i procent op ad y-aksen. Kurveforløbet kan være lineært, kurvet eller U-formet. For de fleste kemiske stoffer forløber dosis-responssammenhængen som en S-formet kurve (figur 3.1.1). Ved de laveste doser er der således ikke observeret effekt, men rent statistisk kan det ikke udelukkes, at en svag effekt måske ville kunne være blevet observeret, hvis der havde været flere dyr i gruppen. Da den S-formede dosis-responskurve gælder for de fleste kemiske stoffers effekter, er denne kurvetype valgt ved illustrationerne af dosis-responssammenhænge i de efterfølgende figurer. For nogle stoffer forløber dosis-responssammenhængen imidlertid anderledes. For eksempel ses for essentielle metaller, at hvis individet får for lidt af et givent essentielt metal, så kan der opstå mangelsymptomer. Omvendt kan der opstå skadelige påvirkninger, hvis individet får for meget af det pågældende essentielle metal. Således har dosis-responssammenhængen for essentielle metallers effekter et U-formet forløb, hvor den højre del af kurven, det vil sige den del, der repræsenterer de toksiske effekter, stadig har det typiske S-formede forløb, som generelt ses for kemiske stoffers effekter. Også i relation til begrebet hormesis ses et U-formet eller omvendt U-formet kurveforløb (se 3.3). Figur 3.1.1 S-formet dosis-respons kurve
Som følge af at et enkelt studie i princippet kan afsløre forskellige typer af effekter for et givent stof, og at forskellige dosis-responssammenhænge kan forventes for de forskellige effekter, kan der i princippet således optegnes en dosis-responskurve for hver af stoffets forskellige effekter, se figur 3.1.2. For nogle typer af effekter skal der ske en væsentlig øgning af dosis, før der ses et respons, og for disse typer af effekter vil den S-formede dosis-responskurve være forholdsvis flad, kurve A i figur 3.1.2. For andre typer af effekter skal der kun en meget lille øgning af dosis til at fremkalde et markant respons, og for disse typer af effekter vil den S-formede dosis-responskurve være stejl, kurve B og C i figur 3.1.2. Figur 3.1.2 Dosis-respons kurver
Hvorvidt den S-formede dosis-responskurve for en given effekt er stejl (figur 3.1.2 kurve B og C) eller flad (figur 3.1.2 kurve A) siger ikke umiddelbart noget om den pågældende effekts alvorlighed. Med henblik på vurdering heraf skal der yderligere inddrages en vurdering af ved hvilke dosisniveauer, de enkelte effekter optræder, det vil sige dosis-responskurvens placering i diagrammet i forhold til y-aksen. Jo kortere afstand fra kurven til y-aksen jo lavere koncentrationer (doser) skal der til for at udløse den givne effekt. Et eksempel til illustration af dette forhold er en sammenligning af kurverne B og C i figur 3.1.2. Kurverne B og C har samme stejlhed, og dermed har de respektive effekter B og C samme relative dosis-responssammenhæng. Men da effekt B optræder ved lavere koncentrationer (doser) end effekt C, så kunne effekt B således betragtes som værende mere kritisk end effekt C. Dette vil også ofte, men langt fra altid være tilfældet, idet typen/alvorligheden af effekt også spiller en rolle i vurderingen af den kritiske effekt (se 3.4). Som eksempel på det sidstnævnte kan nævnes, at hvis effekt C er udvikling af tumorer i leveren og effekt B er luftvejsirritation, så vil effekt C selvsagt vurderes som værende en langt alvorligere effekt end effekt B. Det skal dog understreges, at ved fastsættelse af et luftkvalitetskriterium ville det ikke nødvendigvis være effekt C, der, på trods af alvorligheden, ville blive vurderet som værende den kritiske effekt i relation til fastsættelse af et luftkvalitetskriterium og dermed danne grundlaget for herfor. Et andet eksempel er en sammenligning af kurverne A og C i figur 3.1.2. Kurverne A og C har forskellig stejlhed og dermed har de respektive effekter A og C forskellige relative dosis-responssammenhæng. Umiddelbart kunne man fristes til at betragte effekt C som værende mere kritisk end effekt A på grund af en stejlere dosis-responskurve. Dette vil også kunne være tilfældet ved koncentrationer (doser), der er højere end den koncentration (dosis), der svarer til skæringspunktet mellem kurve A og C. Men ved lavere koncentrationer (doser) end den koncentration (dosis), der svarer til skæringspunktet mellem kurve A og C, vil effekt A kunne betragtes som værende mere kritisk end effekt C. Også i dette tilfælde kan der være andre forhold, der gør sig gældende ved en vurdering af de enkelte effekters alvorlighed. 3.2 Fastlæggelse af nul-effektniveau og laveste effektniveauI vurderingen af det enkelte studie indgår også en udpegning af et nul-effektniveau og det laveste effektniveau for en given effekttype samt for stoffet som sådan i det pågældende studie for så vidt, at det overhovedet er muligt på baggrund af data i studiet. For hver enkelt effekt findes teoretisk set et sandt nul-effektniveau (NEL) og laveste effektniveau (LEL), og ideelt set burde det være disse niveauer, der blev fastlagt for en given effekt. For langt de fleste typer af effekter findes der en tærskel, det vil sige en grænse (koncentration/dosis), hvorunder der ikke ses effekt (se 4.2). For nogle typer af effekter antages det, at der ikke findes en tærskel, for eksempel genotoksiske carcinogener, hvor den tilgrundliggende mekanisme for udvikling af tumorer er en beskadigelse af arvematerialet. Denne problemstilling er uddybet i 3.6. Som beskrevet i 3.1 kan dosis-responssammenhængen for mange typer af effekter optegnes som en S-formet kurve i et diagram med dosis (koncentration) ud ad x-aksen og respons op ad y-aksen. Nul-effektniveauet (NEL) for en given effekt er således skæringspunktet mellem den S-formede kurve og x-aksen (figur 3.2.1). Figur 3.2.1 Dosis-respons kurve med identifikation af nul-effektniveau
Da det sande nul-effektniveau (NEL) og laveste effektniveau (LEL) er teoretiske værdier, kan det således i praksis ikke lade sig gøre at fastlægge disse niveauer. Det næstbedste ville så være at estimere de niveauer, der ligger tættest på det sande nul-effektniveau (NEL) eller laveste effektniveau (LEL). Dette er imidlertid sjældent muligt i praksis, da en estimering af disse niveauer med en vis grad af sikkerhed i givet fald ville kræve et meget stort antal studier af den pågældende effekt samt et særdeles stort antal dyr og koncentrations-(dosis)-niveauer i det enkelte studie. I praksis fastlægges i et givent studie det observerbare nul-effektniveau (NO(A)EL) og det laveste observerbare effektniveau (LO(A)EL) ud fra de konkrete dosisniveauer, der er anvendt i det pågældende studie. Principperne herfor er beskrevet i det efterfølgende afsnit (3.2.1). Det skal dog bemærkes, at en alternativ metode til fastlæggelse af nul-effektniveau, 'benchmark' metoden (se 3.2.2), i visse tilfælde kan give estimater ('benchmark dose'), der ligger tættere på NEL og LEL. Imidlertid giver det klassiske guideline-forsøgsdesign (se 2.2.1) med 3 doserede grupper og en vehikelkontrolgruppe ikke ideelle data til udarbejdelse af gode dosis-responskurver og dermed estimering af 'benchmark dose'. Hertil er studier med 5-10 dosisgrupper nødvendige. 3.2.1 NO(A)EL og LO(A)ELI konsekvens af sondringen mellem effekt og skadelig effekt (se 3.1) skelnes således også mellem to typer af observerbart nul-effektniveau, NOEL og NOAEL, samt to typer af det laveste observerbare effektniveau, LOEL og LOAEL. Såfremt dosisniveauerne er angivet som koncentrationer anvendes ofte betegnelsen 'koncentration' i stedet for 'niveau', det vil sige NOEC og NOAEC samt LOEC og LOAEC. Termerne defineres som følger: NOEL/C (engelsk: No Observed Effect Level / Concentration) er den højeste dosis / koncentration af et stof, som ikke medfører ændringer i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer i forhold til en sammenlignelig kontrolgruppe. NOAEL/C (engelsk: No Observed Adverse Effect Level / Concentration) er den højeste dosis / koncentration af et stof, som ikke medfører påviselige skadelige ændringer i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer. Der kan ved NOAEL/C ses ændringer i ovennævnte parametre, som ikke vurderes at være af skadelig karakter. LOEL/C (engelsk: Lowest Observed Effect Level / Concentration) er den laveste dosis / koncentration af et stof, som medfører en ændring i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer, og som kan påvises i forhold til en sammenlignelig kontrolgruppe. De ændringer, der ses ved LOEL/C, vurderes ikke at være af skadelig karakter. LOAEL/C (engelsk: Lowest Observed Adverse Effect Level / Concentration) er den laveste dosis / koncentration af et stof, som medfører en skadelig ændring i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer og som kan påvises i forhold til en sammenlignelig kontrolgruppe. Det skal understreges at begreberne NO(A)EL og LO(A)EL anvendes med varierende betydning i den videnskabelige litteratur, og at det ikke altid kan afgøres, hvad der menes i de enkelte tilfælde, hvorfor man ofte er nødt til at se nærmere på, om der i det enkelte forsøg er forsøgt en skelnen mellem disse begreber. Med henblik på fastsættelse af kvalitetskriterier for luft, jord og drikkevand tages der oftest udgangspunkt i NOAEL/C eller LOAEL/C, hvorfor disse begreber i det følgende generelt vil være synonymt med observerbart nul-effektniveau henholdsvis det laveste observerbare effektniveau, med mindre der specifikt er tale om NOEL/C eller LOEL/C. Endvidere vil begreberne NOAEL og LOAEL blive anvendt, med mindre der specifikt er tale om koncentrationer. I velgennemførte forsøg er doserne (koncentrationerne) valgt således, at der er mindst én dosis, der resulterer i en observerbar effekt og mindst én dosis, hvor denne effekt ikke observeres. Som beskrevet i 3.1 kan dosis-responssammenhængen for mange typer af effekter optegnes som en S-formet kurve i et diagram med dosis (koncentration) ud ad x-aksen og respons op ad y-aksen. NOAEL og LOAEL for en given effekt vil således ligge et eller andet sted på den S-formede dosis-responskurve, eksempelvis som markeret på kurve A på figur 3.2.2. I praksis er der i det enkelte studie kun få dosisniveauer, og dosis-responskurven vil for eksempel se ud som kurve B på figur 3.2.2. NOAEL for en given effekt vil således være den af de valgte dosisniveauer i studiet, der ikke har medført effekt, og LOAEL i konsekvens heraf det næste dosisniveau i studiet. Dette er illustreret på kurve B på figur 3.2.2 med NOAEL som det nederste punkt på kurven og LOAEL som det næstnederste punkt. Figur 3.2.2 Dosis-respons kurve med identifikation af nul-effektniveauer og laveste effekt niveauer
Som nævnt tidligere (3.1) foretages for det enkelte studie en vurdering af dosis-effekt og dosis-responssammenhænge, ligesom der sondres mellem effekt og skadelig effekt samt mellem systemiske og lokale effekter. Såfremt et enkelt studie afslører forskellige typer af effekter for et givent stof, kan der i princippet optegnes en dosis-responskurve for hver af stoffets forskellige effekter, se figur 3.1.2. Ved fastlæggelse af NOAEL og LOAEL for stoffet som sådan i det pågældende studie skal dernæst foretages en afvejning af de enkelte effekter i forhold til de fastlagte NOAELs og LOAELs. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand vurderes effekter generelt som værende mere kritiske desto lavere doser (koncentrationer), de optræder ved, således at NOAEL for stoffet som sådan i et givent studie ofte, men ikke nødvendigvis altid, vil være identisk med det laveste af de fastlagte NOAELs for enkelteffekter. For de studier, hvor der kan sondres mellem effekter og skadelige effekter, vil NOAEL ikke nødvendigvis være den højeste dosis (koncentration) i studiet, hvor der ikke observeres effekter. Dette forhold er illustreret i figur 3.2.3, hvor kurve B eksempelvis er dosis-responssammenhængen for stigning i aktiviteten af et uspecifikt leverenzym, det vil sige en effekt, og hvor kurve C eksempelvis er dosis-responssammenhængen for irreversible testikelskader, det vil sige en skadelig effekt. I dette tilfælde vil NOAEL for stoffet som sådan i det pågældende studie ligge et eller andet sted på kurve C, mens NOEL vil være at finde på kurve B. Et andet eksempel kunne være, at kurve A er dosis-responssammenhængen for øget (ikke statistisk signifikant) relativ organvægt, og kurve C som ovenfor. I dette tilfælde vil NOAEL for stoffet som sådan i det pågældende studie ligge et eller andet sted på kurve C nedenfor skæringspunktet med kurve A. Figur 3.2.3 Identifikation af nul-effektniveau, sondring mellem effekt og skadelig effekt
En lang række stoffer kan give anledning til både systemiske og lokale effekter. Eksempelvis medfører indånding af mange organiske opløsningsmidler en påvirkning af centralnervesystemet, hvilket er en systemisk effekt, men kan også give anledning til irritation eller skader i luftvejene, hvilket er lokale effekter. I tilfælde af at det i det enkelte studie ikke kan afklares, hvorvidt de systemiske eller de lokale effekter vurderes som værende mest kritiske, fastlægges NOAEL og LOAEL således for begge typer af effekt. Dette forhold er illustreret i figur 3.2.4, hvor kurve A kunne være dosis-responssammenhængen for en systemisk effekt, for eksempel nyreskade, og kurve B dosis-responssammenhængen for en lokal effekt, for eksempel luftvejsirritation. I praksis er det imidlertid ofte således, at datagrundlaget i det enkelte studie ikke er tilstrækkeligt til at fastlægge både NOAEL og LOAEL for stoffet som sådan i det pågældende studie efter de ovenfor beskrevne principper. I konsekvens heraf vil NOAEL således ofte blive fastlagt som den højeste dosis (koncentration) i studiet, hvor der ikke er observeret effekter, og LOAEL som den laveste dosis (koncentration) i studiet, hvor der er observeret effekter. Figur 3.2.4 Identifikation af nul-effektniveau for systemisk effekt og lokal effekt
3.2.2 Andre metoder til fastlæggelse af nul-effektniveauSom beskrevet ovenfor og illustreret på kurve B i figur 3.2.2 vil NOAEL ofte blive fastlagt som den højeste dosis (koncentration) i studiet, hvor der ikke er observeret effekter, og LOAEL som den laveste dosis (koncentration) i studiet hvor der er observeret effekter. Kurveforløbet for dosis-responssammenhængen og den dertil svarende tærskelværdi kan også beregnes ved hjælp af forskellige matematiske modeller (kurvefitning). Der kan anvendes forskellige statistiske modeller til at beskrive sammenhængen mellem dosis og respons og forskellige fordelinger til at beskrive tilfældige udfald. Fælles for disse modeller og fordelinger er, at de afhænger af de indgående parametre, og kurveforløbene må derfor tilpasses efter de aktuelle måledata. Med henblik på fastlæggelse af et udgangspunkt (engelsk: point of departure) for beregning af den tolerable daglige indtagelse (TDI, se kapitel 4) ville det, ud fra statistiske synspunkter, måske være bedre at tage udgangspunkt i den nedre 95% konfidensgrænse for den dosis (koncentration), som medfører effekt hos 5 eller 10% af dyrene i stedet for at tage udgangspunkt i NOAEL eller LOAEL, som beskrevet i det foregående afsnit. Statistisk set vil denne metode være bedre egnet, hvis der kun indgår et lille antal dyr i studiet, og der indbygges derfor automatisk en slags usikkerhedsfaktor. På den anden side er der nogle andre forudsætninger ved valget af beregningsmetode, som måske kan trække i den anden retning. Den hyppigst anvendte alternative metode til fastlæggelse af et udgangspunkt for beregning af TDI er den såkaldte benchmark-metode. Benchmark-metoden indebærer, at en dosis-responskurve beregnes
ud fra det fulde datasæt for hver effektparameter, og der vedtages en ”kritisk effektstørrelse”. På basis af den beregnede kurve defineres benchmark dose (BMD) som den nedre konfidensgrænse for den
dosis, der fører til den givne kritiske effektstørrelse, det vil sige den incidens (for eksempel 5 eller 10%), der estimeres for en given effekt. Imidlertid giver det klassiske guideline-forsøgsdesign med 3 doserede grupper og en vehikelkontrolgruppe ikke ideelle data til udarbejdelse af gode dosis-responskurver. Hertil er studier med 5-10 dosisgrupper nødvendige. Såfremt det totale antal dyr per forsøg ikke øges, vil et øget antal grupper medføre en formindsket gruppestørrelse, hvorved det ikke længere vil være muligt at fastlægge et NOAEL på sædvanlig vis (se 3.2.1). Det kan således ikke i praksis lade sig gøre at lave et forsøgsdesign, som egner sig til begge fremgangsmåder, eller at bruge de to fremgangsmåder på det samme forsøg. Nødvendig videreudvikling af benchmark metoden omfatter forbedret forsøgsdesign, international enighed om størrelsen af kritisk effekt, samt udvikling af specifikke dosis-respons analysemetoder for forskellige typer af forsøgsdata. Som nævnt fastlægges benchmark dose ved modellering af data fra et givent studie. En hvilken som helst model, som passer til de observerede data, vil give et rimeligt estimat af benchmark dose, og valget af model er tilsyneladende ikke kritisk, idet den estimerede værdi for benchmark dose ligger inden for det observerede dosisinterval. Det skal imidlertid understreges, at benchmark-metoden ikke kan anvendes hvis der i det enkelte studie ikke er et tilstrækkeligt antal dosisniveauer, hvor der observeres effekter. Da dette er tilfældet i de fleste dyreeksperimentelle studier, kan benchmark metoden ikke umiddelbart anvendes som en alternativ metode til fastlæggelse af udgangspunkt for beregning af TDI i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand. Derfor anbefales den traditionelle anvendelse af NOAEL eller LOAEL (se 3.2.1) fortsat som udgangspunkt for beregning af TDI med henblik på fastsættelse af kvalitetskriterier for luft, jord og drikkevand. 3.3 HormesisFænomenet hormesis inden for toksikologien har en lang og kontroversiel historie og går helt tilbage til slutningen af 1800 tallet, hvor det blev beskrevet første gang. Hormesis betegner et fænomen, hvor der ved lave doser ses en stimulation og ved høje doser en inhibition, hvilket giver sig udtryk ved en U-formet eller omvendt U-formet dosis-responssammenhæng. (Calabrese & Baldwin 2001a,b, Calabrese 2001). Der er fremsat en hypotese om, at hormesis repræsenterer en overkompensation mod en påvirkning af homeostasen fremkaldt af for eksempel en mild form for stress (Calabrese 2001). Denne hypotese er ifølge Calabrese (2001) i fuld overensstemmelse med de mange rapporter, der i tidens løb er publiceret, idet det beskedne stimulerende respons observeret efter udsættelse for lave doser af nonessentielle toksiske stoffer (f.eks. cadmium, bly, kviksølv, phenol, bestråling osv.) snarere skyldes en overkompensation mod en påvirkning af homeostasen end en direkte stimulerende effekt af det pågældende stof. Hormesis kendetegnes således ved 1) påvirkning af homeostasen, 2) beskeden overkompensation, 3) reetablering af homeostasen og, 4) processens adaptive natur (Calabrese & Baldwin 2001b). Som nævnt er der publiceret mange undersøgelser, der understøtter, at fænomenet hormesis eksisterer. Men i den toksikologiske litteratur er der kun lidt information om, hvor ofte man kan forvente at observere hormesis. Med henblik på at undersøge, hvor mange studier i den publicerede toksikologiske litteratur der opfyldte nogle specifikke kriterier for evidens af hormesis, udarbejdede Calabrese & Baldwin (2001a) en database under anvendelse af nogle meget strikte 'a priori entry' kriterier (veldefineret NOAEL, 2 doser under NOAEL, og det målte end-point kan udvise både stimulerende og inhiberende respons) samt specifikke evalueringskriterier for evidens af hormesis. Af de i alt 20285 lokaliserede artikler, indeholdt 195 artikler (1%) 668 dosis-responssammenhænge, som opfyldte 'a priori entry' kriterierne. Ved anvendelse af evalueringskriterierne viste det sig, at 245 dosis-responssammenhænge (37% af 668) fra 86 artikler (0,4% af de i alt 20285 lokaliserede artikler) opfyldte kriterierne for evidens af hormesis. I alt 73 forskellige stoffer og blandinger fra en bred vifte af kemiske stofklasser var repræsenteret. Ifølge forfatterne viste resultaterne af analysen således, at når forsøgsdesignet opfylder nogle givne strikte 'a priori entry' kriterier, så er hormesis hyppigt forekommende og bredt repræsenteret afhængig af stof, model og end-point. Det synes således, at hormesis er et ret udbredt fænomen. Med henblik på at vurdere hvorvidt hormesis er et generelt biologisk fænomen uafhængigt af stressfaktorer i miljøet, biologisk end-point, og eksperimentelt modelsystem har Calabrese & Baldwin (2001b) en analyse. I denne analyse indgik 1) den ovenfor beskrevne undersøgelse, 2) en vurdering af 17 meget store studier, hvor hvert studie omfattede data for en række stoffer testet i det samme eksperimentelle modelsystem af den samme undersøgelsesgruppe, 3) en vurdering af farmakologiske data for 24 receptorsystemer for hvilke det bifasiske dosis-responssammenhæng karakteristisk for hormesis er blevet klarlagt, og 4) en vurdering af en database med 1600 dosis-responssammenhænge som er i overensstemmelse med hypotesen for hormesis. Ifølge forfatterne viste resultaterne af denne analyse, at hormetiske effekter repræsenterer evolutionært baserede adaptive responser på miljømæssigt inducerede forstyrrelser i homeostasen. På det nuværende grundlag er det vanskeligt at vurdere, hvilken betydning fænomenet hormesis kan have i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand. Ved fastsættelse af kvalitetskriterierne tages der for et givent stof udgangspunkt i det observerede nul-effektniveau (NO(A)EL) eller det laveste observerede effektniveau (LO(A)EL) for de(n) kritiske effekt(er) (se 3.4). Det vil sige, at kvalitetskriterierne baseres på effekter, som vurderes som værende af en egentlig skadelig karakter og ikke i uspecifikke reaktioner, som blot er en følge af individernes reaktion på ændringer i ydre omstændigheder (homeostatiske mekanismer). På baggrund heraf og sammenholdt med at data indikerer, at hormesis snarere skyldes en overkompensation mod en påvirkning af homeostasen end en direkte stimulerende effekt af det pågældende stof, vurderes det således, at hormetiske effekter umiddelbart er irrelevante i relation fastsættelse af kvalitetskriterier for luft, jord og drikkevand. Det skal endvidere bemærkes, at de milde stresspåvirkninger, som kendetegner hormesis, af nogle vurderes at have en positiv effekt på sundheden og levetiden, idet den øgede forsvarsreaktion i cellen forbedrer livskvaliteten ved at fjerne skader og gøre cellen mere aktiv og effektiv (Rattan 2001). 3.4 Kritisk effektSom det sidste led i farlighedsvurderingen af et givent stof i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand vurderes hvilke(n) effekt(er), der anses for at være mest væsentlig(e) i relation til fastsættelsen af det pågældende kvalitetskriterie, det vil sige en identifikation af de(n) kritiske effekt(er). I de indledende trin i farlighedsvurderingen for et givent stof er der foretaget en nøje vurdering af de enkelte studier, principperne herfor er beskrevet i 3.1. Endvidere er der forsøgt fastlagt et NOAEL og et LOAEL for stoffet som sådan i de enkelte studier, principperne herfor er beskrevet i 3.2. Ved identifikation af de(n) kritiske effekt(er) sammenholdes og vurderes alle de informationer, der er samlet sammen under vurderingerne af alle de enkelte studier. Det vurderes, hvor alvorlige de pågældende effekter er og ved hvilke doser (koncentrationer), de pågældende effekter optræder, ligesom der sondres mellem effekter og skadelige effekter, mellem systemiske og lokale effekter, samt hvorvidt effekter observeret hos forsøgsdyr er relevante for mennesker. For eksempel er relevansen for mennesker af visse forsøgsdyrsspecifikke cancerformer omdiskuteret. Som eksempler kan nævnes mononukleære celleleukæmier, specielle nyretumorer som kun ses hos hanrotter, levertumorer hos gnavere som er en følge af peroxisomproliferation, Leydig-celle tumorer, visse tumorer i thyroidea, samt tumorer i formaven hos gnavere. Den for tiden alment accepterede vurdering af relevansen for mennesker af disse cancerformer er beskrevet i 3.7. Alle disse forhold må naturligvis inddrages i vurderingen af de(n) kritiske effekt(er) samt ved fastlæggelse af NOAEL for de(n) kritiske effekt(er). Principperne for den samlende vurdering af alle stoffets effekter med henblik på identifikation af de(n) kritiske effekt(er) er de samme, som beskrevet for vurderingerne i det enkelte studie (se 3.1). Ligeledes er principperne for fastlæggelse af NOAEL for de(n) kritiske effekt(er) de samme, som beskrevet for vurderingerne i det enkelte studie (se 3.2). I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand vurderes effekter i reglen som værende mere kritiske desto lavere koncentrationer, de optræder ved, således at NOAEL for stoffet som sådan ofte, men ikke nødvendigvis, vil være identisk med det laveste af de fastlagte NOAELs fra de enkelte studier. US-EPA (1998) definerer det kritiske end-point som den effekt, der udviser det laveste LOAEL. 3.5 Sensibilisering for kemiske stofferDet har været diskuteret, hvorvidt sensibilisering er en effekttype, hvor der foreligger en tærskel eller ej. Nedenfor gives en kort opsummering af den foreliggende viden i relation til allergi udløst ved eksponering for kemiske stoffer, som den er fremlagt i et særnummer af tidsskriftet Comments on Toxicicology (Volume 7 1999), herunder en vurdering af tærskelværdi. Allergi defineres generelt som de skadelige effekter, der opstår som følge af stimulation af specifikke immunreaktioner. I relation til såkaldt kemisk allergi udløses disse immunreaktioner i følsomme individer ved eksponering for kemiske stoffer. De mest almindelige reaktioner i relation til kemisk allergi er hudsensibilisering, som resulterer i kontaktdermatitis, og luftvejssensibilisering som er associeret med rhinitis og astma. (Kimber & Dearman 1999a). Det skal nævnes, at ikke kun lavmolekylære kemiske stoffer, men også proteiner kan udløse allergiske reaktioner, men i dette afsnit omtales kun sensibilisering udløst af kemiske stoffer. Sensibilisering ved hudkontakt og luftvejssensibilisering som følge af eksponering for et kemisk stof har en del fælles træk. I begge tilfælde er allergenet et lavmolekylært kemisk stof, som stimulerer en specifik immunreaktion. Kemiske stoffer er haptener og som sådan ikke direkte immunogene. For udløsning af en immunreaktion skal det lavmolekylære kemiske stof først associeres med et makromolekyle (protein), hvorved der dannes et immunogent kompleks, som kan genkendes af immunsystemet. Selve den allergiske reaktion forløber i to faser. I den første fase, induktionsfasen, sker der en allergisk sensibilisering som følge af eksponering for et givent lavmolekylært kemisk stof, således at immunsystemet påvirkes til at respondere mere voldsomt ved en efterfølgende eksponering for det samme kemiske stof. Dette er den anden fase, elicitationsfasen (frembrudsfasen), hvor den sekundære immunreaktion initierer en inflammatorisk reaktion på eksponeringsstedet (hud eller luftveje). Disse inflammatoriske reaktioner fremprovokerer de symptomer, som klinisk genkendes som allergisk kontaktdermatitis eller luftvejssensibilisering. De sekundære reaktioner (elicitationsfasen) hos et i forvejen sensibiliseret individ udløses sædvanligvis ved eksponering for langt lavere doser af et givent stof end de doser, der skal til for at sensibilisere individet (induktionsfasen). Men der er også forskelle, som ikke kun begrundes i de to forskellige eksponeringssteder og dermed resulterende kliniske symptomer, men i relation til de immune og inflammatoriske processer, der er et
resultat af induktionsfasen såvel som elicitationsfasen. Disse forskelle fremgår af de efterfølgende to afsnit, som beskriver kontaktallergi og luftvejssensibilisering. Kontaktallergi er en form for forsinket sensibilisering (engelsk: delayed-type hypersensitivity) og som sådan afhængig af stimulation af specifikke T-lymfocyt reaktioner. Reaktionen kan kort beskrives som følger: Et kemisk allergen passerer den yderste forhornede del af overhuden (stratum corneum), og når frem til epidermis, hvor der sker en direkte interaktion med dendritceller i huden. Dendritcellerne transporteres til lymfeknuder, hvor antigenet inducerer og aktiverer T-lymfocytter, som så deler sig og differentierer. Denne del af reaktionen udgør den cellulære basis for sensibilisering. Når det nu sensibiliserede individ igen eksponeres for det pågældende kemiske stof ved hudkontakt, genkender T-lymfocytterne allergenet og responderer ved at frigøre cytokiner og andre molekylære mediatorer, som initierer den kutane inflammatoriske reaktion, der klinisk genkendes som allergisk kontaktdermatitis. Det er bredt accepteret, at kontaktsensibilisering er karakteriseret ved type 1 immunreaktioner og virkninger af associerede cytokiner. (Kimber & Dearman 1999b). Med hensyn til luftvejssensibilisering som følge af eksponering for kemiske stoffer ved man ikke nær så meget om de tilgrundliggende reaktioner. Ved udløsning af luftvejssensibilisering ved udsættelse for proteiner kan reaktionen kort beskrives som følger: Efter eksponering for et givent protein vil et følsomt individ reagere med dannelse af IgE-antistof, som derefter fordeles systemisk og associeres til mastceller, inklusiv mastceller i luftvejene. Ved en efterfølgende eksponering via inhalation for det samme protein vil allergenet bindes til og sammenlænke IgE, der er associeret med mastcellerne i luftvejene. Dette medfører, at mastcellerne degranulerer og frigør inflammatoriske mediatorer, som udløser en reaktion karakteriseret ved vasodilation og bronchokonstriktion. Hvorvidt kemiske allergener inducerer og eliciterer en luftvejssensibilisering via samme reaktionsmønster som proteiner diskuteres fortsat. Usikkerhederne ligger primært i undersøgelser af sammenhængen mellem kemisk luftvejssensibilisering og dannelse af IgE-antistof. For nogle kemiske allergener (f.eks. visse syreanhydrider og reaktive farvestoffer) er der fundet en god korrelation mellem dannelse af IgE-antistof og de kliniske symptomer for luftvejssensibilisering, mens dette ikke er tilfælde for alle undersøgte kemiske allergener (f.eks. visse diisocyanater). Kimber & Dearman har, på baggrund af den foreliggende viden, det synspunkt, at IgE-antistof spiller en vigtig rolle for kemisk luftvejssensibilisering. (Kimber & Dearman 1999b). Det har været diskuteret, hvorvidt sensibilisering er en effekttype, hvor der foreligger en tærskel eller ej. Basketter et al. (1999) har anført, at da kontaktallergener varierer meget med hensyn til den relative potens, så må det også kunne konkluderes, at de udviser stor variation i relation til en tærskelværdi for både induktion og elicitation. At der findes en tærskelværdi, er vist for nogle få kontaktallergener i humane studier. Evidensen for eksistensen af en tærskel for induktion støttes endvidere af, at selvom folk ofte eksponeres for mange kontaktallergener, så er det normalt kun en lille andel, der bliver sensibiliserede. Ligeledes er der mange studier, der viser, at der også findes en tærskel for elicitationsfasen. Derimod er der kun begrænset viden om, hvorvidt der findes en tærskel for induktion og elicitation af luftvejssensibilisering. Ifølge Basketter et al. (1999) er der al mulig grund til at formode, at der skal være en vis kritisk dosis på et relevant eksponeringssted for at kunne inducere luftvejssensibilisering. Tilsvarende formodes at gøre sig gældende for elicitation af luftvejsreaktionerne i sensibiliserede individer. Udover en kritisk dosis er der også andre faktorer, som har betydning. Blandt andet nævnes det, at eksponeringshyppigheden i relation til dosis er vigtig. Endelig er det også anført, at luftvejssensibilisering sandsynligvis kan induceres via andre eksponeringsveje end inhalation, for eksempel ved hudkontakt, og det understreges, at den kritiske dosis for sensibilisering vil variere afhængigt af eksponeringsvejen. I relation til risikovurdering af kemiske allergener er det anført, at denne vurdering kun kan foretages, hvis potens og tærskelværdi er nøje belyst for det givne kemiske allergen. 3.6 Kvalitativ vurdering af kræftfremkaldende stofferFor langt de fleste typer effekter findes der en tærskel, det vil sige en grænse (koncentration eller dosis), hvorunder der ikke ses effekt(er). Tærskelværdien er således den laveste koncentration eller dosis af stoffet, hvor der optræder en effekt (se 4.2). Sådan forholder det sig også for carcinogener, som ikke er genotoksiske, det vil sige, hvor den tilgrundliggende mekanisme for stoffets indflydelse på udvikling af tumorer ikke er en beskadigelse af arvematerialet. For visse typer af effekter findes der muligvis ikke en tærskel. Sådan forholder det sig antageligt for carcinogener, som er genotoksiske, det vil sige, hvor den tilgrundliggende mekanisme for stoffets effekt på udvikling af tumorer er en beskadigelse af arvematerialet. Dette betyder i teorien, at en hvilken som helst eksponering , hvor lav den end måtte være, vil medføre en risiko for udvikling af tumorer, der er større end nul. Det skal dog understreges, at man internationalt i de senere år har diskuteret og fortsat diskuterer, hvorvidt der foreligger er tærskel for genotoksiske effekter eller ej. For de stoffer, hvor den kritiske effekt har en tærskelværdi, fastlægges TDI med udgangspunkt i et nul-effektniveau (NO(A)EL) eller det laveste effektniveau (LO(A)EL) for den kritiske effekter under anvendelse af en usikkerhedsfaktor (se kapitel 4). For genotoksiske carcinogener fastlægges TDI på baggrund af en kvantitativ risikovurdering (se kapitel 5). Med henblik på fastlæggelse af TDI for kræftfremkaldende stoffer er det således af afgørende betydning, hvorvidt det givne stof vurderes som værende et genotoksisk carcinogen eller ej. Ved den kvalitative vurdering af et stofs kræftfremkaldende effekt tages stilling til to spørgsmål: 1) Er det sandsynligt, at stoffet er kræftfremkaldende hos mennesker? Og 2) har stoffet et genotoksisk potentiale? Afhængigt af evidensen for kræftfremkaldende effekt kan stofferne opdeles i forskellige grupper. Således opererer EU-systemet med 3 forskellige kategorier, US-EPA med 5 forskellige kategorier, og IARC med 4 forskellige kategorier. Placeringen af et stof i en af disse kategorier er ikke baseret på en opdeling efter potens, men er derimod en inddeling efter graden af den dokumentation (omfang, kvalitet og relevans), der findes for den kræftfremkaldende effekt. Disse tre forskellige systemer er således funderet på de samme basale principper. Alligevel er de ikke direkte sammenlignelige, blandt andet som følge af forskelle i antallet af kategorier samt forskelle i kriterierne for indplacering i en given kategori. Også med hensyn mutagene effekter opererer EU-systemet med 3 forskellige kategorier, som ligeledes er en inddeling efter graden af den dokumentation (omfang, kvalitet og relevans), der findes for den mutagene effekt. 3.6.1 EUVed klassificering for kræftfremkaldende effekt i EUs arbejdsgruppe for klassificering og mærkning af kemiske stoffer inddeles stofferne i 3 kategorier under hensyntagen til den nuværende viden (MM 2002): Stoffer, der vides at fremkalde kræft hos mennesket, placeres i kategori 1 (Carc1). Der foreligger tilstrækkelig dokumentation for en årsagssammenhæng mellem menneskets udsættelse for stoffet og udvikling af kræft. Stoffer klassificeres i denne kategori på baggrund af epidemiologiske undersøgelser. Stoffer, der bør anses for at fremkalde kræft hos mennesket, placeres i kategori 2 (Carc2). Der foreligger tilstrækkelig dokumentation til at nære stærk formodning om, at stoffets påvirkning af mennesker kan fremkalde kræft, generelt på grundlag af egnede langtidsforsøg i dyr samt andre relevante oplysninger. Der skal enten foreligge positive resultater fra to dyrearter eller klart positiv effekt fra én dyreart støttet af blandt andet data vedrørende genotoksicitet, metaboliske eller biokemiske undersøgelser, forekomst af godartede tumorer, strukturmæssige ligheder med andre kræftfremkaldende stoffer, eller data fra epidemiologiske undersøgelser, som tyder på en forbindelse. Stoffer, der giver anledning til betænkelighed, da de muligvis kan fremkalde kræft hos mennesket, placeres i kategori 3 (Carc3). Der foreligger ikke tilstrækkelige oplysninger til at foretage en tilfredsstillende vurdering. Der er visse tegn fra relevante dyreforsøg, men disse er utilstrækkelige til at placere dem i kategori 2. Kategori Carc3 omfatter 2 underkategorier: a) Stoffer, som er grundigt undersøgt, men hvor dokumentationen for tumorfremkaldende virkninger er utilstrækkelig til klassificering i kategori 2. Yderligere forsøg forventes ikke at frembringe yderligere oplysninger, der kan være relevante for klassificeringen. b) Stoffer, som ikke er tilstrækkeligt undersøgt. De data, der foreligger, er utilstrækkelige, men giver anledning til bekymring for mennesket. Denne klassificering er foreløbig; yderligere forsøg er nødvendige inden der kan træffes en endelig afgørelse. Ved klassificering for mutagene og genotoksiske effekter i EU's arbejdsgruppe for klassificering og mærkning af kemiske stoffer inddeles stofferne ligeledes i 3 kategorier under hensyntagen til den nuværende viden. Et mutagen er et stof, som fremkalder eller øger antallet af mutationer. En mutation er en permanent ændring i arvemassen eller strukturen i det genetiske materiale i en organisme, som medfører en ændring i organismens fænotypiske egenskaber. Ændringerne kan omfatte et enkelt gen, en gengruppe eller et helt kromosom. (MM 2002): Stoffer, der vides at have mutagene virkninger på mennesket, placeres i kategori 1 (Mut1). Der foreligger tilstrækkelig dokumentation for en årsagssammenhæng mellem stoffets påvirkning af mennesket og arvelige skader på det genetiske materiale. For at kunne placere et stof i kategori Mut1 er positiv dokumentation fra epidemiologiske undersøgelser vedrørende human mutation nødvendig. Eksempler på sådanne stoffer kendes ikke til dato. Stoffer, der bør anses for at have mutagene virkninger på mennesket, placeres i kategori 2 (Mut2). Der foreligger tilstrækkelig dokumentation til at nære stærk formodning om, at stoffets påvirkning af mennesket kan resultere i arvelige skader på det genetiske materiale, generelt på grundlag af: egnede langtidsforsøg i dyr, andre relevante oplysninger. For at kunne placere et stof i kategori 2 er der behov for positive resultater fra undersøgelser, som viser a) mutagene virkninger, b) andre cellulære vekselvirkninger i forbindelse med mutagenicitet i pattedyrs kimceller in vivo, eller c) mutagene virkninger i pattedyrs somatiske celler in vivo kombineret med klare beviser for, at stoffet eller en relevant metabolit når frem til kimcellerne. Stoffer, der giver anledning til betænkelighed, da de muligvis har mutagene virkninger, placeres i kategori 3 (Mut3). Der foreligger dokumentation fra mutagenicitetsundersøgelser, men den er utilstrækkelige til at placere stoffet i kategori 2. For at kunne placere et stof i kategori 3 er der behov for positive resultater fra undersøgelser, som viser a) mutagene virkninger, eller b) andre cellulære vekselvirkninger, som er relevante for mutagenicitet i somatiske celler i pattedyr in vivo. 3.6.2 US-EPAVed klassificering af kræftfremkaldende stoffer i US-EPA inddeles stofferne i 5 grupper (US-EPA 1998): Stoffer, der vides at fremkalde kræft hos mennesker, placeres i gruppe A (human carcinogen). Der foreligger fyldestgørende (sufficient) evidens fra epidemiologiske undersøgelser. Stoffer, der anses for sandsynligt kræftfremkaldende hos mennesker, placeres i gruppe B (probable human carcinogen). Der foreligger fyldestgørende evidens fra undersøgelser af forsøgsdyr eller begrænset (limited) evidens hos mennesker. Gruppe B er opdelt i 2 undergrupper: B1 og B2. Stoffer placeres i gruppe B1, når der er begrænset evidens fra epidemiologiske undersøgelser. Stoffer placeres i gruppe B2, når der er fyldestgørende evidens fra undersøgelser af forsøgsdyr og utilstrækkelige (inadequate) eller ingen data fra epidemiologiske undersøgelser. Stoffer, der anses for muligt kræftfremkaldende hos mennesker, placeres i gruppe C (possible human carcinogen). Der foreligger begrænset evidens fra undersøgelser af forsøgsdyr og ingen evidens hos mennesker. Stoffer, der ikke kan indplaceres i andre grupper, placeres i gruppe D (not classifiable). Der foreligger utilstrækkelige data eller ingen data. Stoffer placeres i gruppe E, når der ikke er evidens for kræftfremkaldende effekt i velgennemførte undersøgelser i mindst 2 forskellige arter eller både i epidemiologiske undersøgelser og undersøgelser af forsøgsdyr. 3.6.3 IARCVed klassificering af kræftfremkaldende stoffer i IARC inddeles stofferne i 4 grupper (IARC 1987): Stoffer, der vides at fremkalde kræft hos mennesker, placeres i gruppe 1 (carcinogenic to humans). Der foreligger fyldestgørende (sufficient) evidens for kræftfremkaldende effekt hos mennesker. Stoffer placeres i gruppe 2, når der ikke er fyldestgørende evidens for kræftfremkaldende effekt hos mennesker og/eller fyldestgørende evidens fra undersøgelser af forsøgsdyr. Gruppe 2 er opdelt i 2 undergrupper: 2A og 2B. Stoffer placeres i gruppe 2A (probably carcinogenic to humans), når der er begrænset (limited) evidens fra epidemiologiske undersøgelser og fyldestgørende evidens fra undersøgelser af forsøgsdyr. Undtagelsesvis kan et stof placeres i gruppe 2A udelukkende på baggrund af begrænset evidens fra epidemiologiske undersøgelser eller udelukkende på baggrund af fyldestgørende evidens fra undersøgelser af forsøgsdyr og andre data, som kan understøtte evidens for kræftfremkaldende effekt. Stoffer placeres i gruppe 2B (possibly carcinogenic to humans), når der er begrænset evidens fra epidemiologiske undersøgelser og ikke fyldestgørende evidens fra undersøgelser af forsøgsdyr. Stoffer placeres også i denne gruppe, når der er utilstrækkeligt (inadequate) evidens fra epidemiologiske undersøgelser eller ingen humane data, men fyldestgørende evidens fra undersøgelser af forsøgsdyr. I nogle tilfælde kan stoffer placeres i denne gruppe, når der er utilstrækkeligt evidens fra epidemiologiske undersøgelser eller ingen humane data, men begrænset evidens fra undersøgelser af forsøgsdyr og andre data, som kan understøtte evidens for kræftfremkaldende effekt. Stoffer, der ikke kan indplaceres i andre grupper, placeres i gruppe 3 (not classifiable). Der foreligger utilstrækkelige data eller ingen data. Stoffer placeres i gruppe 4 (probably not carcinogenic to humans), når der er evidens for, at stoffet ikke har kræftfremkaldende effekt i mennesker og heller ikke i forsøgsdyr. Stoffer kan også placeres i denne gruppe, når der er utilstrækkelig evidens fra epidemiologiske undersøgelser eller ingen humane data, men evidens for at stoffet ikke har kræftfremkaldende effekt i forsøgsdyr samt understøttet i en lang række andre data. 3.6.4 Miljøstyrelsens generelle praksis i relation til kvalitetskriterierI relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand tages der ved vurderingen af kræftfremkaldende effekt ikke kun udgangspunkt i det givne stofs placering i en af EU's, IARC's eller US-EPA's grupper. I vurderingen indgår også nyere data samt andre data som for eksempel stoffets fysisk-kemiske egenskaber, om eksponeringsvejene anvendt i dyreforsøgene er relevante for den generelle befolkning, toksikokinetiske forhold, om stoffet er et direkte reagerende carcinogen, hvilken metabolisering der skal ske for at stoffet bliver til et aktivt carcinogen, om forskelle i metabolisering er speciesafhængige, om stoffet har udvist genotoksiske egenskaber i in vivo tests eller kun i in vitro testsystemer, mekanismen (genotoksisk eller non-genotoksisk) for udvikling af tumorer, om typen af tumorer fundet i en dyreart har relevans for mennesker (se 3.7), og om der er epidemiologiske data til støtte for de dyreeksperimentelle fund. Som det fremgår af ovenstående, indgår mange elementer i vurderingen af, hvorvidt et givent stof skal betragtes som kræftfremkaldende, og hvorledes dette skal indgå i fastsættelsen af kvalitetskriterier. På den baggrund kan det være vanskeligt at beskrive en generel og fast procedure på området, da data kan variere meget fra stof til stof. Som tidligere nævnt er epidemiologiske undersøgelser, der klart viser en sammenhæng mellem eksponering for et givent stof og udvikling af tumorer hos mennesker, af afgørende betydning for vurdering af et stof som kræftfremkaldende hos mennesker. Epidemiologiske undersøgelser, der ikke klart viser en sammenhæng, for eksempel hvor det drejer sig om små risikoforøgelser, kan være til støtte i vurderingen af de dyreeksperimentelle fund og dermed bidrage til, at stoffet vurderes som værende kræftfremkaldende hos mennesker. Det skal understreges, at negative resultater i epidemiologiske undersøgelser ikke beviser et fravær af kræftfremkaldende egenskaber. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand inddrages de vurderinger, der måtte være foretaget af EU, IARC og eller US-EPA. Således vil et givent stof, der en indplaceret i EU's carc1 eller carc2, IARC's gruppe 1 eller gruppe 2A/2B, og/eller US-EPA's gruppe A eller B1/B2, som udgangspunkt blive betragtet som et carcinogen, med mindre der er velunderbyggede informationer og fortolkninger af data, der taler imod denne vurdering. Et eksempel herpå er fortolkning af visse typer af tumorer fundet i en dyreart, som vurderes ikke at have relevans for mennesker (se 3.7). Det skal dog understreges, at et stof som enten ikke er vurderet af EU, IARC og/eller US-EPA eller er indplaceret i en lavere kategori (EU carc3, IARC gruppe 3 og/eller US-EPA gruppe C/D), godt kan betragtes som et carcinogen i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand, såfremt der er velunderbyggede data, der taler herfor. Det kan for eksempel være i tilfælde af, at der er publiceret nye undersøgelser siden en eventuel vurdering er foretaget af EU, IARC og/eller US-EPA, eller der er stor strukturlighed med allerede kendte carcinogener. Endvidere skal der tages stilling til, hvorvidt stoffet vurderes som værende et genotoksisk carcinogen eller ej. I vurderingen heraf indgår først og fremmest de resultater, der er opnået ved testning af stoffets mutagene og genotoksiske potentiale i en række forskellige testsystemer, inklusive in vivo undersøgelser. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand tages der som hovedregel udgangspunkt i de vurderinger, der måtte være foretaget af EU, IARC og eller US-EPA. Således vil et givent stof, der i henhold til EU's kriterier er klassificeret for mutagene og/eller genotoksiske egenskaber (mut1, mut2 eller mut3) som udgangspunkt blive betragtet som et muligt humant genotoksisk stof. Det er dog kun et meget lille antal stoffer, der er klassificeres i henhold til EU's kriterier (ingen stoffer er til dato klassificeret mut1), hvorfor vurderingen af et stofs genotoksiske egenskaber må foretages på baggrund af de foreliggende testresultater. Ud fra dette samlede billede vurderes det, om stoffet i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand skal betragtes som et genotoksisk eller non-genotoksisk carcinogen. Denne vurdering har som tidligere nævnt afgørende betydning for valg af procedure for estimering af TDI, det vil sige for valget mellem tærskelværdiprincippet, der anvendes ved estimering af TDI for non-genotoksiske carcinogener (se kapitel 4) og den kvantitative risikovurdering, der anvendes ved estimering af TDI for genotoksiske carcinogener (se kapitel 5). Selv om datagrundlaget for et givent stof ikke er tilstrækkeligt med henblik på at afgøre, hvorvidt stoffet kan vurderes som værende et genotoksisk carcinogen, eller datagrundlaget som sådan er utilstrækkeligt til at kunne foretage en kvantitativ risikovurdering for et stof, der vurderes som værende et genotoksisk carcinogen, kan der selvsagt godt være en bekymring i relation til eventuelle kræftfremkaldende egenskaber for det pågældende stof. I disse tilfælde beregnes TDI med udgangspunkt i et NOAEL eller LOAEL (se kapitel 4), og der kompenseres for usikkerheden vedrørende et eventuelt kræftfremkaldende potentiale ved anvendelse af en større usikkerhedsfaktor, end der ellers ville være blevet anvendt (se 4.4.3). 3.7 Effekter hos forsøgsdyr hvor relevansen for mennesker er omdiskuteretGenerelt antages det, at en toksisk effekt hos forsøgsdyr forårsaget af et kemisk stof viser, at stoffet også er toksisk i mennesket. Dette gælder også udvikling af tumorer, selvom type og placering af tumor ikke altid vil være den samme i dyr og menneske. Dog kan visse effekter hos forsøgsdyr have begrænset relevans med hensyn til at forudsige effekten i mennesker. Tumorer, der induceres via en genotoksisk mekanisme vurderes sædvanligvis som værende relevante for mennesket, også når tumorerne opstår i væv, som ikke findes hos mennesket. Non-genotoksiske mekanismer kan omfatte kronisk celleskade, immunosuppression, øget udskillelse af trofiske hormoner samt receptoraktivering. Desuden kan andre mekanismer, som for eksempel CYP450-induktion, ligge til grund for tumorudvikling. Visse specifikke non-genotoksiske mekanismer anses imidlertid ikke som værende relevante for mennesket. I det følgende omtales en del af de tumortyper og mekanismer, hvor relevansen for mennesket sædvanligvis anses for begrænset, eller hvor det ikke umiddelbart kan antages, at dyrefundene er prædiktive for mennesket. Det skal understreges, at der i hvert enkelt tilfælde foretages en nøje vurdering, og at der kan være stor forskel på de enkelte dyrestammers reaktionsmønster. 3.7.1 Leukæmier (mononucleærcelle type) hos Fischer rottenDenne type leukæmi forekommer kun hos rotten og er kun almindelig hos stammen F-344, hvor den naturlige forekomst har været stigende gennem årene. Der er forskel på hyppigheden hos de to køn, og hyppigheden hos hanner er oppe på 50%, mens hyppigheden hos hunner ligger på omkring 30%, med stor variation fra forsøg til forsøg. Det er vist for nogle genotoksiske carcinogener, at de ikke øger forekomsten af mononukleærcelle-leukæmier hos F-344 rotten, mens en række stoffer, som ikke menes at være kræftfremkaldende hos mennesket, øger forekomsten. Som følge af disse forhold anses en øget forekomst af mononukleærcelle-leukæmier hos F-344 rotten for værende af begrænset relevans for mennesket. (Caldwell 1999). 3.7.2 Nyretumorer hos hanrotterVisse nyretumorer hos hanrotter anses som værende ikke relevante for mennesket, idet der er stærk evidens for, at mekanismen for tumordannelsen er specifik for hanrotter. Der er tale om tumorer, der har en sammenhæng med induceret ophobning af proteinet 2-globulin i nyrecellerne. Dette protein dannes i leveren hos hanrotter, mens hunrotter og andre dyrearter, inklusive mennesket, ikke danner 2-globulin. Binding af et kemisk stof til 2-globulin kan føre til, at proteinet ikke nedbrydes som normalt, men ophobes i nyrecellerne. Ophobningen er skadelig for cellerne og medfører nefropati, øget celleproliferation og endelig nyretumorer. Denne mekanisme forekommer ikke hos mennesket. Såfremt det specifikt kan påvises, at denne mekanisme er den egentlige årsag til forekomst af nyretumorer hos dyrene, anses denne effekt at have begrænset relevans for mennesket. IARC har formuleret en række kriterier for, hvornår nyretumorer hos hanrotter kan anses som værende ikke relevant for mennesket (IARC 1999). 3.7.3 Levertumorer hos mus og rotterLevertumorer er et hyppigt fund i gnavere i cancerstudier. Hos mus er den naturlige forekomst af levertumorer meget høj og stærkt påvirkelig af en række faktorer som for eksempel stress og ernæring. Øget forekomst af levertumorer i et musestudie kan derfor meget vel skyldes andre faktorer end teststoffet. Ligeledes er mus meget tilbøjelige til at udvikle levertumorer efter langvarig stimulering af celledelingen eller langvarig enzyminduktion forårsaget af de høje doser af teststof. Denne mekanisme er ikke relevant for mennesket, hvor eksponeringen som regel er meget lavere, end de doser der anvendes ved studier i forsøgsdyr. Et musestudie, hvor den eneste tumorform er levertumorer, anses ofte for at have begrænset relevans for mennesket. (Williams 1997, Carmichael et al. 1997). En lang række kemiske stoffer, der inducerer peroxisomproliferation i leveren, forårsager udvikling af levertumorer hos rotter og mus. Hos mennesket findes receptoren for peroxisomproliferation (PPAR) også, men i lavere niveau end hos gnavere, hvorfor peroxisomproliferation menes at være sjældent forekommende hos mennesket. Tumorer, der er associeret til peroxisomproliferation, anses således for mindre relevante for mennesket. Imidlertid kendes mekanismen for denne form for udvikling af levertumorer ikke, og sammenhængen mellem peroxisomproliferation og udvikling af levertumorer er ikke entydig. For at afvise relevansen af levertumorer, der er associeret til peroxisomproliferation, kræves et stort datagrundlag. (Williams 1997). 3.7.4 Leydig-celle tumorerLeydigceller findes i testis og danner det hanlige kønshormon, testosteron. I cancerstudier med rotter er Leydig-celle tumorer (hyppigst adenom, sjældent carcinom) et hyppigt fund hos ældre dyr. Der er stor forskel på rottestammer, idet Sprague-Dawley har en baggrundsforekomst på 1-5%, mens Fischer-rotten er næsten oppe på 100%. Fischer-rotten anses derfor for at have en atypisk høj tendens til at udvikle Leydig-celle tumorer. Hos mus er hyppigheden generelt lav. Hyppigheden af denne tumortype er meget lav hos mennesket, hvor kun 2% af alle testestumorer stammer fra Leydigceller. For non-genotoksiske stoffer, der fremkalder Leydig-celle tumorer, og hvor mekanismen er undersøgt, er der typisk tale om en induceret øget koncentration af serum-LH (luteiniserende hormon) og dermed øget stimulation af Leydig-cellerne til vækst og deling, eller øget følsomhed for LH hos Leydig-cellerne. Da mennesket besidder det samme hormonsystem, er mekanismen relevant. Det antages imidlertid, at menneskets Leydigceller er langt mindre følsomme for LH-stimulation. Mænd med endokrine lidelser, der medfører konstant øget LH-niveau , får Leydig-celle adenomer med en frekvens på 2-3%. Til sammenligning ses 100% tumorer hos rotter, der får androgenreceptor antagonisten flutamid. En arvelig defekt hos mænd, der medfører konstant LH-receptoraktivering, giver ikke øget forekomst af Leydig-celle tumorer. Epidemiologiske studier af mennesker udsat for stoffer, der fremkalder Leydig-celle tumorer i rotter, har ikke påvist øget forekomst af denne tumor. Fund af Leydig-celle tumorer i et rottestudie kan således have begrænset relevans for mennesket. (Cook et al. 1999). 3.7.5 Thyreoidea tumorerI cancerstudier med rotter er thyreoidea carcinomer hyppige, og en af mekanismerne for udvikling af carcinomer er øget niveau af det thyreoidea-stimulerende hormon (TSH), som udskilles fra hypofysen som led i et feedback system. Hos mennesket er sådanne tumorer derimod ikke almindelige og synes ikke at have relation til øget TSH. Der er flere måder, hvorpå TSH-udskillelsen kan øges. For eksempel kan en øget enzymaktivitet i leveren medføre denne effekt ved, at nedbrydningen af thyreoideahormon øges. Hos mennesket er thyreoidea hormonsystemet det samme som hos rotten, men langt mere robust. Blandt andet er thyreoideahormon hos mennesket langt fastere bundet til protein i plasma. Rottens thyreoidea er endvidere langt mere aktiv og fungerer på et højere stofskifteniveau end hos mennesket. Såfremt et non-genotoksisk stof fremkalder thyreoidea carcinomer hos rotter, og det er vist, at mekanismen er forstyrrelse af thyreoideas hormonbalance og øget TSH, anses relevansen for mennesket for begrænset (IARC 1999). 3.7.6 Tumorer i urinblærenHos rotter og mus kan tilstedeværelsen af blæresten medføre øget celleproliferation og tumordannelse. Dette kan også ske hos mennesket, men for at et kemisk stof i sig selv danner blæresten, skal det
indtages i meget høje doser, hvilket normalt ikke vil være tilfældet hos mennesket. Tumorer hos rotter og mus forårsaget af blæresten er således relevante for mennesket, men disse udvikles ikke, såfremt
stoffet indtages i mængder, der ligger under tærsklen for udvikling af blæresten. 3.7.7 FormavetumorerMavesækken hos rotter og mus er anatomisk forskellig fra menneskets, idet en del af mavesækken hos disse arter er uden kirtler (kaldet formaven). Epithelet i den kirtelløse del af mavesækken ligner det, der beklæder spiserøret. Selv om mennesket ikke besidder et organ lig gnavernes kirtelløse mave, må spiserøret betragtes som analogt med hensyn til epithel. Ofte vil mekanismen for udvikling af formavetumorer være relateret til hyperplasi, som kræver langvarig kontakt. For et stof, der bevirker udvikling af formavetumorer ved direkte kontakt med epithelet, gælder, at da kontakttiden er meget kortere i spiserøret end i mavesækken, vil tumordannelse hos mennesket formentlig ikke ske. I tilfælde, hvor denne specifikke mekanisme kan dokumenteres, vurderes kræftfremkaldende effekt som følge af formavetumorer hos gnavere ikke som relevant for mennesket. (Kroes & Wester 1986). 3.7.8 Øvrige tumortyper og mekanismerUdover de ovenfor anførte eksempler skal det nævnes, at forekomst af mammatumorer hos mus og milttumorer hos F-344 rotter også i en del tilfælde har været vurderet som værende ikke relevante for mennesket. Phaeochromocytom (tumor i binyremarv), ikke ualmindeligt forekommende hos rotter, men sjældent hos mennesket, kan hos rotten være relateret til hypercalcæmi, hvorimod en sådan relation ikke synes at forekomme hos mennesket. Endvidere er det et velkendt fænomen, at kemiske stoffer, der injiceres i forsøgsdyr, kan fremkalde tumorer lokalt på injektionsstedet, for eksempel i muskulatur. Dette vides at forekomme også for stoffer, der ikke er kræftfremkaldende, når de doseres på anden måde, for eksempel oralt. Sådanne lokale tumorer vil ofte blive vurderet som værende ikke relevante for mennesket. 3.7.9 ReferencerBasketter DA, Evans P, Gerberick GF and Kimber I (1999). Chemical allergy: estimation of potency, thresholds and risk assessments. Comm Toxicol 7, 79-89. Calabrese EJ (2001). Overcompensation stimulation: A mechanism for hormetic effects. Crit Rev Toxicol 31, 425-470. Calabrese EJ and Baldwin LA (2001a). The frequency of U-shaped dose responses in the toxicological literature. Toxicol Sci 62, 330-338. Calabrese EJ and Baldwin LA (2001b). Hormesis: A generalizable and unifying hypothesis. Crit Rev Toxicol 31, 353-424. Caldwell DJ (1999). Review of mononuclear cell leukemia in F-344 rat bioassays and its significance to human cancer risk: A case study using alkyl phthalates. Regul Toxicol Pharmacol 30, 45-53. Carmichael NG, Enzmann H, Pate I and Waechter F (1997). The significance of mouse liver tumor formation for carcinogenic risk assessment: results and conclusions from a survey of ten years of testing by the agrochemical industry. Environ Health Perspect 105, 1196-1203. Cook JC, Klinefelter GR, Hardisty JF, Sharpe RM and Foster PMD (1999). Rodent Leydig cell tumorigenesis: A review of the physiology, pathology, mechanisms and relevance to humans. Crit Rev Toxicol 29, 169-261. Goodman JI (1998). The traditional toxicologic paradigm is correct: Dose influences mechanism. Environ Health Perspect 106, 285-288. IARC (1999). IARC Scientific Publications No. 147. Species differences in thyroid, kidney and urinary bladder carcinogenesis. Edited by C.C. Capen, E. Dybing, J.M. Rice and J.D. Wilbourn. Kimber I and Dearman RJ (1999a). Allergic hypersensitivity induced by chemicals: an introduction. Comm Toxicol 7, 5-7. Kimber I and Dearman RJ (1999b). Mechanisms of sensitization to chemical allergens. Comm Toxicol 7, 9-30. Kroes R and Wester W (1986). Forestomach carcinogens: Possible mechanisms of action. Fd Chem Tox 24, 1083-1089. MM (2002). Bekendtgørelse om klassificering, emballering, mærkning, salg og opbevaring af kemiske stoffer og produkter. Bekendtgørelse nr. 329 af 16. maj 2002, Miljøministeriet, Miljøstyrelsen. Rattan S. (2001). Ungdommens kilde. Aktuel Naturvidenskab 3, 25- 27. US-EPA (1998). Ambient water quality criteria derivation for the protection of human health – Technical Support Document. Final draft. United States Environmental Protection Agency, Office of Water 4304, EPA-822-B-98-005. Williams GM (1997). Chemicals with carcinogenic activity in the rodent liver; mechanistic evaluation of human risk. Cancer Lett 117, 175-188. 4 Fastlæggelse af TDI for stoffer med en tærskelværdi for effekter4.1 Tolerabel daglig indtagelse (TDI) Farlighedsvurderingen munder ud i en identifikation af de(n) kritiske effekt(er) for stoffet, det vil sige hvilke(n) effekt(er), der anses for at være mest væsentlig(e) i relation til fastsættelse af kvalitetskriterier for luft, jord og/eller drikkevand. Det næste skridt i fastsættelse af kvalitetskriterierne er fastlæggelse af den såkaldte tolerable daglige indtagelse (TDI). Ved beregning af TDI skelnes der mellem, hvorvidt der antages at findes en tærskel (engelsk: threshold) for stoffets kritiske effekt(er) eller ej. For langt de fleste typer effekter findes der en tærskel, det vil sige en grænse (koncentration eller dosis), hvorunder der ikke ses effekt(er). Sådan forholder det sig også for carcinogener, som ikke er genotoksiske, det vil sige hvor den tilgrundliggende mekanisme for stoffets indflydelse på udvikling af tumorer ikke er en beskadigelse af arvematerialet. I disse tilfælde fastlægges TDI med udgangspunkt i et nul-effektniveau (NO(A)EL) eller det laveste effektniveau (LO(A)EL) for de(n) kritiske effekt(er) under anvendelse af en usikkerhedsfaktor (UF) (engelsk: Uncertainty Factor). Principperne herfor er beskrevet nærmere i dette kapitel. For nogle typer af effekter findes der muligvis ikke en tærskel. Dette antager man for eksempel for genotoksiske carcinogener, hvor den tilgrundliggende mekanisme for udvikling af tumorer er en beskadigelse af arvematerialet. I disse tilfælde fastlægges TDI på baggrund af en kvantitativ risikovurdering, principperne herfor er beskrevet nærmere i kapitel 5. 4.1 Tolerabel daglig indtagelse (TDI)Den tolerable daglige indtagelse (TDI) er en beregnet størrelse (koncentration eller dosis), som mennesker vurderes at kunne udsættes for (tolerere) gennem et helt livsforløb, uden at der optræder sundhedsskadelige effekter. TDI angives sædvanligvis i enheden mg/kg legemsvægt per dag. Termen TDI anvendes almindeligvis også i de tilfælde, hvor eksponering for stoffet sker via inhalation af luft indeholdende stoffet eller ved hudkontakt, selvom der strengt taget ikke er tale om indtagelse af stoffet i disse tilfælde. Analogt til TDI kan termen tolerabel koncentration (TK) defineres som den koncentration af et stof i luft, jord, eller drikkevand som mennesker vurderes at kunne udsættes for (tolerere) gennem et helt livsforløb uden at der optræder sundhedsskadelige effekter. TK angives i enheden mg/m³ (luft), mg/l (drikkevand), eller i mg/kg (jord). For nogle stoffer fastsættes i stedet for TDI en foreløbig (provisorisk) tolerabel ugentlig indtagelse (engelsk: Provisional Tolerable Weekly Intake – PTWI). PTWI benyttes sædvanligvis i relation til stoffer, hvor eksponeringen kan variere fra dag til dag, og hvor der kan opstå kroniske effekter, som for eksempler visse metaller (bly, cadmium og kviksølv). Derudover anvendes bredt et par andre termer, der er analoge til TDI: 1) den acceptable daglige indtagelse (engelsk: Acceptable Daily Intake - ADI), som anvendes ved vurdering af for eksempel tilsætningsstoffer, pesticidrester samt rester af veterinære lægemidler i levnedsmidler; samt 2) Reference Dose (RfD), som anvendes bredt af den amerikanske miljøstyrelse (US-EPA). De overordnede principper for fastsættelse af ADI og RfD er sammenlignelige med de principper, der beskrives i dette kapitel for fastlæggelse af TDI, mens det for beregningerne tilgrundliggende datamateriale ofte er betydeligt bedre end det datamateriale, der oftest er tilgængeligt i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand. For stoffer, hvor den akutte toksicitet vurderes som værende den kritiske effekt med henblik på fastsættelse af kvalitetskriterier i luft, jord og drikkevand, er det meningsløst er fastsætte en TDI. For sådanne stoffer fastlægges i stedet den maksimale tolerable dosis/koncentration (MTD/K) som udgangspunkt for fastsættelse af kvalitetskriterierne. 4.2 TærskelværdiFor langt de fleste typer af effekter findes der en tærskel, det vil sige en grænse (koncentration eller dosis), hvorunder der ikke ses effekt. Det diskuteres dog fortsat, hvorvidt der eksisterer sådanne tærskler. I teorien kunne man forestille sig, at et enkelt molekyle er tilstrækkeligt til at udløse en toksisk effekt, og den opfattelse er stadig fremherskende med hensyn til mutagene effekter. Men organismen har flere muligheder for at modvirke eventuelle toksiske effekter som for eksempel nedbrydning af stoffet, reparation af en skadelig påvirkning i en celle, udskiftning af skadede celler, eller kompensation på anden vis for en given påvirkning. Tærskelværdien defineres som den koncentration eller dosis hvorunder der ikke observeres effekter. Tærskelværdiens størrelse afhænger af det enkelte stofs potens såvel som af det eksponerede individ. For eksempel vil allergikere reagere på udsættelse for allergifremkaldende stoffer ved langt lavere koncentrationer end ikke-allergikere, det vil sige, at allergikere har en lavere tærskelværdi for sådanne stoffer end ikke-allergikere. Men ved udsættelse for ikke-allergifremkaldende stoffer vil tærskelværdien for en given effekttype (for eksempel nyreskader) alt andet lige ikke nødvendigvis være anderledes hos allergikere sammenlignet med ikke-allergikere. 4.3 Kritisk effekt og nul-effektniveauSom tidligere nævnt, munder farlighedsvurderingen (kapitel 3) ud i en identifikation af de(n) kritiske effekt(er) (se 3.4) for stoffet, det vil sig en udpegning af de(n) effekt(er), der anses for at være mest væsentlig(e) i relation til fastsættelse af kvalitetskriterier for luft, jord og/eller drikkevand. Ved identifikationen af de(n) kritiske effekt(er) sammenholdes og vurderes alle de informationer, der er samlet under vurderingerne af de enkelte studier. Principperne for den samlede vurdering af alle stoffets effekter med henblik på identifikation af de(n) kritiske effekt(er) er de samme, som beskrevet for vurdering af det enkelte studie (se 3.1). Det næste skridt i fastsættelsen af kvalitetskriterier er i teorien en fastlæggelse af tærskelværdien for de(n) kritiske effekt(er). I praksis tager man udgangspunkt i et observeret nul-effektniveau (NO(A)EL), det vil sige den højeste koncentration eller dosis, hvorved de(n) kritiske effekt(er) ikke er blevet observeret og vurderet ud fra alle de informationer, der er samlet sammen. En lang række stoffer er imidlertid ikke undersøgt tilstrækkeligt til, at det er muligt at fastlægge et observeret nul-effektniveau. I disse tilfælde fastlægges det laveste effektniveau (LO(A)EL), hvorved de(n) kritiske effekt(er) er blevet observeret. Principperne for fastlæggelse af nul-effektniveau eller det laveste observerede effektniveau for de(n) kritiske effekt(er) er de samme, som beskrevet for fastlæggelse af nul-effektniveau eller det laveste observerede effektniveau i det enkelte studie (se 3.2). I længerevarende inhalationsstudier med gentagen dosering udsættes forsøgsdyrene oftest for stoffet i et begrænset tidsrum, for eksempel 6 timer om dagen, 5 dage per uge i et antal uger. For mange typer af systemiske effekter anses det i højere grad at være den samlede dosis og ikke stoffets koncentration i luften, der er af betydning for udvikling af disse effekter. Ved fastsættelse af kvalitetskriterier i disse tilfælde foretages en omregning af det fastlagte nul-effektniveau (eller det laveste effektniveau) til et gennemsnitligt døgnniveau (kontinuert eksponering) eller døgndosis ud fra de i studiets aktuelle eksponeringsbetingelser. For lokale effekter (effekter, der optræder lokalt i luftvejene samt direkte effekter på hud og øjne) anses det sædvanligvis at være stoffets koncentration i luften og ikke den samlede dosis som sådan, der er af betydning for udvikling af disse effekter, hvorfor der i disse tilfælde ikke omregnes til en kontinuert eksponering. 4.4 Usikkerhedsfaktorer (UF)Begreberne usikkerhedsfaktor (engelsk: Uncertainty Factor - UF), sikkerhedsfaktor (engelsk: Safety Factor - SF), korrektionsfaktor (engelsk: Adjustment Factor - AF), ekstrapolationsfaktor (engelsk: Extrapolation Factor - EF) samt konverteringsfaktor (engelsk: Conversion Factor - CF) finder anvendelse i relation til fastsættelse af TDI i forskellige sammenhænge. I erkendelse af at faktoren har til formål at afspejle de usikkerheder, der blandt andet kan skyldes mangler i den eksisterende viden, frem for at udtrykke en sikkerhedsmargen, er man internationalt mere og mere gået over til at anvende begrebet usikkerhedsfaktor. Dette begreb vil blive anvendt fremover i rapporten, med mindre der specifikt relateres til et af de andre begreber. Som nævnt tidligere beregnes TDI for stoffer, hvor der er en tærskelværdi for de(n) kritiske effekt(er), med udgangspunkt i det observerede nul-effektniveau (NO(A)EL) eller det laveste observerede effektniveau (LO(A)EL) for de(n) kritiske effekt(er) under anvendelse af en usikkerhedsfaktor (UF). Formålet med usikkerhedsfaktoren er at tage højde for eventuelle mangler i den eksisterende viden og datagrundlaget. I relation til anvendelsen af usikkerhedsfaktoren har der hidtil i en lang række reguleringsmæssige sammenhænge næsten udelukkende været fokuseret på at tage højde for artsforskelle (engelsk: interspecies variation) samt for variationer i menneskers individuelle følsomhed/sårbarhed (engelsk: intraspecies variation, interindividual variation). Traditionelt har der siden midten af 1950'erne været anvendt en samlet sikkerhedsfaktor på 100, når der tages udgangspunkt i data fra forsøgsdyr. Denne procedure blev foreslået i 1954 af Lehman & Fitzhugh til vurdering af `et sikkert niveau' for tilsætningsstoffer eller kontaminanter i levnedsmidler og blev i 1961 adopteret af `the Joint FAO/WHO Expert Committee on Food Additives' (JECFA) og af `the Joint Meeting of Experts on Pesticides Residues' (JMPR) i en lidt modificeret form. Denne 100 faktor var ikke begrundet i kvantitative forhold, men et arbitrært valg og rationalet for størrelsesordenen på 100 kendes ikke. Ifølge Lehman & Fitzhugh (1954) blev 100-faktoren valgt med henblik på at omfatte følgende usikkerheder: 1) intra (human) species variabilitet; 2) inter (dyr til menneske) species variabilitet; 3) tage højde for mere følsomme individer i den humane population som følge af sygdom ved sammenligningen med sunde forsøgsdyr; og 4) mulige synergistiske virkninger af de mange tilsætningsstoffer eller kontaminanter. I de tilfælde, hvor der tages udgangspunkt i velgennemførte undersøgelser i mennesker, har der traditionelt været anvendt en samlet sikkerhedsfaktor på 10, det vil sige, at den traditionelle faktor på 100 i princippet er opdelt i to faktorer på 10 hver til at kompensere for henholdsvis interspecies variation og interindividuel variation. I nyere tid er man internationalt mere og mere gået over til at opdele usikkerhedsfaktoren i flere kategorier, herunder en underopdeling af den enkelte kategori, og i visse tilfælde at anvende en samlet faktor på mere end 100, for eksempel når der tages udgangspunkt i et begrænset datamateriale, ved fravær af et nul-effektniveau, eller ved mangel af kroniske studier (se også 4.4.3). Ved estimering af Reference Dose (RfD) vurderer US-EPA 5 forskellige kategorier af usikkerhed: 1) interindividuel variation (UFH), 2) interspeciesvariation (UFA), 3) subkronisk til kronisk varighed (UFS), 4) LOAEL til NOAEL (UFL) og , 5) datasættes kvalitet og relevans (UFD). Standardværdien for den enkelte kategori er 10, men en samlet usikkerhedsfaktor på over 3000 anvendes generelt ikke. (US-EPA 1998). I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand har sikkerhedsfaktoren (SF) hidtil været opdelt i tre hovedkategorier (MST 1990):
Som nævnt tidligere vil betegnelsen usikkerhedsfaktorer blive anvendt fremover, idet denne term i højere grad svarer til rationalet for at anvende faktorerne. Ved anvendelse af betegnelsen sikkerhedsfaktor kan man fejlagtigt få den opfattelse, at hvis der er anvendt en stor sikkerhedsfaktor for et stof, så er den opnåede beskyttelse (sikkerhed) større for dette stof end for et andet stof, hvor der er anvendt en lavere sikkerhedsfaktor. I det efterfølgende gøres der nærmere rede for de tre usikkerhedsfaktorer, herunder de elementer, der indgår i den enkelte faktor samt rationalet for valg af en arbitrær/pragmatisk standardværdi (engelsk: default value) for de enkelte faktorer. 4.4.1 Usikkerhedsfaktor I (”interspeciesvariation”)4.4.1.1 Biologisk variationRent biologisk og biokemisk minder mennesker om andre pattedyr, også med hensyn til udvikling af forskellige typer af toksiske effekter. I en nylig publiceret undersøgelse (Olson et al. 2000) er overensstemmelsen mellem effekter observeret i dyreforsøg og hos mennesker vurderet for 150 lægemidler med 221 forskellige humane end-points. Resultaterne viste overensstemmelse mellem effekter observeret i mennesker og forsøgsdyr (både gnavere og ikke-gnavere) for 71% af effekterne, det vil sige samme type effekter i samme organsystem. Den største grad af overensstemmelse blev observeret for hæmatologiske, gastrointestinale samt kardio-vaskulære end-points, mens den mindste grad af overensstemmelse blev observeret for dermale effekter. Men selv om testning af stoffer i forsøgsdyr i høj grad vil være prædiktiv for tilsvarende effekter i mennesker, kan mennesker være mere eller mindre følsomme end andre pattedyr over for toksiske stoffer, ligesom der kan være endog store forskelle i de forskellige dyrearters følsomhed. Forskellene i følsomhed afspejler dels forskelle i toksikokinetik (absorption, fordeling, metabolisme samt udskillelse), og dels forskelle i toksikodynamik (interaktionen mellem et toksisk stof og virkningsstedet samt de efterfølgende reaktioner, som leder til udvikling af en toksisk effekt). Den generelle opfattelse er, at det især er forskelle i metabolismen af fremmedstoffer, der spiller den største rolle for interspeciesvariationen (Davidson et al. 1986, Calabrese et al. 1992, Voisin et al. 1990). Hertil kommer, at der findes forskellige former for organer og væv de enkelte arter imellem (for eksempel har gnavere organer og væv, som mennesker ikke har), forskellige former for fordøjelse, samt anderledes struktur af de øvre luftveje. Dette betyder, at visse effekter som udvikles i forsøgsdyr ikke nødvendigvis vil være af relevans for mennesker (se også 3.5). Ekstrapolation af data fra dyr til mennesker kan opfattes som omhandlende to forskellige aspekter: 1) korrektion af dosis for forskelle i kropsstørrelse mellem forsøgsdyr og mennesker (se 4.4.1.2), og 2) andre former for forskelle mellem forsøgsdyr og mennesker, som ikke nødvendigvis afspejles i forskellene i kropsstørrelse (se 4.4.1.3). 4.4.1.2 Korrektion for forskelle i kropsstørrelse mellem dyr og menneskerVed ekstrapolation af data fra dyr til mennesker indgår korrektion af doser anvendt i dyreforsøg for forskelle i kropsstørrelse mellem forsøgsdyr og mennesker, såkaldt allometrisk skalering. Rationalet for skalering er funderet på ligheder i anatomisk henseende samt den universelle karakter i fysiologiske funktioner og i biokemiske reaktioner på tværs af arterne på trods af den store forskel i størrelse, facon og form mellem forskellige arter (Davidson et al. 1986). Hvorvidt dosis bør korrigeres for forskelle i kropsstørrelse ved allometrisk skalering, afhænger af eksponeringsvejen anvendt i dyreforsøget. Dosiskorrektion ved allometrisk skalering anvendes ved udgangspunkt i studier med oral administration og dermal applikation, men ikke med inhalation (se 4.4.1.2.4). Almindeligvis anvendes legemsvægten som et udtryk for kropsstørrelsen, da denne parameter er den letteste at opnå korrekte måleresultater for (Davidson et al. 1986). Korrektion for forskelle i kropsstørrelse mellem dyr og mennesker på basis af legemsvægt bygger på en antagelse om, at legemsvægten er et godt relativt mål for de faktorer, der er bestemmende for koncentrationen af et givent stof i blodet (Vermeire et al. 1999). I dyreforsøg angives den daglige eksponering ofte i enheden mg/kg legemsvægt per dag (mg/kg lgv./dag), og den ækvivalente humane dosis, udtrykt i mg/kg lgv./dag, vil således være identisk med dosis i dyreforsøget. Denne form for dosiskorrektion, hvor der korrigeres forholdsmæssigt med hensyn til kropsvægten på individniveau, vurderes i en vis grad at tage hensyn til forskelle i kropsstørrelse mellem forskellige species (for eksempel dyr og mennesker), men er også blevet kritiseret som værende utilstrækkelig. Mere end 100 forskellige biologiske parametre (morfologiske, fysiologiske, biokemiske, farmakologiske og toksikologiske) har vist sig at kunne udtrykkes som en matematisk funktion af legemsvægten, den såkaldte allometriske ligning, og denne sammenhæng synes at være konsistent på tværs af en lang række species (Davidson et al.1986, Voisin et al. 1990, Calabrese et al. 1992):
For parametrene legemsoverfladeareal (engelsk: body surface area – i det efterfølgende benævnt overfladeareal) samt stofskifte (engelsk: metabolic rate eller caloric requirement/demand) er der fundet en proportional sammenhæng med legemsvægten med eksponenten n lig med 0,67 for overfladeareal og lig med 0,75 for stofskiftet (Davidson et al.1986, Vermeire et al. 1999, Feron et al. 1990). Anvendelse af disse 2 parametre som udgangspunkt for dosiskorrektion for forskelle i kropsstørrelse mellem dyr og mennesker beskrives nærmere i 4.4.1.2.2 (overfladeareal) og 4.4.1.2.3 (stofskifte). Hvorvidt dosis bør korrigeres for forskelle i kropsstørrelse ved allometrisk skalering, afhænger, som tidligere nævnt, af eksponeringsvejen anvendt i dyreforsøget. Således anvendes dosiskorrektion ved allometrisk skalering ved udgangspunkt i studier med oral administration og dermal applikation, men ikke med inhalation. 4.4.1.2.1 Dosiskorrektion på basis af legemsvægt Når korrektion for forskelle i kropsstørrelse mellem dyr og mennesker foretages på basis af legemsvægten (W), er eksponenten n i den allometriske ligning lig med 1, og den ækvivalente humane dosis (Hdosis - udtrykt i samme enhed som Adosis) kan beregnes ud fra dosis i et dyreforsøg (Adosis - udtrykt i f.eks. enheden mg) i henhold til følgende ligning:
Der ligger implicit i dette udtryk, at når dosis i dyreforsøget er udtrykt i enheden mg/kg legemsvægt, er den ækvivalente dosis for mennesker (udtrykt i mg/kg legemsvægt) lig med dosis i dyreforsøget, det vil sige, at der ikke anvendes en skaleringsfaktor, da denne er lig med 1. Dermed kan sammenhængen beskrives som følger:
Dosiskorrektion på basis af legemsvægt illustreres i det følgende eksempel: NOAEL er vurderet til 1 mg i et studie med rotter (vægt 250 g), svarende til 4 mg/kg legemsvægt. Den ækvivalente dosis for mennesket (vægt 70 kg) beregnes til 280 mg (1 mg x [70 kg / 0,25 kg]), svarende til 4 mg/kg legemsvægt (280 mg / 70 kg). 4.4.1.2.2 Dosiskorrektion på basis af overfladeareal Som tidligere nævnt er det blevet kritiseret, hvorvidt anvendelse af legemsvægten som udgangspunkt ved korrektion af dosis for forskelle i kropsstørrelse mellem dyr og mennesker i tilstrækkelig grad tager højde for disse forskelle i kropsstørrelse. De hyppigst anvendte forsøgsdyr er små gnavere, og deres stofskifte (engelsk: metabolic rate), karakteriseret ved iltforbrug og varmeproduktion, er en del anderledes end menneskers, i hvert fald kvantitativt. Små individer har i forhold til legemsvægten et højere iltforbrug end større individer, hvilket betyder, at stofskiftet (udtrykt per kg legemsvægt) stiger i takt med faldende legemsvægt. Derimod har det vist sig, at når stofskiftet udtrykkes per enhed overfladeareal, så er der en direkte proportional sammenhæng mellem forskellige dyrearter og mennesker. (Davidson et al. 1986). Tilbage i 1966 analyserede Freireich et al. (citeret i Davidson et al.1986, Calabrese et al. 1992, Grönlund 1992) interspeciesforskelle i følsomhed for toksiske effekter med udgangspunkt i korrektion af dosis for forskelle i kropsstørrelse mellem individer baseret på legemsvægt (W1,0 i den allometriske ligning) eller på overfladeareal (W0,67 i den allometriske ligning). Standardiseringen blev foretaget for 18 forskellige cancerkemoterapeutika givet til mus, rotte, hamster, hund, abe, og mennesker. Resultaterne af disse analyser ledte til den konklusion, at de toksiske effekter af et givent stof var sammenlignelig på tværs af species, når dosis blev korrigeret på basis af overfladearealet. Dourson & Stara (1983) analyserede anvendelsen af overfladearealet frem for legemsvægten som basis for dosiskorrektion for forskelle i kropsstørrelsen og fandt, at dosiskorrektion på basis af overfladeareal mere præcist afspejlede forskelle i en række biologiske parametre mellem dyrearter sammenlignet med dosiskorrektion på basis af legemsvægt. Når dosis udtrykt på basis af overfladeareal blev omregnet til en tilsvarende dosis udtrykt i mg/kg legemsvægt, så syntes species med høj legemsvægt (f.eks. mennesket) at være mere følsomme for toksiske effekter end species med lavere legemsvægte (f.eks. gnavere). Forskellen i følsomhed var op til en faktor 15 med mennesker i den ene ende af skalaen og mus i den anden ende. Renwick (1999a) har vurderet, at de fleste fysiologiske og mange biokemiske processer korrelerer bedre med overfladearealet end med legemsvægten. For eksempel er der for stoffer, som metaboliseres eller hvor udskillelse (clearance) primært er afhængig af blodgennemstrømningen til eliminationsorganerne, en væsentlig forskel mellem dosis udtrykt på basis af overfladearealet og dosis udtrykt på basis af legemsvægten, når der sammenlignes mellem gnavere og mennesker. Dosiskorrektionen er således tæt relateret til speciesforskelle i basalstofskiftet og blodgennemstrømningen til organerne mellem mennesker og disse species. Med henblik på konvertering af en dosis udtrykt på basis af legemsvægt til en dosis på basis af overfladeareal, bør der ifølge forfatteren anvendes en faktor på 3-4 for rotter og 8-10 for mus. Calabrese et al. (1992) har vurderet, at dosiskorrektion på basis af overfladearealet er en mere konservativ tilgang end dosiskorrektion på basis af legemsvægt. Det er endvidere anført, at jo mere en dyreart nærmer sig de menneskelige dimensioner i relation til legemsvægt og overfladeareal, jo mindre forskel er der mellem dosiskorrektion på basis af overfladeareal og dosiskorrektion på basis af legemsvægt. Ifølge forfatterne synes dosiskorrektion på basis af overfladearealet at tage højde for visse kinetiske forskelle som for eksempel interspeciesforskelle i blodgennemstrømning af organer eller til enzymatiske parametre som Km og Vmax for stoffer, som metaboliseres. Derimod synes det ifølge forfatterne klart, at dosiskorrektion på basis af overfladeareal ikke tager højde for alle forskelle i kinetiske og dynamiske processer. For eksempel tager denne form for dosiskorrektion ikke højde for interspeciesvariation som følge af forskelle i absorptionseffektivitet, tykkelse af epidermis, antal hår per kvadratcentimeter på huden, tilstedeværelse og mængde af tarmflora, den relative dominans af oxidative og conjugative metaboliseringsveje, eller hastigheden af den biliære udskillelse. Vigtigheden af disse faktorer er forskellige fra stof til stof og varierer fra at være stort set ligegyldig til at være kritisk. Ovenstående vurderinger taler således for en anvendelse af overfladeareal (W0,67) som udgangspunkt ved korrektion af dosis for forskelle i kropsstørrelsen i stedet for legemsvægten. US-EPA har tidligere taget udgangspunkt i overfladearealet ved dosiskorrektion, men er nu gået over til at tage udgangspunkt i stofskiftet (W0,75), se nedenfor. Den allometriske ligning der relaterer overfladearealet til legemsvægten kan udtrykkes ved SA = bW0,67, hvor SA er overfladeareal, b er en species-specifik konstant, og W er legemsvægten. Når dosiskorrektion for forskelle i kropsstørrelse mellem dyr og mennesker foretages på basis af overfladearealet, er eksponenten n i den allometriske ligning lig med 0,67, og den ækvivalente humane dosis (Hdosis - udtrykt i samme enhed som Adosis) kan beregnes ud fra dosis i et dyreforsøg (Adosis - udtrykt i f.eks. mg) ud fra følgende ligning:
Er dosis i dyreforsøget (Adosis) udtrykt i enheden mg/kg legemsvægt, beregnes Hdosis (udtrykt i samme enhed som Adosis) ud fra følgende ligning:
Af ligning 2 følger, at når en dosis i et dyreforsøg er udtrykt i enheden mg/kg legemsvægt, er den ækvivalente dosis (udtrykt i mg/kg legemsvægt) for mennesket (vægt 70 kg) lig med dosis i dyreforsøget divideret med en skaleringsfaktor. Skaleringsfaktorerne er således afhængige af legemsvægten for det pågældende species anvendt i et givent dyreforsøg såvel som af legemsvægten sat for mennesket. I forhold til rotter (vægt 250 g) er skaleringsfaktoren [WH / WA]0,33 6,4; mens faktoren for mus (vægt 35 g) tilsvarende er 12,3. Dosiskorrektion på basis af overfladeareal illustreres i det følgende eksempel: Baseret på ligning 1: NOAEL er vurderet til 1 mg (Adosis) i et studie med rotter (vægt 250 g), svarende til 4 mg/kg legemsvægt. Den ækvivalente dosis for mennesket (Hdosis) (vægt 70 kg) beregnes til 43,6 mg (1 mg x [70 kg / 0,25 kg]0,67), svarende til 0,62 mg/kg legemsvægt (43,6 mg / 70 kg). Skaleringsfaktoren er således 6,4 (4 mg/kg legemsvægt / 0,6 mg/kg legemsvægt). Baseret på ligning 2: Den ækvivalente dosis for mennesket (Hdosis) (vægt 70 kg) beregnes til 0,62 mg/kg legemsvægt) (4 mg/kg legemsvægt / [70 kg / 0,25 kg]0,33). Faktoren i den firkantede parentes er lig med skaleringsfaktoren, i dette tilfælde 6,4. 4.4.1.2.3 Dosiskorrektion på basis af stofskifte Feron et al. (1990) har ved korrektion af orale doser for forskelle i kropsstørrelse mellem dyr og mennesker for metaboliserbare stoffer anbefalet at tage udgangspunkt i stofskiftet (W0,75 i den allometriske ligning), da dette blev vurderet at være en mere meningsfuld basis for dosiskorrektionen. Også US-EPA har for nyligt vurderet stofskiftet som værende en mere relevant parameter som udgangspunkt for dosiskorrektion end legemsvægt og overfladeareal, og er gået over til at anvende denne som basis for dosiskorrektion i de seneste år. Ligeledes har Vermeire et al. (1999) vurderet på baggrund af den nuværende viden samt egne analyser, at skalering på basis af stofskiftet med henblik på dosiskorrektion af orale doser for forskelle i kropsstørrelse mellem dyr og er mere korrekt sammenlignet med korrektion på basis af legemsvægt. Når dosiskorrektion for forskelle i kropsstørrelse mellem dyr og mennesker foretages på basis af stofskiftet, er eksponenten n i den allometriske ligning lig med 0,75, og den ækvivalente humane dosis (Hdosis - udtrykt i samme enhed som Adosis) kan beregnes ud fra dosis i et dyreforsøg (Adosis - udtrykt i f.eks. mg) ud fra følgende ligning:
Er dosis i dyreforsøget (Adosis) udtrykt i enheden mg/kg legemsvægt, beregnes Hdosis (udtrykt i samme enhed som Adosis) ud fra følgende ligning:
Af ligning 2 følger, at når en dosis i et dyreforsøg er udtrykt i enheden mg/kg legemsvægt, er den ækvivalente dosis (udtrykt i mg/kg legemsvægt) for mennesket (vægt 70 kg) lig med dosis i dyreforsøget divideret med en skaleringsfaktor. Skaleringsfaktorerne er således afhængige af legemsvægten for det pågældende species anvendt i et givent dyreforsøg såvel som af legemsvægten sat for mennesket. For forskellige species med forskellige legemsvægte kan der således beregnes forskellige skaleringsfaktorer med henblik på korrektion af dosis for forskelle i kropsstørrelse mellem dyr og mennesker baseret på stofskiftet, se tabel 4.4.1.2.3. Det fremgår således, at den ækvivalente dosis for mennesker (udtrykt i mg/kg legemsvægt, gennemsnitlig legemsvægt på 70 kg), bliver ca. 4 gange mindre end dosis for rotter og 6-7 gange mindre end dosis for mus, når doser i dyreforsøg er udtrykt i mg/kg legemsvægt. Dosiskorrektion på basis af stofskiftet illustreres i det følgende eksempel: Baseret på ligning 1: NOAEL er vurderet til 1 mg (Adosis) i et studie med rotter (vægt 250 g), svarende til 4 mg/kg legemsvægt. Den ækvivalente dosis for mennesket (Hdosis) (vægt 70 kg) beregnes til 68,4 mg (1 mg x [70 kg / 0,25 kg]0,75), svarende til 0,98 mg/kg legemsvægt (68,4 mg / 70 kg). Skaleringsfaktoren er således 4,0 (4 mg/kg legemsvægt / 0,98 mg/kg legemsvægt). Baseret på ligning 2: Den ækvivalente dosis for mennesket (Hdosis) (vægt 70 kg) beregnes til 0,98 mg/kg legemsvægt) (4 mg/kg legemsvægt / [70 kg / 0,25 kg]0,25). Faktoren i den firkantede parentes er lig med skaleringsfaktoren, i dette tilfælde 4,0. Tabel 4.4.1.2.3: Skaleringsfaktorer for forskellige dyrearter baseret på stofskiftet som udtryk for kropsstørrelsen. Når dosis for en given species er udtrykt i mg/kg legemsvægt divideres med den pågældende skaleringsfaktor for at få den ækvivalente humane dosis (udtrykt i mg/kg legemsvægt), gennemsnitlig legemsvægt for mennesket sat til 70 kg.
4.4.1.2.4 Dosiskorrektion i inhalationsstudier Ved eksponering via inhalation er forudsætningerne for korrektion af doser i dyreforsøg til ækvivalente doser i mennesker anderledes end ved eksponering via oral administration og dermal applikation. Endvidere er forudsætningerne også anderledes afhængigt af, om der er tale om systemiske og/eller lokale effekter som følge af eksponeringen. For mennesker og dyr er indåndingsvolumen over tid relateret til behovet for ilt og dermed også til det enkelte individs kaloriebehov og dermed stofskiftet. Dette betyder, at et individ automatisk eksponeres for et givent stof i indåndingsluften afhængigt af kaloriebehovet. I praksis betyder dette, at ved eksponering for stoffer, der via inhalation udløser systemiske effekter, er det ikke nødvendigt at foretage en dosiskorrektion for forskelle i kropsstørrelse mellem dyr og mennesker. (Vermeire et al. 1999, Feron et al. 1990, van Genderen 1988). For eksempel vil dosis ved en koncentration på 50 ppm af et stof i indåndingsluften for rotter, mus eller hunde være ækvivalent for mennesker, hvis dosis beregnes i forhold til stofskiftet. 4.4.1.2.4.1 Øvrige forhold, inhalation For stoffer, der udløser lokale effekter i luftvejene (f.eks. irritanter), kan der, på grund af de anatomiske og fysiologiske forskelle i luftvejene mellem dyr og mennesker, ikke siges noget generelt med hensyn til anvendelse af en skaleringsfaktor ved korrektion for forskelle i kropsstørrelse mellem dyr og mennesker. Det skal dog understreges her, at det for lokale effekter sædvanligvis anses at være stoffets koncentration i luften og ikke den samlede dosis som sådan, der er af betydning for udvikling af disse effekter, hvorfor det i disse tilfælde sædvanligvis ikke vil være relevant at korrigere for forskelle i kropsstørrelse. Ved ekstrapolation af doser i inhalationsstudier er der også andre forskelle mellem forsøgsdyr og mennesker, der skal tages i betragtning ved vurdering af resultaterne. Inhalation af luft sker primært gennem næsen, men mennesker og visse dyrearter (hunde og nogle højere aber) kan også trække vejret gennem munden. De øvre luftveje har følgende funktioner: 1) filtrerer, befugter og varmer luften; 2) fjerner uopløselige partikler ved hjælp af den såkaldte mucociliære elevator; 3) har en betragtelig metabolisk kapacitet; og 4) detekterer duftstoffer, sensoriske irritanter og immunogener via lugtesansen, trigeminalsystemet og immunsystemet respektivt. Hos mennesker kan luftvejene betragtes som bestående af fire afsnit. Hvert af disse afsnit har særlige epithelceller, interstitium og forsvarsmekanismer. Nasopharynx (området fra næseborene til stemmebåndene). Trachea og bronchier fører luft til og fra alveolerne. Overfladen er omkring 0,25 til 0,5 m². Tilsammen benævnes disse to første afsnit de konduktive luftveje. Lungeparenkymet indeholder det respiratoriske afsnit og har et overfladeareal på ca. 100 m². Blodgennemstrømningen i lungerne er ca. 5 gange større end i andre væv. Pleurarummet udgør i sig selv et toksikologisk kompartment. Sammenlignet med andre pattedyr har mennesket en relativ simpelt konstrueret næse. Menneskets næse har 3 næsemuslinger (engelsk: nasal turbinates), som er relativt simple i formen sammenlignet med næsemuslingerne i de fleste ikke-primater, hvor disse har komplekse foldninger og forgreninger. Grunden til denne meget komplekse opbygning af næsemuslingerne hos mange dyrearter skyldes, at lugtesansen hos dyr er meget højere udviklet end hos mennesket. Disse forskelle i luftvejene mellem dyr og mennesker betragtes som værende forskelle på detailplan mellem basalt identiske systemer. Endelig har gnavere også evnen til at nedsætte indåndingsfrekvensen og gør det i stor udstrækning ved eksponering for blandt andet irritative og lugtpotente stoffer, hvilket kan medføre en betydelig reduktion i indåndet dosis. Alt i alt bidrager forskellene mellem luftvejene hos dyr og mennesker alle til, at den resulterende eksponeringen af forsøgsdyrene kan være væsentligt forskellig fra en tilsvarende eksponering hos mennesker, selvom dyr og mennesker udsættes for den sammen koncentration af et givent stof i indåndingsluften. Betydningen af disse forskelle skal således tages i betragtning ved vurdering af resultaterne af inhalationsstudier i forsøgsdyr. 4.4.1.2.5 PBPK / PBTK modeller Det overordnede formål med udvikling af disse modeller er at modellere sammenhængen mellem eksponeringen og koncentrationen af det aktive stof (eller den aktive metabolit) i målorganet hos mennesker ud fra data fra dyreforsøg. Hvis der er tale om væsentlige kinetiske forskelle (absorption, fordeling, metabolisme og udskillelse), mellem de enkelte arter, inklusive mennesket, vil det være mest hensigtsmæssigt at gennemføre mere sofistikerede beregninger for at sammenligne de konkrete eksponeringsniveauer. I disse tilfælde kan der anvendes de såkaldte fysiologisk baserede kinetiske modeller (engelsk: Physiologically Based PharmacoKinetic/ToxicoKinetic – PBPK / PBTK). Modellerne opstilles med udgangspunkt i blandt andet tidsmæssige ændringer i stoffets koncentration i blod og urin og indhold af stoffet og dets metabolitter i forskellige organer på forskellige tidspunkter. Disse data kan benyttes til matematiske beregninger (compartment analyser) af den relative fordeling af stoffet og dets metabolitter i kroppen og de dertil knyttede biologiske halveringstider i såkaldte 'compartments'. Fordelen ved denne metode er, at den kan give oplysninger om den interne dosis af et stof i et målorgan, ligesom modellen kan videreudvikles til også at inkludere bestemte reaktioner i cellerne. Blandt ulemperne kan nævnes, at der for de færreste stoffer er tilstrækkelige data til udvikling af modellerne, modellerne tager lang tid at udvikle, modellerne er ikke bedre end deres forudsætninger, det er vanskeligt at beregne betydningen af forskellige usikkerheder, og de fleste tager ikke hensyn til individforskelle. 4.4.1.2.6 Sammenfatning, dosiskorrektion for forskelle i kropsstørrelse Ved ekstrapolation af data fra dyr til mennesker indgår korrektion af doser anvendt i dyreforsøg for forskelle i kropsstørrelse mellem forsøgsdyr og mennesker, såkaldt allometrisk skalering. Rationalet for skalering er funderet på ligheder i anatomisk henseende samt den universelle karakter i fysiologiske funktioner og i biokemiske reaktioner på tværs af arterne på trods af den store forskel i størrelse, facon og form mellem forskellige arter. Hvorvidt korrektion af dosis for forskelle i kropsstørrelse ved allometrisk skalering bør foretages eller ej afhænger af eksponeringsvejen anvendt i dyreforsøget. Dosiskorrektion ved allometrisk skalering anvendes således ved udgangspunkt i studier med oral administration og dermal applikation, men ikke med inhalation. Almindeligvis anvendes legemsvægten (W) som et udtryk for kropsstørrelsen, og korrektion for forskelle i kropsstørrelse mellem dyr og mennesker foretages sædvanligvis på basis af legemsvægt (W1,0). Denne form for dosiskorrektion vurderes i en vis grad at tage hensyn til forskelle i kropsstørrelse mellem dyr og mennesker, men er også blevet kritiseret som værende utilstrækkelig. Dosiskorrektion for forskelle i kropsstørrelse på basis af overfladeareal eller på basis af stofskiftet er blevet vurderet som værende et bedre grundlag end på basis af legemsvægten. På baggrund af den nuværende viden kan det imidlertid ikke afklares, hvorvidt dosiskorrektion på basis af overfladeareal eller på basis af stofskiftet vil være den mest korrekte tilgang. Men internationalt hælder man efterhånden mest til dosiskorrektion på basis af stofskiftet (W0,75) frem for overfladearealet (W0,67), idet det vurderes, at denne parameter tilsyneladende bedre beskriver interindividuelle relationer. Således er US-EPA i de seneste år gået over til at anvende stofskiftet som basis for dosiskorrektion (US-EPA 1998). Skaleringsfaktorer er angivet i 4.4.1.2.2 og 4.4.1.2.3. 4.4.1.3 Rationale for (u)sikkerhedsfaktor ILangt de fleste data vedrørende et stofs toksiske effekter stammer fra dyreforsøg, som oftest er udført med gnavere. Datagrundlaget for et givent stof giver således sjældent kendskab til, hvorvidt mennesker er mere eller mindre følsomme for det pågældende stof end de(t) anvendte forsøgsdyr. Derfor har det internationalt i risikovurderingssammenhænge været accepteret at antage, at mennesker kan være mere følsomme end forsøgsdyr og at kompensere herfor ved anvendelse af en (u)sikkerhedsfaktor, som regel af størrelsesordenen 10. Rationalet for denne størrelsesorden kendes ikke (se indledningen til 4.4). Også i relation til fastsættelse af kvalitetskriterier for luft, jord og vand har denne del (SFI) af den samlede sikkerhedsfaktor historisk været sat til 10. Men såfremt der har foreligget veldokumenteret viden om menneskets følsomhed i forhold til forsøgsdyr, har denne faktor dog kunnet sættes højere eller lavere end standardværdien på 10. (MST 1990). Som nævnt afspejler interspeciesvariationer dels forskelle i toksikokinetik og dels forskelle i toksikodynamik. På baggrund heraf har en yderligere opdeling af den traditionelle 10-faktor været foreslået (Renwick 1993) med henblik på at kunne modificere denne faktor ved at inddrage specifikke toksikokinetiske og/eller toksikodynamiske data. Renwick's analyser af de tilgængelige data vedrørende forskelle i toksikokinetik og toksikodynamik mellem mennesker og de mest anvendte forsøgsdyr (rotte, mus, hund) indikerede, at der tilsyneladende er større forskelle i toksikokinetik end i toksikodynamik. Renwick anbefalede derfor en opdeling af 10-faktoren i en standardværdi på 4 for toksikokinetiske forskelle og en standardværdi på 2.5 for toksikodynamiske forskelle. Denne opdeling af usikkerhedsfaktoren, som har været anvendt i nogle vurderinger og blandt andet er blevet foreslået af WHO (1994), har endnu ikke fundet bred anvendelse, men forventes mere udbredt i fremtiden i takt med den stigende mængde data til belysning heraf. Med henblik på fastsættelse af ADI analyserede Dourson & Stara (1983) den traditionelle 10-faktor og fandt, at 10-faktoren synes at give en rimelig sikkerhedsmargen for interspeciesvariation under forudsætning af, at en antagelse af dosisækvivalens mellem species i forhold til overfladeareal er korrekt. Som beskrevet tidligere (4.4.1.2.2), fandt Dourson & Stara ved en analyse af anvendelsen af overfladearealet ved dosiskorrektion for kropsstørrelsen, at når en dosis udtrykt i en enhed for overfladeareal konverteres til den korresponderende dosis udtrykt i mg/kg legemsvægt, så synes species med høj legemsvægt (f.eks. mennesket) at være mere følsomme for toksiske effekter end species med lavere legemsvægte (f.eks. gnavere). Forskellen i følsomhed var op til en faktor 15 med mennesker i den ene ende og mus i den anden ende. Det blev endvidere konkluderet, at den daværende viden om interspeciesforskelle synes at understøtte en standardværdi på 10 ved ekstrapolation fra dyr til mennesker på baggrund af legemsvægten. Grönlund (1992) undersøgte metoder anvendt ved kvantitativ risikovurdering af ikke genotoksiske stoffer med særlig fokus på valg af usikkerhedsfaktorer. Resultaterne tydede på, at mennesket i de allerfleste tilfælde er mere følsomme end forsøgsdyr, og at den traditionelle 10-faktor for interspeciesvariation i visse tilfælde er for lav til at tage højde for denne variabilitet. Det blev konkluderet, at der ikke var belæg for en standardværdi for interspeciesvariation, som kunne tage højde for alle typer af stoffer og alle forskellige forsøgsdyr. Ligegyldigt hvilken standardværdi der vælges, så vil den altid vise sig at være utilstrækkelig i visse tilfælde. WHO (1994, 1996) anbefalede generelt en standardværdi på 10 for interspeciesvariation, eventuelt opdelt i en kinetisk og en dynamisk delfaktor på henholdsvis 4 og 2,5, som foreslået af Renwick (1993). Det blev endvidere fremhævet, at i de situationer, hvor der foreligger veldokumenteret viden om toksikokinetiske og/eller toksikodynamiske forskelle mellem det givne forsøgsdyr og mennesker, bør der anvendes en dataspecifik faktor i stedet for standardværdi på 10. US-EPA (1998) anvender en usikkerhedsfaktor for interspeciesvariation (UFA) på 1, 3 eller 10, når der ved estimering af RfD tages udgangspunkt i valide resultater fra et langtidsstudie i forsøgsdyr. Standardværdien er 10. Valget af værdien for UFA foretages case-by-case ved en ekspertvurdering. Feron et al. (1990) har konkluderet, at menneskers følsomhed for eksponering for kemiske stoffer sandsynligvis ikke er særligt meget forskellig fra forskellige forsøgsdyrs følsomhed. Forfatterne anførte endvidere, at ekstrapolation af dosis fra dyr til mennesker på basis af legemsvægt giver anledning til en systematisk fejl og anbefalede at foretage dosiskorrektion på baggrund af kaloriebehovet, det vil sige legemsvægt0,75, og en samtidig reduktion af den traditionelle 10-faktor for interspeciesvariation. ECETOC (1995) har ligeledes konkluderet, at interspeciesvariationen kan beskrives udelukkende ved at anvende en dosiskorrektion baseret på kaloriebehovet (legemsvægt0,75) for doser angivet i mg/kg legemsvægt (se afsnit 4.4.1.2.3) og at den arbitrære ekstrapolationsfaktor for interspeciesvariation i konsekvens heraf kan sættes til mindre end 10. Med udgangspunkt i data for rotter vil ekstrapolationsfaktoren således være omkring 4 (se tabel 4.4.1.2.3). Calabrese et al. (1992) har imidlertid påpeget, at dosiskorrektion baseret på overfladeareal (legemsvægt0,67) ikke tager højde for alle former for interspeciesforskelle og derfor bør vurderes selvstændigt og ikke umiddelbart træde i stedet for usikkerhedsfaktoren for interspeciesvariation. Tilsvarende betragtninger gør sig sandsynligvis gældende for dosiskorrektion baseret på kaloriebehovet. Renwick (1999) konkluderede på baggrund af den nuværende viden, at det er naivt at forvente, at en enkel 10-faktor vil kunne tage højde for alle forskellene mellem forskellige forsøgsdyr og mennesker. Som et argument herfor har Renwick anført, at 10-faktoren anvendes ved vurderinger foretaget med udgangspunkt i dyreeksperimentelle studier, hvor korrektion af dosis for forskelle i kropsstørrelse mellem dyr og mennesker er foretaget på basis af legemsvægt, og dosis er dermed udtrykt i mg/kg legemsvægt, mens de fleste fysiologiske og mange biokemiske processer korrelerer bedre med overfladearealet. Ifølge Vermeire et al. (1999) kan interspeciesvariationen opdeles i metaboliske forskelle og i øvrige forskelle. Med henblik på at tage højde for metaboliske forskelle korrigeres dosis (ved oral og dermal applikation af det givne stof) for forskelle i kropsstørrelse mellem forsøgsdyr og mennesker (allometrisk skalering, se 4.4.1.2). Med henblik på at tage højde for øvrige interspeciesforskelle anvendes ofte en usikkerhedsfaktor af en given standardværdi. Forfatterne har analyseret den del af interspeciesvariationen, der vurderes ikke at være relateret til allometrisk skalering. Analysen blev foretaget med udgangspunkt i data for 184 stoffer undersøgt i forskellige species (mus, rotter og hunde) og via forskellige administrationsveje (oral og inhalation). Nul-effektniveauer blev udvalgt fra studier, hvor de tre species var eksponeret for samme stof, via samme administrationsvej, og med samme eksponeringsvarighed. To forskellige eksponeringsvarigheder blev endvidere analyseret, subakut og subkronisk. For orale nul-effektniveauer blev der foretaget dosiskorrektion på basis af stofskiftet og følgende ratios blev beregnet: NOAELmus/NOAELrotte, NOAELmus/NOAELhund og NOAELrotte/NOAELhund. For inhalationsdata blev følgende ratio beregnet: NOAELmus/NOAELrotte. Disse ratios blev vurderet med udgangspunkt i deres fordelinger. Af tabel 4.4.1.3 fremgår antallet af ratios, de geometriske gennemsnit og geometriske standardafvigelse, samt 90 og 95 percentilerne for fordelingerne af de enkelte ratios. I relation til dermal applikation var datagrundlaget for spinkelt til at kunne indgå i analysen. De analyserede data tyder på, at fordelingerne for disse ratios kan beskrives som værende log-normalfordelte. Hvis interspeciesvariationen udelukkende er afhængig af metaboliske forskelle, og hvis NOAELs er perfekte estimater af det sande nul-effektniveau (hvilket som bekendt ikke er tilfældet), er det geometriske gennemsnit og den geometriske standardafvigelse for fordelingen af de enkelte ratios lig med 1. Resultaterne af analyserne viste, at det geometriske gennemsnit for fordelingen for ratio NOAELmus/NOAELrotte og ratio NOAELmus/NOAELhund, men ikke for ratio NOAELrotte/NOAELhund, var tættere på 1, når der var foretaget dosiskorrektion på basis af stofskiftet sammenlignet med det geometriske gennemsnit for fordelingen for de tilsvarende ratios, når der ikke var foretaget dosiskorrektion på basis af stofskiftet (se tabel 4.4.1.3). Disse observationer støtter således i nogen grad det synspunkt, at interspeciesvariationen langt hen ad vejen skyldes metaboliske forskelle. Med henblik på at tilnærme de øvrige interspeciesforskelle (de forskelle der ikke skyldes metaboliske forskelle) ved ekstrapolation fra dyr til mennesker, blev gennemsnittet af fordelingsparametrene anvendt, det vil sige et geometrisk gennemsnit på omkring 1 og en geometrisk standardafvigelse på 6 (se tabel 4.4.1.3). Baseret på disse analyser, kan usikkerhedsfaktoren til korrektion for de interspeciesforskelle, der ikke skyldes metaboliske forskelle, karakteriseres som værende tilnærmelsesvis lognormal fordelt med et geometrisk gennemsnit på omkring 1 og en geometrisk standardafvigelse på 6. Baseret på denne fordeling blev værdier for denne usikkerhedsfaktor beregnet til 10, 19 og 65 for 90, 95 og 99 percentilerne respektivt. Disse resultater tyder således på, at den del af interspeciesvariationen, der ikke er relateret til metaboliske forskelle, i en række tilfælde ikke ligger inden for den traditionelle 10-faktor for interspeciesvariationen. Tabel 4.4.1.3. Fordelingsparametre for ratio mellem forskellige NOAELs, se tekst ovenfor. Modificeret fra Vermeire et al. (1999).
4.4.1.4 Sammenfatning (u)sikkerhedsfaktor IRent biologisk og biokemisk minder mennesker om andre pattedyr, også med hensyn til udvikling af forskellige typer af toksiske effekter. Men selv om testning af stoffer i forsøgsdyr i høj grad vil være prædiktiv for tilsvarende effekter i mennesker, kan mennesker være mere eller mindre følsomme end andre pattedyr over for toksiske stoffer. Forskellene i følsomhed afspejler dels forskelle i toksikokinetik og dels forskelle i toksikodynamik. Den generelle opfattelse er, at det især er forskelle i metabolismen af fremmedstoffer, der spiller den største rolle for interspeciesvariationen. Hertil kommer, at der findes forskellige former for organer og væv de enkelte species imellem (for eksempel har gnavere organer og væv, som mennesker ikke har), forskellige former for fordøjelse, samt anderledes struktur af de øvre luftveje, hvilket betyder, at visse effekter som udvikles i forsøgsdyr ikke nødvendigvis vil være relevante for mennesker. Langt de fleste data vedrørende et stofs toksiske effekter stammer fra dyreforsøg, og er som oftest udført med gnavere. Datagrundlaget for et givent stof giver således sjældent kendskab til, hvorvidt mennesker er mere eller mindre følsomme for det pågældende stof end de(t) anvendte forsøgsdyr. Derfor har det internationalt i risikovurderingssammenhænge været accepteret at antage, at mennesker kan være mere følsomme end forsøgsdyr og at kompensere herfor ved anvendelse af en (u)sikkerhedsfaktor, som regel af størrelsesordenen 10. Rationalet for denne størrelsesorden kendes ikke. Ekstrapolation af data fra dyr til mennesker kan opfattes som omhandlende to forskellige aspekter: 1) korrektion af dosis for forskelle i kropsstørrelse mellem forsøgsdyr og mennesker, såkaldt allometrisk skalering, og 2) andre former for forskelle mellem forsøgsdyr og mennesker, som ikke nødvendigvis afspejles i forskellene i kropsstørrelse. Hvorvidt dosis bør korrigeres for forskelle i kropsstørrelse ved allometrisk skalering afhænger af eksponeringsvejen anvendt i dyreforsøget. Dosiskorrektion ved allometrisk skalering anvendes således ved udgangspunkt i studier med oral administration og dermal applikation, men ikke med inhalation. Almindeligvis anvendes legemsvægten (W) som et udtryk for kropsstørrelsen, og korrektion for forskelle i kropsstørrelse mellem dyr og mennesker foretages sædvanligvis på basis af legemsvægt. Denne form for dosiskorrektion vurderes i en vis grad at tage hensyn til forskelle i kropsstørrelse mellem dyr og mennesker, men er også blevet kritiseret som værende utilstrækkelig. Dosiskorrektion for forskelle i kropsstørrelse på basis af overfladeareal (W0,67) eller på basis af stofskiftet (W0,75) er blevet vurderet som værende et bedre grundlag end på basis af legemsvægten. På baggrund af den nuværende viden kan det imidlertid ikke afklares, hvorvidt dosiskorrektion på basis af overfladeareal eller på basis af stofskiftet vil være den mest korrekte tilgang. Men internationalt hælder man efterhånden mest til dosiskorrektion på basis af stofskiftet frem for overfladearealet, idet det vurderes, at denne parameter tilsyneladende bedre beskriver interindividuelle relationer. Ved ekstrapolation af doser fra dyr til mennesker på basis af stofskiftet tages der i en vis grad hensyn til forskelle i forskellige arters stofskifte og dermed metaboliske omsætning og iltforbrug. I konsekvens heraf har der været fremført holdninger om at kompensere for interspeciesvariation udelukkende ved at korrigere dosis for forskelle i kropsstørrelse mellem det anvendte forsøgsdyr og mennesker på basis af stofskiftet. Således har ECETOC (1995) anbefalet, at den arbitrære ekstrapolationsfaktor for interspeciesvariation kan sættes til mindre end 10. Feron et al. (1990) har vurderet, at menneskers følsomhed for kemiske stoffer sandsynligvis ikke er særligt meget forskellig fra forskellige forsøgsdyrs følsomhed og har ligeledes anbefalet at foretage dosiskorrektion på basis af stofskiftet og en samtidig reduktion af den traditionelle 10-faktor for interspeciesvariation. Calabrese et al. (1992) har imidlertid sat spørgsmålstegn ved en generel anvendelse af overfladearealet til korrektion af dosis for forskelle i kropsstørrelse mellem dyr og mennesker og har påpeget, at dosiskorrektion på basis af overfladeareal ikke tager højde for alle de mest relevante faktorer i relation til interspeciesvariation. Forfatterne anbefalede således, at dosiskorrektion på basis af overfladeareal derfor bør vurderes selvstændigt og ikke umiddelbart træde i stedet for en anvendelse af usikkerhedsfaktoren for interspeciesvariation. Tilsvarende betragtninger vurderes sandsynligvis at gøre sig gældende for dosiskorrektion baseret på kaloriebehovet. Ifølge Vermeire et al. (1999) kan interspeciesvariationen opdeles i metaboliske forskelle og i øvrige forskelle. Forfatterne har analyseret den del af interspeciesvariationen, der ikke vurderes som værende relateret til metaboliske forskelle, og resultaterne af disse analyser tyder på, at denne del af interspeciesvariationen i en række tilfælde ikke ligger inden for den traditionelle 10-faktor for interspeciesvariation. Sammenfattende understøtter de tilgængelige data, at der anvendes en standardværdi på 10 for interspeciesvariation ved udgangspunkt i dyreeksperimentelle studier, hvor korrektion af dosis for forskelle i kropsstørrelse mellem dyr og mennesker foretages på basis af legemsvægt. Men det ligger også klart, at denne 10-faktor ikke vil kunne tage højde for alle typer af stoffer, effekter og forsøgsdyr. Data indikerer endvidere, at korrektion af orale og dermale doser i dyreeksperimentelle studier for forskelle i kropstørrelse på basis af stofskiftet (legemsvægt0,75) giver et mere korrekt dosisestimat hos mennesker end ved korrektion foretaget på basis af legemsvægt. Denne procedure vurderes at finde bredere anvendelse internationalt i fremtiden. I relation til fastsættelse af kvalitetskriterier for luft, jord og vand anbefales det fortsat at foretage dosiskorrektion på basis af legemsvægt og i konsekvens heraf en standardværdi på 10 for interspeciesvariation (UFI), eventuelt opdelt i en kinetisk og en dynamisk delfaktor, som foreslået af Renwick (1993) og af WHO (1994). Såfremt der foreligger veldokumenteret viden om toksikokinetiske og/eller toksikodynamiske forskelle mellem det givne forsøgsdyr og mennesker, anbefales det at tage udgangspunkt i denne viden med henblik på fastsættelse af en dataspecifik faktor i stedet for anvendelse af en standardværdi på 10. Anbefalingerne skal ses i lyset af, at der er data, der tyder på, 1) at kompensation for interspeciesvariation udelukkende ved allometrisk skalering på basis af stofskiftet muligvis ikke tager højde for alle relevante faktorer i relation til interspeciesvariation, samt 2) at den del af interspeciesvariationen, der ikke omfattes af allometrisk skalering, i en række tilfælde ikke ligger inden for den traditionelle 10-faktor for interspeciesvariation. Det vurderes, at usikkerhederne i den anbefalede procedure sandsynligvis vil være relativt lav i forhold til øvrige usikkerheder i risikovurderingsproceduren. Det skal dog understreges, at det for visse stoffer og effekter samt for visse forsøgsdyr vurderes, at der sandsynligvis vil være større forskelle i følsomheden mellem forsøgsdyr og mennesker, og som dermed falder uden for denne 10-faktor. Omvendt vurderes det også, at for visse stoffer og effekter samt for visse forsøgsdyr vil der sandsynligvis være mindre forskelle i følsomheden mellem forsøgsdyr og mennesker, således at den traditionelle 10-faktor giver anledning til en mere konservativ vurdering. 4.4.2 Usikkerhedsfaktor II (”intraspecies- / interindividuel variation”)4.4.2.1 Biologisk variationPå baggrund af den biologiske variation, der findes mellem mennesker, er der også forskelle mellem enkelte individers følsomhed for et givent stof. Faktorer som alder, køn, graviditet, genotype, helbred, og livsstil kan være medvirkende til en øget biologisk følsomhed, som afspejler dels forskelle i toksikokinetik og dels i toksikodynamik. Den interindividuelle variation i biologisk følsomhed er nærmere belyst i Beltoft et al. (2001). Data viser, at der kan være interindividuelle forskelle både i enzymniveau og i enzymaktivitet, hvilket kan føre til interindividuelle forskelle i metabolisering af et givent stof. Hertil kommer, at der findes genetiske polymorfismer for en række enzymer, der er involveret i omsætning af fremmede stoffer, for eksempel for enzymer der tilhører cytochrom P450-familien, glutathion-S-transferase, paraoxonase, og acetylcholinesterase. Genetisk polymorfisme er arveligt betinget, og begrebet anvendes, hvor et enzym forekommer i flere former, som ikke nødvendigvis har samme kapacitet med hensyn til at omsætte et givent substrat. Den sundhedsmæssige betydning af variationer i enzymniveau og enzymaktivitet samt genetisk polymorfiske enzymer kendes ikke i detaljer. (Beltoft et al. 2001, Grandjean 1997). Det ufødte barn samt spædbørn har vist sig at være mere følsomme overfor en række kemiske stoffer end voksne generelt. I denne aldersgruppe er hurtig vækst af organer og væv en vigtig faktor for den øgede følsomhed. Nogle organer, som for eksempel hjernen, udvikler sig langt op i barneårene og vil således have en øget sårbarhed overfor toksiske stoffer. Også udviklingen af reproduktionssystemet og immunforsvaret fortsætter i barnealderen og kan således medvirke til en øget sårbarhed hos denne aldersgruppe. Endvidere er receptorer og andre former for 'molekylære mål' også under udvikling hos spædbørn, hvilket kan medføre aldersrelaterede forskelle i resultatet af receptor-stof-interaktioner, herunder direkte modsatte effekter af et givent stof hos spædbørn og voksne. De toksikokinetiske forskelle gør sig primært gældende hos spædbørn. I relation til legemsvægten er det nyfødte barns overfladeareal næsten tre gange så stort som hos voksne. Da stofskiftet
tilnærmelsesvist er proportionalt med overfladearealet, har det nyfødte barn relativt et meget højere stofskifte og respiration, hvilket kan medføre øget optagelse ved eksponering for luftbårne stoffer. Også
den relative optagelse gennem tarmen er øget på grund af tarmepithelets øgede permeabilitet samt et højere pH i mavesækken. Både lever og nyrer er umodne på fødselstidspunktet, hvilket spiller en rolle for
metabolisering samt udskillelse af toksiske stoffer. Dette sidste aspekt kan både være en fordel og en ulempe afhængigt af, om det givne stof i sig selv udløser en toksisk effekt, eller om der skal ske en
omdannelse før udløsning af effekt. Hos ældre personer kan der forekomme et øget respons i visse væv og organer på grund af en mindre 'reservekapacitet' (engelsk: functional capacity in reserve) eller ændret receptorfølsomhed. Hvis en tidlig eksponering for fremmede stoffer har medført skader på sådanne væv og organer, kan disse skader afdækkes ved, at der med alderen ikke længere er nok kapacitet til at kompensere for dem. En sådan mekanisme spiller muligvis en rolle for visse degenerative sygdomme i nervesystemet som for eksempel parkinsonisme og amyotrof lateral sclerose. Et andet eksempel er lungefunktionen, som ganske langsomt mindskes med alderen, hvilket kan medføre en øget følsomhed for stoffer, som påvirker lungerne. Også evnen til at reparere inducerede skader kan være nedsat med alderen. For eksempel har nogle ældre vist sig at have en ringere modstandskraft mod peroxidation, fordi det antioxidative forsvar mindskes. Endvidere kan toksikokinetiske aspekter spille en rolle for ældres øgede følsomhed. Hos ældre ses blandt andet ændringer i P450-enzymsystemets evne til at omsætte toksiske stoffer. Også evnen til at
nedbryde og udskille toksiske stoffer kan være nedsat på grund af nedsat funktion af lever og nyrer. Det skal bemærkes, at et øget respons som følge af ændringer i receptorfølsomhed, reparationsmekanismer og reservekapacitet også kan være sygdomsinducerede. Interindividuelle forskelle mellem de to køn er også rapporteret. Som eksempel kan nævnes forskelle i enzymniveauer og enzymaktiviteter. For stoffer der ophobes i knoglevævet (bly og cadmium), kan der, som følge af det knogletab der ses efter overgangsalderen, måles en øget koncentration i blodet ved frigørelse af disse stoffer fra knoglerne. (Beltoft et al. 2001, Grandjean 1997). Kronisk syge og svækkede personer vil i mange tilfælde udvise større følsomhed overfor fremmede stoffer end den gennemsnitlige befolkning. Personer, der er sensibiliserede overfor et bestemt stof, har som reglen antistoffer i blodet mod dette stof og vil oftest reagere ved en efterfølgende eksponering for det samme stof ved lavere koncentration end den gennemsnitlige befolkning. Et andet eksempel er, at leversygdom kan føre til øget følsomhed, idet leverens kapacitet til at metabolisere fremmede stoffer vil være nedsat. Der har også været rapporteret en ændret receptorfølsomhed for visse stoffer i relation til udvikling af levercirrhose. Ligeledes kan nyreskader resultere i øget følsomhed, idet nyrernes evne til at udskille fremmedstoffer i så fald vil være nedsat. Endelig kan eksponering for fremmede stoffer tænkes at mindske organismens styrke til at modstå andre skadelige påvirkninger udefra. Måske vil der ikke umiddelbart opstå en påviselig skade, men den mindskede modstandskraft vil kunne komme til udtryk i form af øget følsomhed ved en efterfølgende eksponering. (Grandjean 1997, Beltoft et al. 2001). Der er også påvist markante forskelle i følsomhed i forbindelse med luftforureningens sundhedsskadelige effekter. Personer med kroniske luftvejslidelser og personer med hjerte-karlidelser er fundet at være særligt følsomme, idet øget sygelighed og dødelighed som følge af luftforureningen er særligt udtalt i disse grupper. Endvidere påvirkes småbørn og ældre i relativt større omfang end den øvrige normalbefolkning af luftforureningen. (Larsen et al. 1997, Nielsen et al. 2001). I de senere år har begrebet 'Multiple Chemical Sensitivity' (MCS) været diskuteret bredt i videnskabelige kredse. Begrebet omfatter et syndrom med nedsat tolerance og særlig følsomhed ved udsættelse for kemiske stoffer, således at en lang række effekter ses ved meget lave eksponeringer, som ellers ikke anses for problematiske. Syndromet MCS anføres at kunne opstå i tilslutning til en initial, massiv kemisk eksponering og er efterfølgende karakteriseret ved, at der opstår anfaldsvise gener fra flere organer i forbindelse med eksponering for kemiske lugte i lav koncentration. Lugte fra flere, ikke-beslægtede kemiske stoffer kan udløse generne, som forsvinder efter ophør med lugteksponering, men som kan fremkaldes ved fornyet eksponering. (MST 2001). 4.4.2.2 Rationale for (u)sikkerhedsfaktor IILangt de fleste data vedrørende et stofs toksiske effekter stammer som bekendt fra dyreforsøg. Datagrundlaget for et givent stof giver således sjældent kendskab til, hvorvidt det enkelte individ er mere eller mindre følsomt for det pågældende stof end den gennemsnitlige befolkning. Derfor har det internationalt i risikovurderingssammenhænge været accepteret at kompensere for interindividuel variation ved anvendelse af en (u)sikkerhedsfaktor, som regel af størrelsesordenen 10. Rationalet for denne størrelsesorden kendes ikke (se indledningen til 4.4). Også i relation til fastsættelse af kvalitetskriterier for luft, jord og vand har denne del (SFII) af den samlede sikkerhedsfaktor oftest været sat til 10. Men såfremt der har foreligget veldokumenteret viden om en specifik gruppes særlige følsomhed i forhold til den gennemsnitlige befolkning, har denne faktor dog kunnet sættes højere eller lavere end standardværdien på 10. (MST 1990). Som nævnt tidligere (4.4.2.1) afspejler interindividuelle variationer dels forskelle i toksikokinetik og dels forskelle i toksikodynamik. På baggrund heraf har en yderligere opdeling af den traditionelle 10-faktor været foreslået (Renwick 1993) med henblik på at kunne modificere denne 10-faktor ved inddragelse af specifikke toksikokinetiske og/eller toksikodynamiske data. Renwick's analyser af de tilgængelige data vedrørende forskelle i toksikokinetik og toksikodynamik mellem mennesker indikerede, at der ikke er belæg for den skæve opsplitning af 10-faktoren, som det er tilfældet ved interspeciesvariation (se 4.4.1.3). Renwick anbefalede derfor en opdeling af 10-faktoren i to standardværdier på 3,16 (100,5) for dels toksikokinetiske forskelle såvel som for toksikodynamiske forskelle. Denne opdeling af (u)sikkerhedsfaktoren, som blandt andet er blevet foreslået af WHO (1994), har endnu ikke fundet bred anvendelse i den internationale verden. Den interindividuelle variation mellem mennesker og mellem forsøgsdyr er nærmere belyst i Beltoft et al. (2001) med henblik på at vurdere størrelsesordenen af usikkerhedsfaktor II. De centrale studier summeres kort i det efterfølgende. Calabrese (1985) har undersøgt variationer i humant respons indenfor 1) metabolisme af fremmedstoffer, 2) binding af toksiske stoffer til for eksempel hæmoglobin og DNA, 3) aktiviteten af udvalgte enzymer, og 4) risiko for udvikling af sygdomme i den humane population. Resultaterne af disse undersøgelser indikerede, at den interindividuelle variation for 80 til 95% af populationen lå inden for en faktor 10, mens variationen for den resterende del af populationen var højere end en faktor 10 og i nogle tilfælde op til flere størrelsesordener højere. Hattis et al. (1987) vurderede den humane variabilitet for tre centrale farmakokinetiske parametre (halveringstiden for elimination (T½), arealet under plasmakoncentrationskurven (AUC), samt den maksimale koncentrationen i blodet (Cmax)). Vurderingerne blev foretaget med baggrund i 101 datasæt for 49 specifikke stoffer, hovedsageligt lægemidler, indsamlet i perioden 1979 til 1985. Analyserne viste, at en 10-folds forskel i disse parametre ville svare til 7-9 standardafvigelser, eller mere end 99,99% i en normalfordelt population. For kun et af de undersøgte stoffer (et lipofilt stof) var der en større variation i AUC og Cmax, hvor en 10-folds forskel ville svare til 2,5 standardafvigelser for disse parametre i den humane population, eller 98% i en normalfordelt population. På baggrund heraf blev det konkluderet, at den traditionelle 10-faktor for interindividuel variation er en ret konservativ standardværdi. Renwick og Lazarus (1998) analyserede den traditionelle 10-faktor med henblik på en evaluering af den tidligere foreslåede opdeling af 10-faktoren i en kinetisk og dynamisk delfaktor på 3,16 (Renwick 1993) såvel som tilstrækkeligheden (engelsk: adequacy) af denne 10-faktor. Data vedrørende kinetik og dynamik blev tabuleret, og gennemsnitlige variationskoefficienter blev bestemt for forskellige studier, som relaterede til et generelt (engelsk: common) end-point eller for multiple doser, som relaterede til det samme (engelsk: same) end-point. Data blev også analyseret med udgangspunkt i den rapporterede standardafvigelse og den beregnede geometriske standardafvigelse for hver parameter med henblik på at definere en såkaldt 'Z-score' og dermed at estimere den del af en normalfordelt eller log-normalfordelt population, som ville være omfattet af delfaktoren på 3,16. For vurdering af variationer i kinetikken blev data for 60 stoffer analyseret. Disse 60 stoffer repræsenterede en række forskellige metaboliseringsveje eller clearance. Den gennemsnitlige variationskoefficient for de kinetiske parametre var 38% med et minimum på 9% og et maksimum på 114%. I relation til variationer i dynamikken blev der analyseret koncentrations-effekt data (primært in vivo plasmakoncentrations-respons data fra behandling af patienter i klinikken) for 49 stofrelaterede effekter (primært ændringer i kardio-vaskulære eller centralnervesystemiske parametre). Den gennemsnitlige variationskoefficient for de dynamiske parametre var 51% med et minimum på 8% og et maksimum på 137%. Ifølge forfatterne støttede resultaterne af denne analyse således en opdeling af 10-faktoren med lige vægtning til kinetik og dynamik med en delfaktor på 3,16 (100,5) for kinetiske såvel som for dynamiske variationer. Data blev også analyseret med henblik på at vurdere, hvor stor en del af en population der ikke blev beskyttet af en delfaktor på 3,16 for kinetik henholdsvis dynamik. Antallet af individer per million i en population, der ikke var omfattet af en delfaktor på 3,16, var direkte proportional med standardafvigelsen på estimatet. For kinetikken var det gennemsnitlige antal af individer, der ikke var omfattet af en faktor på 3,16 fra gennemsnitsestimatet, 685 af en million (ca. 0,07%) ved normalfordeling og 8564 (ca. 0,9%) ved log-normalfordeling. For dynamikken var tallene 2930 af en million (ca. 0,3%) ved normalfordeling og 18896 (ca. 2%) ved log-normalfordeling. Sandsynligheden for at det samme individ falder udenfor både med hensyn til kinetik og til dynamik blev også analyseret under antagelse af, at kinetiske og dynamiske 'risikofaktorer' er uafhængige variable. Gennemsnitlig 2 individer af en million ville ikke være omfattet af den kombinerede faktor på 10 (3,16 gange 3,16) ved normalfordeling og 162 af en million ved log-normalfordeling. På baggrund af disse resultater konkluderede forfatterne, at den traditionelle 10-faktor er et hensigtsmæssig valg som standardværdi for de undersøgte stoffer og biologiske effekter, idet 10-faktoren vil dække mere end 99,9% af populationen uafhængigt af valg af fordeling (normalfordeling versus log-normalfordeling). Endvidere blev de ovenfor beskrevne analyser også foretaget med henblik på at undersøge, hvorvidt visse specifikke undergrupper af den samlede population ikke ville være omfattet, hvis delfaktoren på 3,16 blev anvendt på gennemsnittet for hovedparten af den samlede population. Følgende sammenligninger blev foretaget: 1) Undergruppen børn i forhold til voksne; 2) etniske forskelle; og 3) forskelle i metabolisme som følge af polymorfiske enzymer. Da størstedelen af de relevante data omhandlede kinetiske forhold for lægemidler hos kaukasiske voksne, blev denne gruppe anvendt som referencegruppe ved analyserne, og analyserne blev således kun foretaget i relation til de kinetiske aspekter af den interindividuelle variation. For undergruppen børn konkluderede forfatterne, at børn omfattes i tilstrækkelig grad af delfaktoren på 3,16 for kinetik anvendt på gennemsnittet for voksne og begrundede det med, at børn med hensyn til variationer inden for kinetik ikke generelt udgør en speciel gruppe. Forskellige etniske grupper kan udvise forskelle i både kinetik og dynamik, som følge af genetiske forskelle eller miljømæssige faktorer. I følge forfatterne er en delfaktor på 3,16 tilstrækkelig til at dække de fleste etniske grupper i den samlede population. Dog kan der være forskelle mellem forskellige etniske grupper på grund af forskelle i metabolisme som følge af polymorfiske enzymer, se efterfølgende. Genetisk polymorfisme inden for enzymer, der er involveret i metabolisering af fremmedstoffer, har stor betydning for den interindividuelle variation, og for eksempel personer med en meget langsom metabolisering af et givent stof vil ikke nødvendigvis være dækket af en delfaktor på 3,16 for forskelle i kinetik og dermed heller ikke nødvendigvis af den samlede faktor på 10 for interindividuel variation. På baggrund af alle de foretagne analyser konkluderede forfatterne, at en faktor 10 for interindividuel variation synes at være et hensigtsmæssig valg som standardværdi, idet denne 10-faktor vil dække mere end 99,9% af populationen. Men det er også understreget, at der er identificeret en række tilfælde, hvor denne 10-faktor er utilstrækkelig, for eksempel i relation til genetisk polymorfisme inden for enzymer, der er involveret i metabolisering af fremmedstoffer. I et forsøg på at give et videnskabeligt rationale for den traditionelle 10-faktor for interindividuelle forskelle med henblik på fastsættelse af ADI, analyserede Dourson og Stara (1983) data for akut toksicitet samlet af Weil (1972) for 490 kemikalier indgivet som enkeltdosis til rotter. For 92% af kemikalierne var en faktor på 10 tilstrækkelig til at dække tre standardafvigelser og dermed omfatte 99,9% af populationen. Forfatterne konkluderede, at analysen var en indirekte støtte til 10-faktoren for interindividuel variation. Støtten blev vurderet som værende indirekte, fordi effekterne (dødelighed versus nul-effektniveau) ikke er direkte sammenlignelige. Endvidere var analysen baseret på data fra rotter, og forsøgsdyr er generelt en mere homogen gruppe end den humane population. Grönlund (1992) undersøgte metoder anvendt ved kvantitativ risikovurdering af ikke genotoksiske stoffer med særlig fokus på valg af usikkerhedsfaktorer og konkluderede, at de humane data indikerede, at en 10-faktor for interindividuel variation omfattede størstedelen af populationen, men ikke alle. WHO (1994, 1996) har foreslået en standardværdi på 10 for interindividuel variation, eventuelt opdelt i en kinetisk og en dynamisk delfaktor på 3,16 til hver, som foreslået af Renwick (1993). Men WHO understregede også, at denne faktor ikke for alle stoffer vil yde en tilstrækkelig grad af beskyttelse og nævnte specifikt eksemplet med polymorfiske enzymer. Endvidere blev det anbefalet, at i de tilfælde, hvor der foreligger veldokumenteret viden om den interindividuelle variabilitet i toksikokinetik og i toksikodynamik, bør den traditionelle 10-faktor erstattes med en dataspecifik faktor. I nogle tilfælde kan denne faktor sættes til 1, hvis de tilgængelige data for et givent stof belyser følsomheden for alle identificerede følsomme undergrupper af populationen. US-EPA (1998) anvender en usikkerhedsfaktor for interindividuel variation (UFH) på 1, 3 eller 10, når der ved estimering af RfD tages udgangspunkt i valide data fra studier af gennemsnitsbefolkningen ved langtidseksponering. Standardværdien er 10. Valget af værdien for UFH foretages case-by-case ved en ekspertvurdering. Ifølge ECETOC (1995), må den interindividuelle variation blandt mennesker forventes at være større end inden for en given dyreart, og at en faktor mellem 1 og 10 vil være tilstrækkelig til at tage højde herfor. ECETOC's vurdering var derimod, at der ikke er noget videnskabeligt grundlag med henblik på en bestemt numerisk standardværdi og anbefalede en standardværdi på 3 (det geometriske gennemsnit af 1 og 10). Dourson et al. (1996) vurderede, at en standardværdi på 10 for interindividuel variation tilsyneladende er tilstrækkelig, når udgangspunktet for vurderingen er et observeret nul-effektniveau (NOAEL), der antages at dække den gennemsnitlige populationen. Hvis NOAEL estimeres for en kendt, følsom undergruppe, bør 10-faktoren justeres i henhold til denne viden. Kalberlah & Schneider (1998 - citeret i Vermeire et al. 1999) har på baggrund af de tilgængelige humane data foreslået en standardværdi på 25 for interindividuel variation inden for den almene humane population, bestående af en delfaktor på 8 til at tage højde for toksikokinetiske variationer og enzympolymorfier og en delfaktor på 3 til at tage højde for toksikodynamiske variationer. Burin & Saunders (1999) konkluderede med baggrund i undersøgelser af den humane variabilitet i respons (kliniske undersøgelser af lægemidler), at en faktor mellem 1 og 10 vil være tilstrækkelig til at beskytte mere end 99% af den humane population, herunder forskellige undergrupper i populationen, inklusive børn. Dette betyder ifølge forfatterne, at 10-faktoren kan reduceres, hvis intentionen er at beskytte op til 99% af populationen. Vermeire et al. (1999) konkluderede, at det for nuværende ikke er muligt at foreslå en databaseret fordeling for usikkerhedsfaktor II, som det er blevet foreslået for usikkerhedsfaktor I (se afsnit 4.4.1.3). Forfatterne foreslog derfor indtil videre at fastholde den traditionelle 10-faktor og antog, at denne faktor vil yde beskyttelse for hovedparten af den generelle befolkning. 4.4.2.3 Sammenfatning (u)sikkerhedsfaktor IIPå baggrund af den biologiske variation, der findes mellem mennesker, er der også forskelle mellem enkelte individers følsomhed for et givent stof. Faktorer som alder, køn, graviditet, genotype, helbred, og livsstil kan være medvirkende til en øget biologisk følsomhed, som afspejler dels forskelle i toksikokinetik og dels i toksikodynamik. For eksempel har det ufødte barn samt spædbørn vist sig at være mere følsomme overfor en række kemiske stoffer end voksne generelt, men der er også enkelte eksempler på, at det modsatte kan være tilfældet. I denne aldersgruppe er hurtig vækst af organer og væv en vigtig faktor for den øgede følsomhed. For nogle organer, som for eksempel hjernen, reproduktionssystemet og immunforsvaret, fortsætter udviklingen i barnealderen og kan således medvirke til en øget sårbarhed hos denne aldersgruppe. De toksikokinetiske forskelle gør sig primært gældende hos spædbørn og kan resultere i enten øget eller nedsat følsomhed i forhold til voksne. Hos ældre personer kan der forekomme et øget respons i visse væv og organer på grund af en mindre reservekapacitet eller ændret receptorfølsomhed. Også evnen til at reparere inducerede skader kan være nedsat med alderen. Endvidere kan toksikokinetiske aspekter også spille en rolle for ældres øgede følsomhed, for eksempel en nedsat evne til at nedbryde og udskille toksiske stoffer. Interindividuelle forskelle mellem de to køn er også rapporteret, som for eksempel forskelle i enzymniveauer og enzymaktiviteter. Kronisk syge og svækkede personer vil i mange tilfælde også udvise større følsomhed overfor fremmede stoffer end den gennemsnitlige befolkning. For eksempel vil sensibiliserede personer oftest reagere ved eksponering for det sensibiliserende stof ved lavere koncentration end den gennemsnitlige befolkning. Et andet eksempel er, at leversygdom kan føre til øget følsomhed, idet leverens kapacitet til at metabolisere fremmede stoffer vil være nedsat. Ligeledes kan nyreskader resultere i øget følsomhed, idet nyrernes evne til at udskille fremmedstoffer i så fald vil være nedsat. Der er også påvist markante forskelle i følsomhed i forbindelse med luftforureningens sundhedsskadelige effekter, hvor personer med kroniske luftvejslidelser og personer med hjerte-karlidelser er fundet at være særligt følsomme. Endvidere påvirkes småbørn og ældre i relativt større omfang end den øvrige normalbefolkning af luftforureningen. Langt de fleste data vedrørende et stofs toksiske effekter stammer som bekendt fra dyreforsøg. Datagrundlaget for et givent stof giver således sjældent kendskab til, hvorvidt det enkelte individ er mere eller mindre følsomt for det pågældende stof end den gennemsnitlige befolkning. Derfor har det internationalt i risikovurderingssammenhænge været accepteret at kompensere for interindividuel variation ved anvendelse af en (u)sikkerhedsfaktor, som regel af størrelsesordenen 10. Rationalet for denne størrelsesorden kendes ikke. Der er publiceret en række analyser med henblik på at belyse den interindividuelle variation mellem mennesker og dermed en vurdering af størrelsesordenen af usikkerhedsfaktor II. Sammenfattende understøtter disse analyser en standardværdi på 10 for interindividuel variation, idet denne 10-faktor vurderes at tage højde for den interindividuelle variation inden for størstedelen (mere end 99%) af den humane population. Men det skal bemærkes, at der i disse analyser er identificeret tilfælde, hvor den interindividuelle variation ligger udenfor 10-faktoren. I relation til fastsættelse af kvalitetskriterier for luft, jord og vand anbefales det fortsat at anvende en standardværdi på 10 for interindividuel variation (UFII), eventuelt opdelt i en kinetisk og en dynamisk delfaktor, som foreslået af Renwick (1993) og af WHO (1994). Såfremt der foreligger veldokumenteret viden om en specifik gruppes særlige følsomhed i forhold til den gennemsnitlige befolkning, anbefales det at tage udgangspunkt i denne viden med henblik på fastsættelse af en dataspecifik faktor i stedet for anvendelse af en standardværdi på 10. Det skal dog understreges, at det for visse stoffer og effekter samt for visse undergrupper eller enkelte individer i populationen vurderes, at der vil være større forskelle i den interindividuelle variation, og som dermed falder uden for denne 10-faktor. 4.4.3 Usikkerhedsfaktor III – (overordnet vurdering af datagrundlaget)I relation til anvendelsen af en (u)sikkerhedsfaktor ved estimering af den tolerable daglige indtagelse (TDI) med udgangspunkt i et nul-effektniveau for de(n) kritiske effekt(er) har der oprindeligt i de fleste reguleringsmæssige sammenhænge næsten udelukkende været fokuseret på at kompensere for interspeciesvariation samt for interindividuel variation. Ved udgangspunkt i velgennemførte humane studier har der traditionelt været anvendt en samlet sikkerhedsfaktor på 10 med henblik på at tage højde for den interindividuelle variation (se 4.4.2). Ved udgangspunkt i velgennemførte studier i forsøgsdyr har der traditionelt været anvendt en samlet sikkerhedsfaktor på 100, det vil sige i princippet en opdeling af 100-faktor i to delfaktorer på hver 10 til at kompensere for henholdsvis interspeciesvariation (se 4.4.1) og interindividuel variation. Der har endvidere i enkelte tilfælde været anvendt en samlet sikkerhedsfaktor på mere end 100, hvor det eksisterende datagrundlag har været vurderet som værende ikke helt tilstrækkelig til en sikker estimering af TDI. Som eksempler herpå kan nævnes estimering af TDI med udgangspunkt i et datasæt, hvor kvaliteten ikke er fuldt tilstrækkelig med deraf følgende usikkerhed omkring fastlæggelsen af nul-effektniveauet (se 4.4.3.1), hvor den anvendte administrationsvej ikke er relevant (“route-to-route” ekstrapolation - se 4.4.3.2), hvor der ikke kan fastlægges et nul-effektniveau for de(n) kritiske effekt(er) (“LOAEL-to-NOAEL” ekstrapolation - se 4.4.3.3), hvor der ikke er kroniske studier (“duration of exposure” - se 4.4.3.4), eller hvor de observerede effekter skønnes som særligt alvorlige (“nature and severity of toxicity” - se 4.4.3.5). Det skal dog understreges, at datagrundlaget kan være så ringe, at det ikke giver mening at estimere en TDI på det foreliggende grundlag. I relation til fastsættelse af kvalitetskriterier for luft, jord og vand har denne del (SFIII) af den samlede sikkerhedsfaktor typisk varieret fra 1 til 100 afhængigt af datagrundlaget for det pågældende stof. (MST 1990). 4.4.3.1 Vurdering af kvaliteten af datasætEn del af usikkerhedsfaktor III skal søge at tage højde for, hvis kvaliteten af de foreliggende undersøgelser og omfanget af datasættet ikke lever op til det, som vurderes at være nødvendigt for at kunne foretage en tilstrækkelig sikker estimering af TDI. Eksempler herpå kan være mangel på visse typer undersøgelser, dyreforsøg hvor et stof kun er blevet undersøgt i få forsøgsdyr, ved et enkelt eller kun få dosisniveauer, eller hvor der er store afstande mellem dosisniveauerne med deraf følgende usikkerhed omkring fastlæggelsen af nul-effektniveauet. WHO (1994) har anført, at kvaliteten samt fuldstændigheden af datasæt er meget varierende, og dermed vil en (u)sikkerhedsfaktor til at tage højde herfor også variere. For eksempel anbefales det at inddrage en faktor på 1 ved datasæt, som vurderes at være tilstrækkeligt for det givne stof, mens en faktor op til 100 kan inddrages afhængigt af graden af datasættets kvalitet. Hvis der er tale om mindre mangler, anbefales en faktor af størrelsesorden 3-5, mens en ekstra faktor på 10 anbefales ved større mangler, som for eksempel manglende reproduktionsstudier og/eller cancerstudier. WHO (1996) har i relation til guidelines for drikkevandskvalitet anført, at mangler i studier eller i det samlede datasæt har medført, at der ikke kunne fastlægges et NOAEL, og at studierne var af kortere eksponeringstid end ønskeligt. Til at tage højde for disse usikkerheder har der været anvendt en usikkerhedsfaktor af størrelsesorden op til 10. I denne rapport er disse delelementer af usikkerhedsfaktor III beskrevet i 4.4.3.3 (”LOAEL-to-NOAEL”) og 4.4.3.4 (”duration of exposure”). US-EPA (1998) anvender en usikkerhedsfaktor (UFD) på 1, 3 eller 10, når der ved estimering af RfD tages udgangspunkt i et datasæt, som vurderes som værende ikke komplet. Denne usikkerhedsfaktor skal tage højde for, at et enkelt studie ikke vil kunne afsløre alle mulige former for skadelige effekter. Et komplet datasæt inkluderer 2 velgennemførte langtidsstudier i pattedyr med den relevante eksponeringsvej i forskellige species, hvoraf det ene studie skal være udført med en gnaver. Derudover skal der være et velgennemført multigenerationsreproduktionsstudie i pattedyr med en relevant eksponeringsvej samt 2 velgennemførte studier til undersøgelse af påvirkninger under fosterudviklingen (engelsk: developmental toxicity) i forskellige pattedyr og med en relevant eksponeringsvej. Ved estimering af RfD skal der som minimum foreligge et velgennemført 90-dages studie i et pattedyr og med den relevante eksponeringsvej. Valget af værdien for UFD foretages case-by-case ved en ekspertvurdering. Standardværdien er 10. En faktor på 3 kan anvendes, hvis der ikke er større mangler i datasættet. Ifølge Vermeire et al. (1999) bør justeringsfaktoren ligge højere end 1 i de tilfælde, hvor kvaliteten af selve datasættet vurderes som værende utilstrækkelig, og denne faktor kan være helt op til 100. Denne justeringsfaktor kan ifølge forfatterne kun fastlægges på baggrund af en ekspertvurdering. I praksis anvendes dosis-responskurven sjældent som udgangspunkt for estimering af nul-effektniveauet for de(n) kritiske effekt(er), da datagrundlaget som oftest er utilstrækkeligt hertil. I stedet tages der som regel udgangspunkt i et observeret nul-effektniveau (NO(A)EL), det vil sige den højeste koncentration eller dosis, hvorved de(n) kritiske effekt(er) ikke er blevet observeret og vurderet ud fra alle de informationer, der er samlet sammen. Dette er beskrevet mere detaljeret i 4.3. Dourson et al. (1996) har anført, at hvis der kun foreligger data fra et enkelt kronisk studie til fastlæggelse af nul-effektniveau, vil der være en risiko for at yderligere undersøgelser vil afsløre et lavere nul-effektniveau. Usikkerhederne heri bør derfor afspejles i en (u)sikkerhedsfaktor, og forfatterne har foreslået en 3- eller 10-faktor med baggrund i en antagelse om, at den kritiske effekt af et givent stof vil blive afsløret ved testning i et forholdsvis lille antal forskellige toksicitetsstudier. WHO (1994) har anført, at man alternativt til anvendelse af en (u)sikkerhedsfaktor til at kompensere for usikkerheder ved fastlæggelse af et NO(A)EL kan estimere en benchmark dose ved en matematisk modellering af dosis-responsdata (se 3.2.2). Fordelene ved denne fremgangsmåde er, at man udnytter alle eksperimentelle data og tager højde for dosis-responskurvens form, og at benchmark dose, i modsætning til NO(A)EL, ikke afhænger af de valgte forsøgsdesign. Imidlertid giver det klassiske guideline-forsøgsdesign med 3 doserede grupper og en vehikelkontrolgruppe ikke ideelle data til udarbejdelse af gode dosis-responskurver. Så i praksis vil denne metode kun kunne anvendes i begrænset omfang. Ifølge Vermeire et al. (1999) er der ikke noget videnskabeligt belæg for en bestemt standardværdi til at tage højde for usikkerhederne ved estimering af et nul-effektniveau i et givent studie. 4.4.3.2 ”Route-to-route” ekstrapolationDet er af og til tilfældet i reguleringsmæssige sammenhænge, at der mangler data for den relevante eksponeringsvej for mennesker. Der kan være tale om, at datasættet omfatter forsøg, hvor dyrene er blevet eksponeret for et stof på en for mennesker usædvanlig måde, for eksempel ved injektion i bughulen eller via en administrationsvej, der ikke er relevant for det medie, for hvilket kvalitetskriteriet skal fastsættes, som for eksempel hvis der som grundlag for fastsættelse af et luftkvalitetskriterium kun findes orale studier. I disse tilfælde er det nødvendigt at vurdere, om der kan foretages en ”route-to-route” ekstrapolation. ECETOC (1995) vurderede på baggrund af akutte toksicitets data, at ”route-to-route” ekstrapolation fra orale data (LD50-værdier) til inhalation (LC50-værdier) er utroligt svært, idet de tilgængelige data har vist en meget stor variation. Det blev derfor anbefalet, at denne form for ekstrapolationer ikke bør foretages. For hudkontakt er det anført, at absorption via denne eksponeringsvej ofte vil være lavere end ved oral administration af et givent stof på grund af hudens barrierefunktion. Ved ekstrapolation af orale data foreslås således at antage en hudabsorptionsfraktion på mellem 10 og 50%, men at vurderingerne skal foretages case-by-case. Også i relation til ”route-to-route” ekstrapolation på baggrund af data fra kroniske studier vurderede ECETOC, at der ikke i de tilgængelige data er belæg for en bestemt standardværdi og anbefalede derfor en vurdering heraf case-by-case. En hollandsk undersøgelse (Wilschut et al. 1998 - citeret i Vermeire et al. 1999) har analyseret ekstrapolation af NOAEL fra orale data til en korresponderende værdi for inhalation eller for dermal eksponering. For hvert stof blev der beregnet en ekstrapolationsfaktor (defineret som den faktor, der anvendes ved ”route-to-route” ekstrapolation for at tage højde for forskelle i systemisk toksicitet afhængigt af eksponeringsvej) på basis af data for absorption og akut toksicitet. Denne faktor blev anvendt til estimering af et såkaldt ”ekstrapoleret nul-effektniveau”. For ekstrapolation fra oralt NOAEL til NAEC efter inhalation (n = 28) viste analyserne, at den ekstrapolerede værdi (NAEC) ofte var højere end den observerede værdi efter inhalation (NOAEC), således at stoffet blev vurderet som værende mindre toksisk efter ekstrapolation ved sammenligning med den eksperimentelt fundne værdi. Fordelingen for forholdene (ratios) mellem ekstrapoleret nul-effektniveau og observeret nul-effektniveau kan beskrives som værende log-normalfordelt. Med baggrund i 95 percentilen i denne log-normalfordeling blev der fundet usikkerhedsfaktorer fra 75 til 201 for de forskellige ekstrapolationsmetodikker. For ekstrapolation fra oralt NOAEL til NAEL efter hudkontakt (n = 25) viste analyserne, at den ekstrapolerede værdi (NAEL) ofte var lavere end den observerede værdi ved hudkontakt (NOAEL), således at stoffet blev vurderet som værende mere toksisk efter ekstrapolation ved sammenligning med den eksperimentelt fundne værdi. Med baggrund i 95 percentilen i log-normalfordelingen for forholdene (ratios) mellem ekstrapoleret nul-effektniveau og observeret nul-effektniveau blev der fundet usikkerhedsfaktorer fra 2.7 til 35 for de forskellige ekstrapolationsmetodikker. Forfatterne konkluderede, at udvikling af videnskabeligt baserede principper for ”route-to-route” ekstrapolationer er en meget svær opgave, da det tilgrundliggende datamateriale ikke findes. Endvidere blev det konkluderet, at der ikke er noget videnskabeligt belæg for at foretage ”route-to-route” ekstrapolationer. Ifølge Vermeire et al. (1999) er der for nuværende to forskellige tilgange med henblik på vurdering af ”route-to-route” ekstrapolation: 1) Anvendelse af usikkerhedsfaktorer, eller 2) igangsættelse af studier med relevant eksponeringsvej indtil validerede ”route-to-route” ekstrapolationsmetodikker er tilgængelige. Endvidere er det anført, at valget mellem disse to tilgange er et regulatorisk spørgsmål, og at ”route-to-route” ekstrapolation udelukkende må bero på en ekspertvurdering. 4.4.3.3 “LO(A)EL to NO(A)EL” ekstrapolationI en række tilfælde kan der med baggrund i et givent datasæt ikke fastlægges et nul-effektniveau NO(A)EL, selvom det antages, at et sådant findes. I disse tilfælde tages der ved estimering af TDI udgangspunkt i det laveste observerede effektniveau (LO(A)EL) (se 3.2), og der inddrages ofte en (u)sikkerhedsfaktor til at kompensere herfor. Forholdet mellem NOAEL og LOAEL er afhængigt af hældningen på dosis-responskurven, antallet af individer i den enkelte dosisgruppe samt af intervallet mellem doser i det givne studie (ECETOC 1995). Ved ekstrapolation fra LOAEL til NOAEL har faktorer af størrelsesorden 1 til 10 været anvendt (ECETOC 1995, Dourson & Stara 1983) afhængigt af alvorligheden (engelsk: severity) af den effekt, hvorpå LOAEL baseres (Dourson & Stara 1983). For eksempel har der været anvendt en faktor på 10 for LOAEL fastlagt på baggrund af levercellenekrose, mens en faktor på 3 har været anvendt for LOAEL fastlagt på baggrund af ”fatty infiltration” i leveren (Dourson & Stara 1983). Dourson & Stara (1983) analyserede forholdet mellem LOAEL og NOAEL for subkronisk (n = 27) og kronisk (n = 25) eksponering. Resultaterne for subkronisk eksponering viste, at NOAEL var maksimalt 5 gange lavere end det korresponderende LOAEL og med størstedelen af værdierne liggende på 2 til 3. For kronisk eksponering var LOAEL maksimalt 10 gange højere end det korresponderende NOAEL i 2 af tilfældene, mens resten af LOAELs var maksimalt 5 gange højere end de korresponderende NOAELs. Sammenholdt var NOAELs i alle sammenligninger højst 10 gange lavere end de korresponderende LOAELs og i 96% af tilfældene højst 5 gange lavere. Forfatterne konkluderede på baggrund heraf, at en faktor på mellem 1 og 10 synes at tage højde for ekstrapolation fra LOAEL til NOAEL. WHO (1994) har anbefalet, at i de tilfælde, hvor der ikke kan fastlægges et nul-effektniveau, kan der foretages en ekstrapolation fra det laveste observerede effekt niveau under anvendelse af en passende sikkerhedsfaktor, såfremt datagrundlaget vurderes at være af en tilstrækkelig kvalitet. Det er endvidere anført, at usikkerhedsfaktorer af størrelsesorden 3, 5 eller 10 har været anvendt ved ekstrapolation fra et LOAEL til et NOAEL afhængigt af, hvilke(n) effekt)er) der har været tale om samt dosis-responsrelationen. ECETOC (1995) har anført, at hvis intervallet mellem doser er stort (for eksempel 10), og hvis de effekter der observeres ved LOAEL vurderes som værende af minimal karakter (dvs. at NOAEL ligger tæt på LOAEL), så kan det i vurderingen være mere relevant at tage udgangspunkt i LOAEL i stedet for NOAEL under inddragelse af en (u)sikkerhedsfaktor på 3 (standardværdi). ECETOC konkluderede, at på trods af det begrænsede datagrundlag til vurdering af denne problemstilling, så synes en faktor på 2-3 at være i overensstemmelse med det tilgængelige grundlag. En faktor på 3 blev anbefalet som standardværdi og bør anvendes i størsteparten af tilfældene. En faktor på 2 kan dog anvendes i de tilfælde, hvor den relevante effekt vurderes som værende af mindre betydning. Dourson et al. (1996) har på baggrund af den tilgængelige viden konkluderet, at en faktor på 10 eller mindre synes at tage højde for de usikkerheder, der ligger i at ekstrapolere fra LOAEL til NOAEL. Størrelsen af usikkerhedsfaktoren bør i følge forfatterne generelt afhænge af alvorligheden (engelsk: severity) af den effekt, der ligger til grund for fastlæggelsen af LOAEL, således at ved mere alvorlige effekt bør faktoren være højere, idet NOAEL i disse tilfælde formodes at ligge længere væk fra LOAEL. Og modsat, ved mindre alvorlige effekter behøves en mindre faktor, idet LOAEL formodes at ligge tættere på NOAEL Pieters et al. (1998) beregnede forholdet mellem LOAEL og NOAEL (ratio) fra subakutte, subkroniske og kroniske studier og foretog statistiske analyser af fordelingen for disse ratio. Ifølge forfatterne er størrelsen af ratio udelukkende afhængig af afstanden mellem doserne i de for analyserne tilgrundliggende studier. Under antagelse af log-normalfordelte fordelinger for ratio blev det geometriske gennemsnit, den geometriske standardafvigelse, 95 percentilen samt 95% konfidensintervallet beregnet. Resultaterne af analyserne af subakutte studier (95 ratio) viste, at det geometriske gennemsnit for fordelingen af ratio var 3,5 med en geometrisk standardafvigelse på 1,8 og en 95 percentil på 9 med et 95% konfidensinterval på 8-11. Resultaterne af analyserne af subkroniske studier (226 ratio) viste, at det geometriske gennemsnit for fordelingen af ratio var 4,3 med en geometrisk standardafvigelse på 2,2 og en 95 percentil på 16 med et 95% konfidensinterval på 14-19. Resultaterne af analyserne af kroniske studier (175 ratio) viste, at det geometriske gennemsnit for fordelingen af ratio var 4,5 med en geometrisk standardafvigelse på 1,7 og en 95 percentil på 11 med et 95% konfidensinterval på 10-12. Forfatterne konkluderede, at selv om resultaterne kunne tolkes som støtte for en usikkerhedsfaktor på 10, er der ikke belæg for anvendelsen af sådan en faktor. I stedet anbefalede forfatterne at anvende dosis-respons modellering, hvilket unødvendiggør anvendelsen af en usikkerhedsfaktor for ekstrapolation fra LOAEL til NOAEL. Vermeire et al. (1999) har konkluderet, at sådan som de fleste dyreforsøg er designet, er der på ingen måde belæg for at ekstrapolere fra et LOAEL til et NOAEL. Forfatterne anbefalede derfor, at korrektion herfor foretages ved ekspertvurdering baseret på dosis-responskurvens form samt typen og alvorligheden af den observerede effekt ved det givne LOAEL. US-EPA (1998) anvender en usikkerhedsfaktor (UFL) på 1, 3 eller 10, når der ved estimering af RfD tages udgangspunkt i LOAEL i stedet for NOAEL. Standardværdien er 10. Valget af værdien for UFL foretages case-by-case ved en ekspertvurdering. 4.4.3.4 Fravær af langtidsundersøgelser (”duration of exposure”)For stoffer, hvor der kun findes subakutte eller subkroniske studier, er det ikke muligt at estimere et nul-effektniveau for livstidseksponering. I subakutte studier er der som regel færre forsøgsdyr i eksponeringsgrupperne end i subkroniske studier, hvor der ligeledes er færre forsøgsdyr end i kroniske studier. På baggrund heraf kan det forventes, at et nul-effektniveau estimeret i et subakut studie er højere end et nul-effektniveau estimeret i et subkronisk studie, som igen kan forventes at være højere end et nul-effektniveau estimeret i et kronisk studie. McNamara (1976 – citeret fra ECETOC 1995, Dourson & Stara 1983, Grönlund 1992) undersøgte forholdet mellem NOAELs fra subkroniske studier og kroniske studier (NOAELsubkronisk / NOAELkronisk) for 41 forskellige stoffer (pesticider, tilsætningsstoffer, lægemidler). Data stammede primært fra studier i rotter og hunde, men udført forskellige steder under anvendelse af forskellige eksponeringstider og teknikker. Resultaterne viste, at forholdene var 1 eller mindre for 34 ud af 41 stoffer; forholdene for de resterende stoffer var alle mindre end 3. Ifølge Dourson & Stara (1983) indikerer disse McNamara's undersøgelser, at en dosisreduktion på 3 eller derunder vil være tilstrækkelig med henblik på at estimere NOAEL for kronisk eksponering ud fra et korresponderende NOAEL for subkronisk eksponering. Dourson & Stara (1983) analyserede forholdet (ratio) mellem subkronisk og kronisk eksponeringsvarighed for enten NOAELs (n = 30), LOAELs (n = 22), eller kombinationen heraf (n = 52) med udgangspunkt i studier i rotter og hunde. Eksponeringsvarigheden i subkroniske studier varierede fra 30 til 210 dage, og de kroniske studier var alle af 2 års varighed. Disse ratio kan ifølge forfatterne tolkes som den reduktion i subkroniske NO(A)ELs eller LOAELs, der skal til for at ekstrapolere til tilsvarende kroniske NO(A)ELs eller LOAELs. Resultaterne viste, at ratio mellem subkronisk og kronisk eksponeringsvarighed (ratiosubkronisk/kronisk) i omkring 96% af tilfældene var mindre end 10, og at forholdene var 2 eller mindre i omkring 50% af tilfældene. Forfatterne konkluderede på baggrund heraf, at en faktor på 10 synes at tage højde for usikkerhederne ved at estimere en ADI fra et subkronisk effektniveau i mangel af et kronisk effektniveau. Woutersen et al. (1984 – citeret fra ECETOC 1995) sammenlignede nul-effektniveauer for 82 kemikalier, som var testet i korttidsstudier og i subkroniske studier under samme omstændigheder. Resultaterne viste, at ratio var lig med eller mindre end 10 i alle tilfældene, en faktor på 4 dækkede 80% af tilfældene, og en faktor på 1 blev fundet i 56% af tilfældene. Lewis (1993 – citeret fra Dourson et al. 1996) analyserede ratio mellem subkroniske og kroniske NOAELs med baggrund i peer-reviewed litteratur samt data fra U.S. National Toxicology Program. For 14 af 18 kemikalier var forholdet 3,5 eller mindre, og for 3 af de resterende kemikalier var forholdet 10 eller mindre. Ifølge ECETOC (1995) er datagrundlaget for vurderinger af ekstrapolation af nul-effektniveauer fra korttidsstudier til længerevarende studier meget begrænset. Det er anført, at faktoren for ekstrapolation af NOAEL fra subkronisk til kronisk varighed afhænger af det givne stof. Faktoren kan sættes til 1 for stoffer, som ikke udløser kumulative effekter eller akkumuleres i kroppen, mens en højere faktor kan inddrages for stoffer, der udviser sådanne egenskaber. Det er endvidere anført, at yderligere ekstrapolation kan være nødvendig, hvis varigheden af de tilgængelige korttidsstudier ligger i området fra 14 til 28 dage, da det, ifølge ECETOC, er generelt accepteret, at nul-effektniveauer fra subakutte studier ikke kan erstatte nul-effektniveauer fra subkroniske studier. ECETOC konkluderede på baggrund af de tilgængelige videnskabelige data, at en provisorisk standardværdi af størrelsesorden 2-3 synes hensigtsmæssig ved ekstrapolation af NOAEL fra subkronisk til kronisk varighed. Dourson et al. (1996) anførte, at de tilgængelige analyser af ratio mellem subkroniske og kroniske NOAELs og LOAELs generelt udviser et værdi mellem 2 og 3, og at kun en lille del af de undersøgte kemikalier udviser forskelle på mere end en faktor 10. På baggrund heraf har forfatterne anbefalet, at den traditionelle 10-faktor for ekstrapolation af NOAEL/LOAEL fra subkronisk til kronisk varighed bør vurderes grundigt og sandsynligvis kun i sjældne tilfælde bør sættes så højt som 10. Kramer et al. (1996) analyserede anvendelsen af en konverteringsfaktor (CF) med henblik på at estimere et NOAEL for kronisk varighed ud fra toksicitetsdata i korttidsstudier. Fordelingerne af ratio mellem (sub)akutte og kroniske toksicitetsdata blev evalueret for 332 stoffer. CF blev defineret som den øvre 95% konfidensgrænse for 95 percentilen for den relevant fordeling af ratio. Med udgangspunkt i NOAELsubakut blev en CF på 87 estimeret (baseret på 87 ratio), og med udgangspunkt i LD50-værdier en CF på 17000 (baseret på 244 ratio). Pieters et al. (1998) foretog en statistisk analyse af NOAELsubkronisk / NOAELkronisk for 149 ratio fra orale studier. Resultaterne viste, at det geometriske gennemsnit for fordelingen af ratio var 1,7 med en geometrisk standardafvigelse på 5,6 og en 95 percentil på 29 med et 95% konfidensinterval på 20-46. Forfatterne konkluderede, at der med baggrund i disse resultater ikke er belæg for at reducere den traditionelt anvendte 10-faktor. Ifølge Vermeire et al. (1999) anvendes i mangel af kroniske studier en ekstra justeringsfaktor på sædvanligvis 10 med henblik på at ekstrapolerere et subakut eller subkronisk nul-effektniveau til et kronisk nul-effektniveau. Forfatterne har analyseret fordelingsfunktionen for justeringsfaktoren i en række orale studier, det vil sige forholdet mellem orale nul-effektniveauer fra subakutte/subkroniske studier og kroniske studier. Resultaterne viste, at justeringsfaktoren for at ekstrapolere fra subkronisk til kronisk er tilnærmelsesvis log-normalfordelt med et geometrisk gennemsnit på omkring 2 og en geometrisk standardafvigelse på omkring 4. Baseret på denne fordeling blev standardværdier for 90, 95 og 99 percentilerne beregnet til 12, 20 og 50 respektivt; en 10-faktor svarede til 88 percentilen. Det geometriske gennemsnit var signifikant højere (4) for at ekstrapolere fra subakut til kronisk end for at ekstrapolere fra subkronisk til kronisk med en geometrisk standardafvigelse på omkring 4. Baseret på denne fordeling blev standardværdier for 90, 95 og 99 percentilerne beregnet til 24, 39 og 101 respektivt. US-EPA (1998) anvender en usikkerhedsfaktor (UFS) på 1, 3 eller 10, når der ved estimering af RfD tages udgangspunkt i dyreeksperimentelle studier med kortere eksponeringstid (ikke kroniske studier). Standardværdien er 10. En værdi på 3 i stedet for 10 kan for eksempel anvendes, når der tages udgangspunkt i et 1-års studie i rotter, idet NOAELs fra 1-årige studier vurderes at ligge tættere på NOAELs for kroniske studier end NOAELs fra 90-dages studier. Valget af værdien for UFS foretages case-by-case ved en ekspertvurdering. 4.4.3.5 Type og alvorlighed af effekter (“nature and severity of toxicity”)Det har været fremført, at der også bør tages højde for typen samt alvorligheden af de observerede effekter (for eksempel cancer, mutagenicitet/genotoksicitet, reproduktionsskader og sensibilisering) ved at inddrage dette element i usikkerhedsfaktor III. WHO (1994) har fremført, at vurdering af typen af effekter allerede indgår som et element i fastlæggelsen af NO(A)EL og/eller LO(A)EL, og at der således til en vis grad er taget højde herfor ved fastlæggelse af disse. En række internationale instanser, blandt andet JECFA og JMPR, har inddraget en ekstra usikkerhedsfaktor af størrelsesorden op til 10 i de tilfælde, hvor nul-effektniveauet fastlægges for en kritisk effekt, der betragtes som værende af alvorlig grad samt irreversibel. Som eksempler nævnes teratogenicitet og carcinogenicitet, hvor den tilgrundliggende mekanisme for stoffets indflydelse på udvikling af tumorer ikke er en beskadigelse af arvematerialet. Den ekstra faktor anvendes især i de tilfælde, hvor der er observeret en flad dosis-responskurve. (WHO 1994). WHO (1996) har i relation til guidelines for drikkevandskvalitet anført, at det i nogle situationer har været nødvendigt at anvende en ekstra usikkerhedsfaktor som følge af ”nature and severity of effect”. Som eksempler nævnes studier, hvor end-point var fostermisdannelser eller forekomst at tumorer, hvor 'guideline value' i sidstnævnte tilfælde blev fastlagt på baggrund af tærskelværdiprincippet. Til at tage højde for disse usikkerheder har en usikkerhedsfaktor af størrelsesorden op til 10 været anvendt. Renwick (1995) har evalueret brugen af en (u)sikkerhedsfaktor rettet specifikt mod typen af toksisk effekt. Baggrunden for anvendelsen af denne faktor var at tage højde for non-genotoksiske carcinogener og teratogener. I enkelte tilfælde er faktoren blevet inddraget specifikt på NOAEL for denne type af effekter. Men i langt de fleste tilfælde er faktoren blevet anvendt, hvor udgangspunktet har været et NOAEL for en helt anden type effekt og somme tider også for et andet species. Forfatteren konkluderede, at såfremt der skal inddrages en (u)sikkerhedsfaktor for typen af toksisk effekt, så skal den anvendes på NOAEL for den givne type af effekt, og ikke på NOAEL for en helt anden type effekt. Vermeire et al. (1999) har konkluderet, at det som udgangspunkt kan antages, at det ikke er nødvendigt at foretage yderligere korrektion for type og alvorlighed af effekter, men at en eventuel korrektion herfor må bero på en ekspertvurdering. 4.4.3.6 Sammenfatning for usikkerhedsfaktor IIII tilfælde af at det ved estimering af TDI er nødvendigt at tage udgangspunkt i et datasæt, der vurderes som værende ikke fuldt ud tilstrækkeligt til en sikker estimering af TDI, er det blevet mere og mere udbredt internationalt at anvende en samlet usikkerhedsfaktor, der af størrelsesorden er højere end den traditionelle 100-faktor, der kompenserer for interspecies og interindividuel variation. I denne del (UFIII) af den samlede usikkerhedsfaktor indgår elementer som for eksempler kvaliteten af datasættet, route-to-route” ekstrapolation, “LOAEL-to-NOAEL” ekstrapolation , “duration of exposure”, og “nature and severity of toxicity”. Det skal dog understreges, at ved omfattende mangler kan datagrundlaget være så ringe, at det ikke giver mening at estimere en TDI på det foreliggende grundlag. De enkelte delelementer af usikkerhedsfaktor III er beskrevet nedenfor med henblik på at vurdere, hvorvidt de tilgængelige data giver belæg for at pege på en specifik størrelsesorden af en standardværdi for det enkelte delelement af usikkerhedsfaktor III. En del af usikkerhedsfaktor III skal søge at tage højde for, hvis kvaliteten af de foreliggende undersøgelser ikke lever op til det, som vurderes at være nødvendigt for at kunne foretage en tilstrækkelig sikker estimering af TDI (se 4.4.3.1). De tilgængelige data giver intet belæg for at pege på en specifik størrelsesorden for standardværdi til at kompensere for usikkerheder som følge af kvaliteten af undersøgelserne. For eksempel har det været anbefalet at anvende en faktor på 1 ved datasæt, som vurderes at være fuldt tilstrækkeligt for det givne stof, mens der ved mindre eller større mangler har været anbefalet en faktor af størrelsesorden 3-5 eller 10 respektivt. Det har også været anført, at det måske i nogle tilfælde kan være relevant at anvende faktorer på helt op til 100. En vurdering heraf må således bero på en ekspertvurdering og foretages case-by-case. Vedrørende vurdering af usikkerheden ved fastlæggelse af NO(A)EL (se 4.4.3.1) kan der heller ikke peges på en konkret størrelsesorden for en standardværdi til at kompensere herfor, og denne vurdering må ligeledes bero på en ekspertvurdering og foretages case-by-case. Alternativt er det blevet anbefalet at estimere en benchmark dose ved en matematisk modellering af dosis-responsdata, hvorved usikkerheden ved fastlæggelse af NO(A)EL minimeres. Fordelene herved er, at alle eksperimentelle data udnyttes og tager højde for dosis-responskurvens form, og at benchmark dose, i modsætning til NO(A)EL, ikke afhænger af de valgte forsøgsdesign. Imidlertid giver det klassiske guideline-forsøgsdesign ikke ideelle data til dosis-responskurver. Så i praksis vil denne metode næppe finde udbredt anvendelse foreløbig. Ved mangel af data for den relevante eksponeringsvej (se 4.4.3.2) kan det blive nødvendigt at foretage en “route-to-route” ekstrapolation. Data indikerer, at ved ekstrapolation fra oralt NOAEL til NOAEC efter inhalation vil en ekstrapoleret værdi ofte være højere end en observeret værdi, mens ved ekstrapolation fra oralt NOAEL til NOAEL efter hudkontakt vil en ekstrapoleret værdi ofte være lavere end en observeret værdi. Med baggrund i de tilgængelige data er der intet belæg for at pege på en bestemt størrelsesorden for standardværdi til at kompensere for de usikkerheder, der ligger i at foretage en “route-to-route” ekstrapolation. Det er således blevet konkluderet, at der ikke er noget videnskabeligt belæg for at foretage ”route-to-route” ekstrapolationer og anbefalet, at “route-to-route” ekstrapolation må bero på en ekspertvurdering og foretages case-by-case. I en række tilfælde kan der med baggrund i et givent datasæt ikke fastlægges et NO(A)EL, selvom det antages, at et sådant findes. I disse tilfælde tages der ved estimering af TDI udgangspunkt i LO(A)EL, og en faktor af størrelsesorden 1 til 10 (afhængigt af den effekt hvorpå LOAEL baseres) har været anvendt med henblik på at tage højde for eventuelle usikkerheder ved at ekstrapolere fra LOAEL til NOAEL (se 4.4.3.3). Der er i de sidste 20-25 år foretaget flere analyser af forholdet mellem NOAEL og LOAEL, herunder også under hensyntagen til eksponeringsvarigheden. Analyserne tenderer til at blive mere sofistikerede med tiden, og de senere analyser vurderes i højere grad at tage højde for diverse usikkerheder i analyserne og dermed sandsynligvis at være mere pålidelige end de tidlige analyser. Med baggrund i analyserne synes der at være belæg for en standardværdi af størrelsesorden 10. Det skal dog nævnes, at de to nyeste referencer har anbefalet enten at anvende dosis-respons modellering, hvorfor der så ikke skulle være grund til at anvende en (u)sikkerhedsfaktor (Pieters et al. 1998), eller at basere korrektionen for ekstrapolation fra LOAEL til NOAEL ved en ekspertvurdering af dosis-responskurvens form samt typen og alvorligheden af den effekt, der ligger til grund for fastlæggelsen af LOAEL (Vermeire et al. 1999). For stoffer, hvor der kun findes subakutte eller subkroniske studier, er det ikke muligt at estimere et NOAEL for livstidseksponering, og det er blevet anført, at NOAEL i et subakut eller subkronisk studie kan forventes at være højere end et nul-effektniveau estimeret i et kronisk studie. Ved fravær af kroniske studier har der været anvendt en (u)sikkerhedsfaktor på sædvanligvis 10 med henblik på at tage højde herfor (se 4.4.3.4). Ligeledes har der er i de sidste 20-25 år været foretaget flere analyser af forholdet mellem NOAELs og LOAELs opnået i studier af forskellige eksponeringsvarigheder. Som nævnt ovenfor tenderer analyserne til at blive mere sofistikerede med tiden, og de senere analyser vurderes i højere grad at tage højde for diverse usikkerheder i analyserne og dermed sandsynligvis at være mere pålidelige end de tidlige analyser. Med baggrund i analyserne synes der at være belæg for en standardværdi af størrelsesorden minimum 10. Den nyeste analyse (Vermeire et al. 1999) viste, at en standardværdi på 10 dækkede i 88% af tilfældene, når der var tale om ekstrapolation fra subkroniske data. Ved ekstrapolation fra subakutte data var 90 percentilen 24. Det har været fremført, at typen samt alvorligheden af en given effekt (engelsk: nature and severity of toxicity - se 4.4.3.5) også er et delelement af usikkerhedsfaktor III. For eksempel har blandt andet JECFA og JMPR anvendt en faktor af størrelsesorden op til 10 i tilfælde af, at NOAEL fastlægges for en kritisk effekt, som vurderes at være af alvorlig grad samt irreversibel (f.eks. teratogenicitet og non-genotoksisk carcinogenicitet med ”shallow” dosis-responsrelation). Andre har konkluderet, at det som udgangspunkt kan antages, at det ikke er nødvendigt at foretage yderligere korrektion herfor, idet der allerede skulle være taget højde for dette delelement ved fastlæggelse af NOAEL og/eller LOAEL for den kritiske effekt. Det har også været fremført, at hvis der skal foretages korrektion for type af effekt, så skal korrektionen foretages på NOAEL for den givne effekt, og ikke på NOAEL for et helt anden type effekt eller en anden species. Det er således ikke muligt på baggrund af den nuværende viden at give en generel anbefaling om, hvorvidt der bør foretages yderligere korrektion for type og alvorlighed af en given effekt, ej heller at pege på en specifik størrelsesorden for standardværdi til at kompensere for eventuelle usikkerheder herfor. En vurdering heraf må således bero på en ekspertvurdering og foretages case-by-case. Sammenfattende kan det konkluderes, at det på baggrund af de tilgængelige data ikke er muligt at pege på en specifik størrelsesorden for en standardværdi, hverken for de enkelte delelementer af usikkerhedsfaktor III eller for usikkerhedsfaktor III som sådan. I relation til fastsættelse af kvalitetskriterier for luft, jord og vand anbefales det derfor fortsat at fastlægge usikkerhedsfaktor III på baggrund af en ekspertvurdering og case-by-case under hensyntagen til det givne stofs samlede datasæt. Baggrunden for vurderingen af de enkelt delelementer i usikkerhedsfaktor III bør være så transparent som overhovedet mulig. 4.4.4 Samlet usikkerhedsfaktorI praksis vurderes de enkelte elementer (UFI, UFII og UFIII) i den samlede usikkerhedsfaktor hver for sig, som beskrevet ovenfor, hvorefter den samlede usikkerhedsfaktor beregnes som produktet af hver af de enkelte usikkerhedsfaktorer. Ved denne procedure kan man imidlertid let opnå en meget høj samlet usikkerhedsfaktor, og jo flere delelementer, der indgår i den samlede usikkerhedsfaktor, jo mere konservativ bliver den estimerede TDI. Især vil de enkelte delelementer i usikkerhedsfaktor III bidrage til en høj samlet usikkerhedsfaktor. Derfor skal der foretages endnu en vurdering af den samlede usikkerhedsfaktor med henblik på rimeligheden af denne faktor i forhold til det givne datasæt. I de fleste tilfælde vurderes det, at en samlet usikkerhedsfaktor med en værdi på højere end 10.000 ikke vil være rimelig, selvom produktet af de enkelte usikkerhedsfaktorer har en højere værdi. Gaylor et al. (1999) har for alvorlige, irreversible effekter foreslået en standardværdi for en samlet usikkerhedsfaktor af størrelsesorden 10.000 ved udgangspunkt i data fra dyreforsøg. For reversible, biologiske effekter blev foreslået en standardværdi på 1000. Ved estimering af Reference Dose (RfD) vurderer US-EPA 5 forskellige kategorier af usikkerhed: 1) interindividuel variation (UFH), 2) interspeciesvariation (UFA), 3) subkronisk til kronisk varighed (UFS), 4) LOAEL til NOAEL (UFL) og , 5) datasættes kvalitet og relevans (UFD). Standardværdien for den enkelte kategori er 10, men værdier på 1 eller 3 kan også vælges for den enkelte kategori. Valget af værdien foretages case-by-case ved en ekspertvurdering. Den samlede usikkerhedsfaktor er afhængig af værdierne for de enkelte kategorier. Ved usikkerheder i en, to eller tre af kategorierne anvendes generelt en samlet usikkerhedsfaktor på 10, 100, eller 1000 respektivt. Ved usikkerheder i fire kategorier er den samlede usikkerhedsfaktor sjældent over 3000, da man har den holdning, at hvis det tilgrundliggende datasæt er så ringe, at den samlede usikkerhedsfaktor bliver højere end 3000, så er datagrundlaget for ringe med henblik på estimering af en RfD. Også den samlede usikkerhedsfaktor vurderes case-by-case ved en ekspertvurdering, og det er understreget, at denne skal være så fleksibel, at den tager højde for usikkerhederne i de 5 enkelte kategorier og datagrundlaget som sådan. Udover usikkerhedsfaktorerne anvender US-EPA også en såkaldt modificerende faktor (engelsk: Modifying Factor MF). MF anvendes til at tage højde for eventuelle usikkerheder, som ikke vurderes i
relation til den samlede usikkerhedsfaktor UF. Som eksempel er nævnt, at jo færre antal dyr i dosisgrupperne jo større er sandsynligheden for, at der ikke observeres en given skadelige effekt ved en
specifik dosis, men som ville blive afsløret i en større population. Et andet eksempel er, at MF kan anvendes, hvis et givent stof kun er undersøgt i en enkelt species. Værdien for MF er større end nul og
mindre end lig med 10, og standardværdien er 1. WHO (1996) har i relation til guidelines for drikkevandskvalitet anført, at den samlede usikkerhedsfaktor ikke bør være højere end 10.000, idet den resulterende TDI i så fald vil være så upræcis, at den er meningsløs. Hvis vurderingen af den samlede usikkerhedsfaktor resulterer i en meget høj værdi (f.eks. over 10.000), bør datagrundlaget vurderes som værende af så ringe kvalitet, at det ikke giver mening at estimere en TDI på det foreliggende grundlag, og dermed heller ikke et kvalitetskriterie for jord, vand eller luft. Omvendt vil det i mange situationer med kontaminanter i miljøet være nødvendigt at foretage en eller anden form for regulering baseret på en sundhedsmæssig vurdering, hvorfor det er vigtigt at foretage en så objektiv vurdering af den samlede usikkerhedsfaktor som muligt på baggrund af det foreliggende datagrundlag. Ender man op med at være nødt til at fastlægge TDI med baggrund i en meget høj samlet usikkerhedsfaktor, bør usikkerheden ved estimatet nøje understreges, og der bør tages hensyn hertil i det videre forløb ved fastsættelse af kvalitetskriterier. 4.5 Fastlæggelse af TDIDen tolerable daglige indtagelse (TDI) er en beregnet størrelse (koncentration eller dosis), som mennesker vurderes at kunne udsættes for (tolerere) gennem et helt livsforløb, uden at der forventes sundhedsskadelige effekter (se 4.1). TDI estimeres med udgangspunkt i et NO(A)EL eller LO(A)EL for de(n) kritiske effekt(er) (se 4.3) under anvendelse af en samlet usikkerhedsfaktor UF (se 4.4). TDI beregnes, som angivet i nedenstående ligning og angives sædvanligvis i enheden mg/kg legemsvægt per dag. 4.6 ReferencerBeltoft V, Nielsen E, Meyer O and Ladefoged O (2001). Individual variations in biological susceptibility to xenobiotics: a review of the current knowledge. Rapport udarbejdet for MST, marts 2001. Burin GJ and Saunders DR (1999). Addressing human variability in risk assessment – the robustness of the intraspecies uncertainty factor. Regul Toxicol Pharmacol 30, 209-216. Calabrese EJ (1985). Uncertainty factors and interindividual variation. Regul Toxicol Pharmacol 5, 190-196. Calabrese EJ, Beck BD and Chappell WR (1992). Does the animal-to-human uncertainty factor incorporate interspecies differences in surface areas? Regul Toxicol Pharmacol 15, 172-179. Davidson IWF, Parker JC and Beliles RP (1986). Biological basis for extrapolation across mammalian species. Regul Toxicol Pharmacol 6, 211-237. Dourson ML and Stara JF (1983). Regulatory history and experimental support of uncertainty (safety) factors. Regul Toxicol Pharmacol 3, 238-244. Dourson ML, Pelter SP and Robinson D (1996). Evolution of science-based uncertainty factors in noncancer risk assessment. Regul Toxicol Pharmacol 24, 108-120. ECETOC (1995). Assessment factors in human health risk assessment. Technical Report No. 68. European Centre for Ecotoxicology and Toxicology of Chemicals, Brussels August 1995. Feron VJ, van Bladeren PJ and Hermus RJJ (1990). A viewpoint on the extrapolation of toxicological data from animals to man. Fd Chem Toxicol 28, 783-788. Gaylor DW, Kodell RL, Chen JJ and Krewski D (1999). A unified approach to risk assessment for cancer and noncancer endpoints based on benchmark doses and uncertainty/safety factors. Regul Toxicol Pharmacol 29, 151-157. Grandjean P (1997). Farlig forurening. Fra risikovurdering til forebyggelse. Sundhedsstyrelsen. Nyt Nordisk Forlag Arnold Busck, København. Grönlund MH (1992). Kvantitativ riskbedömning av icke-genotoxiska substanser – en metodikstudie. IMM-rapport 4/92. Institutet för miljömedicin. Karolinska instituttet (in Swedish). Hattis D, Erdreich L and Ballew M (1987). Human variability in susceptibility to toxic chemicals – a preliminary analysis of pharmacokinetic data from normal volunteers. Risk Anal 7, 415-426. Kramer HJ, van den Ham WA, Slob W and Pieters MN (1996). Conversion factors estimating indicative chronic no-observed-adverse-effect levels from short-term toxicity data. Regul Toxicol Pharmacol 23, 249-255. Larsen PB, Larsen JC, Fenger, J og Jensen SS (1997). Sundhedsmæssig vurdering af luftforurening fra vejtrafik. Miljøprojekt nr. 352. Miljø- og Energiministeriet, Miljøstyrelsen. Lehman AJ and Fitzhugh OG (1954). 100-fold margin of safety. Assoc Food Drug Off U.S.Q. Bull 18, 33-35. MST (2001). Personlig meddelelse. MST (1990). Begrænsning af luftforurening fra virksomheder. Vejledning fra Miljøstyrelsen Nr. 6 1990. Miljøministeriet, Miljøstyrelsen. Nielsen E, Thorup I, Schnipper A, Hass U, Meyer O, Ladefoged O, Larsen JC, Østergaard G, Sørensen TL and Larsen PB (2001). Children and the unborn child. Exposure and susceptibility to chemical substances – an evaluation. Environmental Project No. 589. Miljøstyrelsen, Miljø- og Energiministeriet. Pieters MN, Kramer HJ and Slob W (1998). Evaluation of the uncertainty factor for subchronic-to-chronic extrapolation: Statistical analysis of toxicity data. Regul Toxicol Pharmacol 27, 108-111. Renwick AG (1999). Data-derived safety factors for the evaluation of food additives and environmental contaminants. Food Addit Contam 10, 275-305. Renwick AG (1999a). Subdivision of uncertainty factors to allow for toxicokinetics and toxicodynamics. Hum Ecol Risk Assess 5, 1035-1050. Renwick AG (1995). The use of an additional safety or uncertainty factor for nature of toxicity in the estimation of acceptable daily intake and tolerable daily intake values. Regul Toxicol Pharmacol 27, 3-20. Renwick AG and Lazarus NR (1998). Human variability and noncancer risk assessment – an analysis of the default uncertainty factor. Regul Toxicol Pharmacol 27, 3-20. US-EPA (1998). Ambient water quality criteria derivation for the protection of human health – Technical Support Document. Final draft. United States Environmental Protection Agency, Office of Water 4304, EPA-822-B-98-005. Van Genderen H (1988). General conclusions of the chairman (presented at the workshop May 2-3, 1988 at the National Institute of Public Health and Environmental Protection, Ba Bilthoven, The Netherlands). Regul Toxicol Pharmacol 8, 431-436. Vermeire T, Stevenson H, Pieters MN, Rennen M, Slob W and Hakkert BC (1999). Assessment factors for human health risk assessment: a discussion paper. Crit Rev Toxicol 29 (439-490). Voisin EM, Ruthsatz M, Collins JM and Hoyle PC (1990). Extrapolation of animal toxicity to humans: interspecies comparisons in drug development. Regul Toxicol Pharmacol 12, 107-116. Weil CS (1972). Statistics vs safety factors and scientific judgement in the evaluation of safety for man. Toxicol Appl Pharmacol 21, 454-463. WHO (1996). 12. Chemical and physical aspects: introduction. In: Guidelines for drinking-water quality, second edition. Volume 2 Health criteria and other supporting information. World Health Organization, Geneva, 121-131. WHO (1994). Assessing human health risks of chemicals: Derivation of guidance values for health-based exposure limits. Environmental Health Criteria 170, World Health Organization, Geneva. 5 Fastlæggelse af TDI for genotoksiske carcinogener5.1 Kvantitativ vurderinger For langt de fleste typer af effekter findes der en tærskel, det vil sige en grænse (koncentration eller dosis), hvorunder der ikke ses effekt(er). Sådan forholder det sig antageligt for non-genotoksiske carcinogener, hvor den tilgrundliggende mekanisme for stoffets indflydelse på udvikling af tumorer er epigenetisk, det vil sige ikke via en beskadigelse af arvematerialet. I disse tilfælde fastlægges TDI med udgangspunkt i et nul-effektniveau (NO(A)EL) eller det laveste effektniveau (LO(A)EL) for de(n) kritiske effekt(er) under anvendelse af en usikkerhedsfaktor (UF). Principperne herfor er beskrevet nærmere kapitel 4. For visse typer af effekter findes der muligvis ikke en tærskel. Sådan forholder det sig antageligt for genotoksiske carcinogener, hvor den tilgrundliggende mekanisme for udvikling af tumorer er en beskadigelse af arvematerialet. Dette betyder i teorien, at en hvilken som helst eksponering , hvor lav den end måtte være, vil medføre en risiko for udvikling af tumorer, der er større end nul. Det skal dog understreges, at man internationalt i de senere år har diskuteret og fortsat diskuterer, hvorvidt der foreligger er tærskel for genotoksiske effekter eller ej. Under antagelse af at der for genotoksiske carcinogener ikke findes en tærskel, kan der således ikke anvendes et NO(A)EL eller LO(A)EL som udgangspunkt for estimering af TDI. I stedet foretages en kvantitativ risikovurdering. Principperne for den kvantitative risikovurdering uddybes i dette kapitel. I 5.3 er gennemgået fire forskellige kvantitative risikovurderingsmetoder/-modeller. Ved estimering af TDI for genotoksiske carcinogener er der flere forhold, der skal afklares, inden selve beregningen kan foretages: 1) kvalitativ vurdering (se 3.6), 2) kvantitativ vurdering (se 5.1 og 5.3), og 3) tolerabel risiko (se 5.2). Den administrative praksis i Danmark er baseret på, at man i videst muligt omfang skal undgå eksponering for genotoksiske carcinogener, og at disse stoffer bør erstattes med andre mindre farlige stoffer. I de tilfælde, hvor det ikke er muligt at finde alternativer kan det være nødvendigt at foretage en risikoberegning. (MST 1990). I relation til fastsættelse af kvalitetskriterier for genotoksiske carcinogener i luft, jord og drikkevand anvender Miljøstyrelsen den såkaldte one-hit model ved den kvantitative risikovurdering til beregning af TDI (afsnit 5.3.1). 5.1 Kvantitativ vurderingerVed den kvantitative vurdering (potensvurderingen) af genotoksiske carcinogener tages stilling til spørgsmålet: Hvor stor en dosis skal der til, for at det medfører en vis hyppighed af kræfttilfælde? Dyreforsøg med carcinogener har vist, at der kan være endog meget store forskelle mellem forskellige stoffer med hensyn til de doser, der skal til for at inducere tumorer i forsøgsdyr, det vil sige, at potensen med hensyn til carcinogen effekt hos forsøgsdyr varierer meget fra stof til stof. Der er ingen grund til at tro, at det skulle forholde sig anderledes for mennesker i den henseende. Med henblik på en kvantitativ risikovurdering af humane genotoksiske carcinogener foretrækkes velgennemførte og valide epidemiologiske undersøgelser. Såfremt sådanne foreligger for et givent stof, for eksempel epidemiologiske data vedrørende påvirkning af mennesker i arbejdsmiljøet, kan disse anvendes kvantitativt til at omregne fra arbejdstidseksponering til livstidseksponering. Det må imidlertid erkendes, at det er yderst sjældent, at sådanne epidemiologiske undersøgelser findes, blandt andet mangler der ofte præcise oplysninger om eksponeringsniveauer, der kan associeres med en forhøjet hyppighed af tumorer hos arbejderne. Derfor bygger potensvurderingen oftest på resultater fra langtidsdyreforsøg. Ligesom ved beregning af TDI for effekter, hvor det antages, at der findes en tærskelværdi, er der ved den kvantitative vurdering i relation til kræftfremkaldende effekter for genotoksiske carcinogener en række faktorer, som gør, at det kan være meget vanskeligt at fastlægge en bestemt værdi. Når et genotoksisk carcinogen gives i forskellige doser til grupper af forsøgsdyr, vil der typisk ved de laveste doser ses ingen eller kun en lille stigning i hyppigheden af tumorer, mens hyppigheden stiger med stigende dosis. Der vil altid være en vis hyppighed af tumorer, selv om dyrene ikke er blevet eksponeret for teststoffet, det vil sige forekomst af spontane tumorer. Lige meget hvor mange dyr der indgår i et enkelt studie for derved at gøre studiet mere robust overfor forekomsten af spontane tumorer og for at øge studiets følsomhed, kan det således rent principielt aldrig udelukkes, at stoffet kan være carcinogent. Optegnes dosis-responssammenhængen (for f.eks. hyppigheden af tumorer i %) i et diagram med logaritmen til dosis ud ad x-aksen og respons (hyppighed af tumorer) op ad y-aksen, fås for de fleste genotoksiske carcinogener en S-formet kurve (figur 3.1.1.). For at udtrykke graden af det kræftfremkaldende respons (potensen) benyttes en dosisdeskriptor, for eksempel TD5, der defineres som den dosis/koncentration der er associeret med en 5% forekomst (incidens) af tumorer i forsøgsdyr eller i epidemiologiske studier. Dosisdeskriptoren anvendes som udgangspunkt for estimering af forholdet mellem eksponering og potens (engelsk: Exposure/ Potency Indeks EPI), det vil sige, den estimerede daglige eksponering hos mennesker divideret med TD5. Et EPI på 10-6 repræsenterer således en forskel på 106 mellem den humane eksponering og den eksponering på den nedre del af dosis-responskurven, hvorpå potensestimatet er baseret. Værdien på 5% er arbitrært valgt, og valg af en anden værdi vil ikke påvirke de relative potenser for en række stoffer. Dosisdeskriptoren TD5 anvendes af de canadiske myndigheder i relation til vurdering af prioriterede stoffer i henhold til den canadiske miljøbeskyttelseslov. Dosisdeskriptoren TD50 er blevet valgt som det
mest praktiske kvantitative estimat for potensen af genotoksiske carcinogener af den britiske Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. I EU sammenhæng er dosisdeskriptoren TD25 blevet foreslået som en parameter til potensvurdering med henblik på fastsættelse af specifikke koncentrationsgrænser for indhold af kræftfremkaldende stoffer i præparater og produkter i henhold til klassifikation og mærkning af præparater (se 5.3.4). US-EPA anvender dosisdeskriptoren LED10 (se 5.3.3). Der kan anvendes forskellige statistiske modeller til at beskrive sammenhængen mellem dosis og respons og forskellige fordelinger til at beskrive tilfældige udfald. Fælles for disse modeller og fordelinger er, at de forudsætter kendskab til en række parametre, der indgår i beskrivelsen af den enkelte model. Men som ofte kendes disse parametre ikke eksakt på baggrund af det foreliggende datamateriale, hvorfor kurveforløbene derfor bedst muligt må tilpasses efter de aktuelle måledata. En række modeller er blevet udviklet med henblik på at ekstrapolere fra tumorhyppigheden observeret ved de relativt høje doser anvendt i forsøgsdyr til de som regel meget lavere niveauer, der er relevante i relation til befolkningens eksponering for kemiske stoffer via miljøet (engelsk: low-dose risk extrapolation). Eksempler på udvalgte modellers anvendelse er nærmere beskrevet i 5.3. Miljøstyrelsen har siden 1990 ved estimering af en TDI for genotoksiske carcinogener anvendt den såkaldte one-hit model (se 5.3.1), med mindre der er specifikke forhold, som taler for en anvendelse af andre modeller. En hidtil meget anvendt matematiske model er den såkaldte lineære multistadiemodel (engelsk: Linearised Multi-Stage (LMS) model - se 5.3.2), som blandt andet har været anvendt af US-EPA gennem mange år og af WHO (1996) i relation til guidelines for drikkevandskvalitet. Andre metoder/modeller til kvantitativ risikovurdering er blevet foreslået i de seneste år. For eksempel har US-EPA (1996) fremsat forslag til nye retningslinier for kvantitativ risikovurdering af genotoksiske carcinogener og anbefalet som standard at anvende lineær ekstrapolation fra en benchmark dose (afsnit 3.2.2) med udgangspunkt i dosisdeskriptoren LED10 (afsnit 5.3.3). Inden for EU har der været fremsat forslag om at anvende en mere simplificeret metode baseret på dosisdeskriptoren T25 fra dyreforsøg som basis for den kvantitative risikovurdering (afsnit 5.3.4). 5.2 Tolerabel risikoVed vurdering af tolerabel risiko tages stilling til spørgsmålet: Hvilken statistisk risiko kan anses for tolerabel eller acceptabel? Der findes hverken nationalt eller internationalt faste regler for, hvilken livstidsrisiko der kan tolereres eller accepteres, da dette i høj grad er et politisk/administrativt spørgsmål. Den tolerable livstidsrisiko kan derfor være forskellig hos forskellige myndigheder og i relation til forskellige befolkningsgrupper (den generelle befolkning eller arbejdere). En livstidsrisiko på én ud af en million (10-6) blev først indført som basis for grænseværdier i 1979 som del af amerikansk lovgivning om forurening af fødevarer. En livstidsrisiko for at udvikle tumorer på 10-6 betyder, at livslang eksponering for den pågældende dosis eller koncentration kan medføre, at én ud af en million eksponerede individer i princippet kan udvikle én tumor, men ikke at det nødvendigvis sker. En livstidsrisiko på mellem 10-6 og 10-7 anses for at være tolerabel, og en livstidsrisiko, der er mindre end 10-7, anses for at være acceptabel. Miljøstyrelsens hidtidige administrative praksis i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand har været en livstidsrisiko på 10-6 (MST 1990). Tilsvarende tages der inden for EU udgangspunkt i en dosis svarende til en 10-6 livstidsrisiko, når der fastsættes drikkevandskvalitetskrav eller grænseværdier for udeluft for genotoksiske carcinogener (MST 2002). WHO (1996) opererer i relation til guidelines for drikkevandskvalitet med en livstidsrisiko på 10-5. Dog anføres i de enkelte stofmonografier også de koncentrationer, der er associeret med livstidsrisici på 10-4 og 10-6 med den begrundelse, at det er op til det enkelte land selv at vælge hvilken livstidsrisiko, der anses for at være tolerabel. I relation til guidelines for luftkvalitet har WHO (2000) valgt at beregne det risikoestimat (engelsk: incremental unit risk estimate), som er den risiko, der er associeret med livstidseksponering for en given koncentration (1 µg/m³) af et genotoksisk carcinogen i luften. I de enkelte stofmonografier anføres endvidere de koncentrationer, der er associeret med livstidsrisici på 10-4, 10-5 og 10-6 med samme begrundelse, som anført for drikkevand, se ovenfor. US-EPA (1998) anvender i forbindelse med fastsættelse af 'Maximum Contaminant Level Goals' (MCLG) i henhold til 'Safe Drinking Water Act' for kræftfremkaldende stoffer placeret i gruppe C (se 3.6.2) en livstidsrisiko på 10-6 til 10-5. For stoffer placeret i gruppe A eller B fastsættes MCLG til nul. For disse stoffer fastsættes derudover 'Maximum Contaminant Levels' (MCL) med udgangspunkt i en livstidsrisiko på 10-6 til 10-4. MCLG er udelukkende fastsat på baggrund af sundhedsmæssige betragtninger, mens der ved fastsættelse af MCL også tages hensyn til økonomiske og teknologiske elementer. I US-EPA's database IRIS anføres for carcinogene stoffer et risikoestimat (engelsk: unit risk) for inhalation af luft og for indtagelse via drikkevand. Endvidere anføres de koncentrationer i luft henholdsvis drikkevand, der er associeret med livstidsrisici på 10-4, 10-5 og 10-6. (IRIS 2002). 5.3 Kvantitativ risikovurdering, eksempler på metoder/modellerDer er udviklet forskellige modeller til beregning af, hvor stor en risiko for udvikling af tumorer en given eksponering for et genotoksisk carcinogen udgør. I alle modeller anvendes en eller anden form for statistisk ekstrapolation fra dosisniveauer med kendte eksperimentelle værdier til de som regel meget lavere dosisniveauer, der oftest er relevante i relation til den generelle befolknings eksponering for kemiske stoffer i miljøet. Modellerne beskriver relationen mellem den administrerede daglige dosis (eller koncentration) og den resulterende tumorhyppighed. I de fleste dyreforsøg, herunder OECD- og EU guidelineforsøg for carcinogen effekt, anvendes som regel kun få dosisniveauer (oftest 2 eller 3). I de fleste dyreforsøg vælges doserne således, at responset vil ligge mellem 10 og 90%, men de få doser betyder, at der kun er nogle få punkter til beregning af dosis-responskurven, og derfor vil de fleste datasæt inden for dette interval (10-90% respons) stort set passe lige godt ind i forskellige matematiske modeller. Det er således som oftest ikke muligt at skelne mellem forskellige modellers anvendelighed ud fra de givne dyreeksperimentelle data. Problemerne opstår imidlertid, når der skal ekstrapoleres ned til lavdosisområdet, hvor der ikke findes dyreeksperimentelle data til at beregne dosis-responskurven. Det har således vist sig, at modellerne ikke nødvendigvis giver samme resultat i lavdosisområdet, hvorfor risikoestimaterne kan variere meget afhængigt af den valgte model og i nogle tilfælde op til flere størrelsesordener. Det er derfor af stor betydning, hvilken model der vælges, når risici og tolerable grænser for genotoksiske carcinogener skal beregnes. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand kan anvendelse af forskellige modeller ved ekstrapolering fra de doser, der resulterer i en målelig hyppighed af tumorer hos forsøgsdyr (for eksempel 10 dyr med tumorer per 100 dyr, eller 10-1) til doser, der vurderes at være tolerable (10-6), medføre store forskelle i den beregnede tolerable dosis og dermed i det beregnede kvalitetskriterium. Der er udviklet en række matematiske modeller, der er egnede til at ekstrapolere virkningen af genotoksiske carcinogener, det vil sige stoffer, der udøver deres virkning gennem et eller flere `hit' (kritiske, genotoksiske skader). Til denne gruppe hører one-hit modellen (afsnit 5.3.1), som Miljøstyrelsen hidtil har anvendt i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand. Også den hidtil mest anvendte matematiske model, den såkaldte lineære multistadiemodel (engelsk: Linearised MultiStage (LMS) model) (afsnit 5.3.2), hører til denne gruppe af modeller og har blandt andet været anvendt af US-EPA gennem mange år og af WHO (1996) i relation til guidelines for drikkevandskvalitet. Andre metoder/modeller til kvantitativ risikovurdering er blevet foreslået i de seneste år. For eksempel har US-EPA i 1996 fremsat nye retningslinier for kvantitativ risikovurdering af genotoksiske carcinogener og anbefalet som standard at anvende lineær ekstrapolation fra en benchmark dose (afsnit 3.2.2) med udgangspunkt i dosisdeskriptoren LED10 (afsnit 5.3.3). Inden for EU har der været fremsat forslag om at anvende en mere simplificeret metode baseret på dosisdeskriptoren T25 fra dyreforsøg som basis for den kvantitative risikovurdering (afsnit 5.3.4). Også anvendelse af såkaldt biologisk baseret modellering (engelsk: biologically-based modelling) er blevet introduceret. Disse modellerne forsøger at inkorporere biologiske fænomener, der betragtes som værende essentielle i udvikling af tumorer ved eksponering for genotoksiske carcinogener, for eksempel cellekinetik og mutationsrater. Det ligger implicit i disse modeller, at der kræves data vedrørende disse forskellige biologiske parametre, og sådanne data er svære at kvantificere og findes kun yderst sjældent. Således kan disse modeller på nuværende tidspunkt ikke anvendes i den kvantitative risikovurdering i reguleringsmæssigt øjemed, og modellerne vil derfor ikke blive omtalt yderligere i denne rapport. 5.3.1 One-hit modellenI relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand anvender Miljøstyrelsen one-hit modellen, med mindre der er specifikke forhold, som taler for at anvende andre modeller. Begrundelsen for at benytte one-hit modellen er: 1) Modellen undervurderer i mindre grad end de andre modeller risikoen ved lavere koncentrationer, og 2) den er let at anvende (MST 1990). Nedenstående gennemgang er primært baseret på følgende referencer: MST (1990), MST (1992), LST (1990). Hit-modeller er egnede til at ekstrapolere virkningen af genotoksiske carcinogener, der udøver deres virkning gennem et eller flere 'hit', det vil sige kritiske, genotoksiske skader. Dette betyder, at et bestemt antal 'hit' skal ramme en celle, før denne bliver til en malign celle (kræftcelle). Det ligger implicit i hit-modellerne, at alle 'hit' sker i én bestemt celle, der først begynder at dele sig og udvikle sig til en tumor, når den har opnået det nødvendige antal 'hit'. Dette er imidlertid i dårlig overensstemmelse med eksperimentelle data, der viser, at proliferation af de celler, der har fået det første 'hit' (de initierede celler), til præneoplastiske læsioner væsentligt forøger risikoen for, at andet 'hit' netop rammer en initieret celle. One-hit modellen forudsætter, at kun et enkelt 'hit' skal ramme en celle, for at den kan udvikles til en malign celle. Således oversimplificerer one-hit modellen processen, mens de højere hit-modeller lægger en stram restriktion på muligheden for, at mere end én kritisk skade rammer samme celle. Den simple lineære one-hit model har vist sig at give omtrent samme resultater som den lineære multistadiemodel (afsnit 5.3.2) i lavdosisområdet. Inden for de generelle usikkerheder på beregningerne, gør det således ikke den store forskel, hvilken af de to modeller der anvendes i den kvantitative risikoberegning. Beregninger ved brug af one-hit modellen, som det praktiseres i relation til fastsættelse af kvalitetskriterier i luft, jord og drikkevand, er dog betydeligt simplere end ved brug af den lineære multistadiemodel, da sidstnævnte model kun kan anvendes med computerbaserede beregningsprogrammer. Ved benyttelse af one-hit modellen til beregning af TDI i relation til udvikling af systemiske tumorer med udgangspunkt i data fra dyreforsøg med oral administration eller dermal applikation af stoffet kan nedenstående formel (figur 5.3.1.1) anvendes. Ved denne beregning af TDI foretages korrektion af dosis (sidste led i nævneren) for forskelle i kropsstørrelse mellem dyr og mennesker på basis af overfladearealet (se 4.4.1.2). Dosis D skal angives i enheden mg/kg legemsvægt per dag. Hvis eksponeringen i et givent studie er udtrykt som en koncentration af stoffet i for eksempel foder, drikkevand eller luft, foretages der en omregning til enheden mg/kg legemsvægt per dag under anvendelse af standardforudsætningerne (se 6.2). Figur 5.3.1.1 Beregning af TDI i relation til udvikling af systemiske tumorer med udgangspunkt i et studie med oral administration eller dermal applikation af et stof
Ved beregning af TDI (enhed: mg/kg legemsvægt per dag) eller den tolerable koncentration (TK) (enhed: mg/m³) for genotoksiske carcinogener med udgangspunkt i data fra dyreforsøg, hvor dyrene er eksponeret for stoffet via inhalation bør one-hit modellen modificeres på følgende måde: Sidste led i nævneren i formlen i figur 5.3.1.1 bør udgå, idet der ikke bør foretages korrektion af dosis for forskelle i kropsstørrelse mellem dyr og mennesker, da mennesker og dyrs indåndingsvolumen over tid er relateret til behovet for ilt og dermed også til det enkelte individs kaloriebehov, hvorfor der implicit er taget højde for forskelle i metabolisk kapacitet mellem dyr og mennesker (se 4.4.1.2). Ved beregningen af TDI (enhed: mg/kg legemsvægt per dag) skal dosis D angives i enheden mg/kg legemsvægt per dag, hvorfor der foretages en omregning af koncentrationen i indåndingsluften (enhed mg/m³) til dosis D (enhed mg/kg legemsvægt per dag) under anvendelse af standardforudsætningerne (se 6.2). Ved beregning af TK er dosis D lig med koncentrationen i indåndingsluften (enhed mg/m³). De øvrige parametre er de samme, som angivet under figur 5.3.1.1. Den modificerede one-hit model kan også anvendes i forbindelse med genotoksiske carcinogener, der fremkalder lokale tumorer i luftvejene, idet man her antager, at det er stoffets koncentration i luften, der er af betydning for udviklingen af tumorer. Når one-hit modellen anvendes på inhalationsdata skal det i de konkrete tilfælde vurderes, om eksponeringen af dyrene har medført nedsættelse af deres indåndingsfrekvens, da dette i givet fald vil betyde, at dyrene har været udsat for lavere koncentrationer end den nominelle koncentration i inhalationskammeret. Endvidere skal der i de konkrete beregninger også tages hensyn til, om retentionen af stoffet ved de forholdsvis høje eksponeringsniveauer ved inhalationsforsøg er sammenlignelig med retentionen ved de miljømæssigt betydeligt lavere niveauer. En række forudsætninger skal være opfyldt for at one-hit modellen kan benyttes med tilstrækkelig sikkerhed (MST 1990, 1992):
5.3.2 Den lineære multistadiemodelDen lineære multistadiemodel (engelsk: Linearised MultiStage (LMS) model) har gennem mange år været US-EPA's foretrukne matematiske model til kvantitativ risikovurdering af genotoksiske carcinogener og har også været anvendt af WHO (1996) i relation til guidelines for drikkevandskvalitet. Nedenstående gennemgang er blandt andet baseret på følgende referencer: LST (1990), Lovell & Thomas (1996), Grandjean (1997). I relation til kemiske stoffer vurderes en multi-hit model at give bedre mening rent biologisk end one-hit modellen, idet den vurderes som værende i bedre overensstemmelse med den eksisterende viden om genotoksiske carcinogeners virkningsmekanisme(r). En multi-hit model forudsætter, at mindst to reaktioner er nødvendige for at fremkalde tumorer, og at sandsynligheden for hver af dem beskrives ved en Poisson fordeling. Det ligger implicit i hit-modellerne, at alle 'hit' sker i én bestemt celle, der først begynder at dele sig og udvikle sig til en tumor, når den har opnået det nødvendige antal 'hit'. Dette er imidlertid i dårlig overensstemmelse med eksperimentelle data, der viser, at proliferation af de celler, der har fået det første 'hit' (de initierede celler), til præneoplastiske læsioner væsentligt forøger risikoen for at andet 'hit' netop rammer en initieret celle. Således lægger hit-modellerne en stram restriktion på muligheden for, at mere end én kritisk skade rammer samme celle. Blandt andet for at tage højde for disse problemer med hit-modellerne blev de såkaldte multistadiemodeller udviklet. Den originale Armitage-Doll multistage model (publiceret i 1954) antog, at en normal celle skal igennem en serie irreversible, genetiske ændringer eller stadier, mutationer, i en bestemt rækkefølge, før den bliver til en malign celle og fortsætter med at udvikle sig til en tumor. Rent matematisk antog modellen, at et genotoksisk carcinogen påvirker mindst ét af disse stadier. Multistagemodellen blev valgt til regulatoriske formål, fordi modellen syntes at have paralleller til den biologiske forklaring af genotoksiske carcinogeners virkningsmekanisme. Implicit i modellen ligger, at der kræves data vedrørende biologiske fænomener, der betragtes som værende essentielle i kræftprocessen, som for eksempel cellekinetik og mutationsrater. Sådanne parametre bestemmes ikke i de gængse guideline-forsøg, og det har derfor været nødvendigt at simplificere modellen med henblik på anvendelse til regulatoriske formål. Den lineære multistadiemodel (LMS) blev udviklet fra den originale multistagemodel ved at tilpasse et polynomium til de observerede tumorhyppigheder. LMS forudsætter, at alle typer af cancer forårsaget af genotoksiske carcinogener har en fælles mekanisme, og at et hvilket som helst genotoksisk carcinogen påvirker denne del af kræftprocessen. LMS er praktisk taget lineær i lavdosisområdet, og denne lineære del benyttes til ekstrapolation mod nul. Som udgangspunkt i den kvantitative beregning anvendes data fra et langtidsstudie i gnavere. En `best fit' kurve tilpasses data fra studiet ved at anvende et computerbaseret kurvefitningsprogram (der er indtil flere forskellige muligheder). Ud fra `best fit' kurven estimeres forskellige parametre (kaldet Maximum Likelihood Estimates - MLE) baseret på den statistiske procedure, der er anvendt ved tilpasning af kurven. Disse estimater vurderes som værende `best fit estimates' og anvendes som udgangspunkt ved ekstrapolation til lavdosisområdet. En af MLE-parametrene (den såkaldte lineære parameter q1 i LMS) er i lavdosisområdet stort set ækvivalent med hældningen på kurven for sammenhængen mellem dosis og tumorhyppighed. Den øvre 95% konfidensgrænse for estimatet af q1 kaldes q1* og er central i US-EPA's anvendelse af LMS i den kvantitative risikovurdering, idet q1* repræsenterer en øvre grænse eller `worst case' estimat for dosisresponssammenhængen ved lave doser. Parameteren q1* benævnes også `unit carcinogenic risk' eller `carcinogenic potency factor'. Estimaterne af parametrene q1 og q1* anvendes til at estimere enten den risiko, der er associeret med en specifik dosis, eller den dosis der er associeret med en specifik forøgelse i risiko. Den dosis, der er associeret med en 10-6 livstidsrisiko, kaldes af US-EPA for `virtually safe dose' (VSD). Multistadiemodellerne har forskellige forudsætninger, og det er ikke muligt at afgøre, hvorvidt disse er opfyldt. For eksempel har det vist sig, at nogle af de forudsætninger, der ligger implicit i LMS, ikke længere er biologisk realistiske. Som eksempler herpå kan nævnes, at forløbet gennem de forskellige stadier ikke nødvendigvis er irreversibelt, at rækkefølgen for de forskellige processer kan variere, at `ventetiden' i de forskellige stadier sandsynligvis ikke er statistisk uafhængig, at cellerne muligvis ikke går gennem de forskellige stadier uafhængigt af hinanden, og at blandt andet celledeling kan påvirke forløbet. Endvidere kan de kinetiske forhold ved lave doser være anderledes end ved de højere doser, der normalt anvendes i dyreforsøg. US-EPA anvendte oprindeligt one-hit modellen, men valgte siden hen LMS, fordi LMS med dens flere variable parametre giver en bedre kurvefitning. I praksis har det imidlertid vist sig, at LMS giver omtrent samme resultater som den mere simple lineære one-hit model i lavdosisområdet, dog tenderer one-hit modellen til at være en anelse mere konservativ end LMS. Inden for de generelle usikkerheder på beregningerne giver det ofte således ikke den store forskel, hvilken af de to modeller der anvendes i den kvantitative risikoberegning. Ligesom ved one-hit modellen har US-EPA ved anvendelse af LMS anvendt dosiskorrektion for forskelle i kropsstørrelse mellem dyr og mennesker baseret på overfladeareal, når der blev taget udgangspunkt i studier med oral administration af stoffet. 5.3.3 Kvantitativ risikovurdering baseret på dosisdeskriptoren LED10I 1996 fremsatte US-EPA (1996) forslag til nye retningslinier for kvantitativ risikovurdering af kræftfremkaldende stoffer. Et revideret forslag til retningslinier er publiceret i 1999 (US-EPA 1999), og denne udgave er gældende, indtil de endeligt reviderede retningslinier publiceres, hvilket forventes af finde sted i løbet af 2002 US-EPA 2001). Nedenstående gennemgang er blandt andet baseret på følgende reference: US-EPA (1998), US-EPA (1999), Dragsted (1997). En af ændringerne i forhold til de tidligere retningslinier er, at LMS ikke længere anbefales som standard ved den kvantitative risikovurdering af genotoksiske carcinogener, men i stedet anbefales det i de tilfælde, hvor der ikke er noget bevis for nonlinearitet i dosis-responssammenhængen, som standard at anvende lineær ekstrapolation fra en benchmark dose (afsnit 3.2.2) med udgangspunkt i dosisdeskriptoren LED10. En anden ændring er, at der ikke længere kun tages udgangspunkt i den observerede hyppighed af tumorer i det valgte dyreforsøg, men også andre effekter relateret til cancer. En tredje ændring er, at korrektion af orale doser for forskelle i kropstørrelse mellem dyr og mennesker nu anbefales på basis af stofskiftet frem for på basis af overfladeareal (afsnit 4.4.1.2). I de nye retningslinier fremføres det, at respons i et dyreforsøg kan inkludere tumorer eller andre effekter relateret til cancer. Eksempler på sidstnævnte, som sammenfattes under begrebet `precursor data' er ændringer i DNA, chromosomer, andre essentielle makromolekyler, effekter på `growth signal transduction', inducering af fysiologiske eller hormonelle ændringer, effekter på celleproliferation, eller andre effekter som spiller en rolle i kræftprocessen. Det første kvantitative trin er en vurdering af dosis-responssammenhængen inden for det dosisområde, der er anvendt i det valgte dyreforsøg. Der er skitseret to muligheder: 1) udvikling af en biologisk baseret eller en case-specifik model, og 2) kurvefitning af tumor- eller `precursor' data. Den biologisk baserede model anbefales som `first choice', men det erkendes også, at der kun for et fåtal af stoffer vil være tilstrækkelige data til at udvikle denne type af modeller. Derfor foretages vurdering af dosis-responssammenhængen oftest på baggrund af kurvefitning af tumor- eller `precursor' data. Som standard udgangspunkt (engelsk: point of departure) anbefales at anvende den nedre 95% konfidensgrænse for den dosis, der er associeret med 10% ekstra risiko, benævnt LED10. Denne metode er faktisk identisk med 'benchmark dose' metoden (afsnit 3.2.2), det vil sige, at LED10 er identisk med BMD10. Der anvendes et computerbaseret kurvefitningsprogram til beregning af LED10. Rationalet for valget af 10% responset er, at et 10% respons er lige omkring grænsen for følsomheden med henblik på at kunne opnå statistisk signifikante forskelle i de fleste langtidsstudier i gnavere. Rationalet for valget af den nedre 95% konfidensgrænse er, at der hermed tages højde for den eksperimentelle usikkerhed. Sådanne usikkerheder inkluderer blandt andet faktorer som antal doser i studiet, afstand mellem de anvendte dosisniveauer, antal dyr per dosisgruppe, nøjagtighed af dosisbestemmelser (engelsk: precision and accuracy), nøjagtighed (engelsk: accuracy) for de patologiske fund, samt valg af lavdosisekstrapolation. Det næste kvantitative trin er ekstrapolation til lavdosisområdet. Der kan anvendes forskellige ekstrapolationsmetoder. I henhold til de nye retningslinier afhænger valget af ekstrapolationsmetode af det pågældende stofs virkningsmåde (engelsk: mode of action) og er således kædet sammen med konklusionerne i farlighedsvurderingen. Også i dette trin anbefales de biologisk baserede modeller som `first choice', men i erkendelse af at der kun yderst sjældent vil være et tilstrækkeligt datagrundlag for anvendelse af sådanne modeller anbefales derfor 3 forskellige fremgangsmåder (engelsk: default extrapolation approaches): 1) `default linear extrapolation approach', 2) `default non-linear approach', eller 3) `both linear and non-linear approaches'. Den sidstnævnte fremgangsmåde anvendes i de tilfælde, hvor der er tale om mere end en enkelt virkningsmåde for udvikling af tumorer i forskellige væv og organer. De 2 andre fremgangsmåder beskrives nærmere i det efterfølgende. `Default linear extrapolation approach', som i realiteten er en erstatning for LMS, anvendes i følgende tilfælde: 1) stoffer som er genotoksiske via en reaktion med DNA; 2) stoffer hvor der er evidens som understøtter en anden virkningsmåde end reaktion med DNA, og hvor der kan antages at være linearitet i lavdosisområdet; og 3) stoffer hvor der mangler information vedrørende virkningsmåde. Sammenfattende betyder ovenstående, at `default linear extrapolation approach' anvendes i alle de tilfælde, hvor der ikke er data, der direkte understøtter anvendelsen af `default `non-linear approach', se senere. `Default linear extrapolation approach' vurderes generelt af US-EPA som værende et konservativt `approach'. Ved selve ekstrapolationen til lavdosisområdet tages der udgangspunkt i LED10, som er estimeret ved kurvefitning af de observerede data i det valgte dyreforsøg. Er stoffet givet via oral administration eller dermal applikation, foretages der en dosiskorrektion for forskelle i kropsstørrelse mellem dyr og mennesker ved at omregne LED10 bestemt i dyreforsøget til den tilsvarende humane LED10 ved allometrisk skalering på basis af stofskiftet (afsnit 4.4.1.2) på følgende måde: Human LED10 = (LED10) [(BWdyr) / (BWhuman)]0,25 hvor BW er legemsvægten. Der foretages også en dosiskorrektion, hvis eksponeringsperioden (le) har været kortere end forsøgets varighed (Le) ved at gange dosis med forholdet mellem le og Le. En tilsvarende korrektion foretages også, hvis dyrene er blevet doseret færre dage per uge, for eksempel kun i 5 af ugens 7 dage. Selve ekstrapolationen foretages ved at tegne en ret linie mellem den korrigerede humane LED10 og nul (enten nul dosis eller nul respons). Dette udtrykkes matematisk ved ligningen y = mx, hvor y er respons eller incidens, m er liniens hældning (også kaldet `cancer potency factor'), og x er dosis. Hældningen `m' beregnes udfra ligningen m = 0,10 / LED10. Dernæst beregnes den risikospecifikke dosis RSD for en specifik livstidsrisiko for cancer udfra ligningen RSD = livstidsrisiko / m. Livstidsrisikoen for cancer ligger i henhold til US-EPA's retningslinier i intervallet mellem 10-4 og 10-6. `Default non-linear approach' anvendes i de tilfælde, hvor der er tilstrækkelig evidens til at understøtte en antagelse om, at kurveforløbet er nonlineært i lavdosisområdet, for eksempel hvor responset falder meget hurtigt i forhold til dosis. Denne metode anvendes også for kræftfremkaldende stoffer, hvor den tilgrundliggende mekanisme for udvikling af tumorer er non-genotoksisk, det vil sige, i de tilfælde hvor der er en tærskel for udvikling af tumorer. Med udgangspunkt i `point of departure', som kan være LED10, NOAEL eller LOAEL, foretages en `Margen of Exposure' (MoE) analyse ved at sammenligne `point of departure' med det humane eksponeringsniveau. Andre faktorer som `nature of the response', interindividuel variation, speciesforskelle og biopersistens vurderes også i MoE analysen, og der kompenseres for diverse typer af usikkerhed ved at inddrage en sikkerhedsfaktor. Det vil i praksis sige, at den nonlineære metode er lig den benchmarkmetode, som US-EPA anvender for de toksiske effekter, hvor der findes en tærskelværdi. 5.3.4 Kvantitativ risikovurdering baseret på dosisdeskriptoren T25Inden for EU har der været fremsat forslag om at anvende en mere simplificeret metode baseret på dosisdeskriptoren T25 som basis for den kvantitative risikovurdering. T25-metoden kræver ikke anvendelse af computerbaserede beregningsprogrammer. Nedenstående gennemgang er blandt andet baseret på følgende reference: Dybing et al. (1997), Sanner et al. (2001), Roberts et al. (2001). T25 defineres som den kroniske dosis (enhed: mg/kg legemsvægt per dag), som vil give 25% af forsøgsdyrene tumorer i et specifikt væv, efter korrektion for den spontane hyppighed, indenfor den standardiserede levetid for det pågældende species. T25 beregnes med udgangspunkt i et langtidscancerstudie. Den laveste dosis, der giver en signifikant forøgelse af forsøgsdyr med tumorer i et specifikt væv, anvendes i reglen ved beregningen af T25. Men hvis der er en højere hyppighed ved en højere dosis, der giver en lavere T25, anvendes sidstnævnte, med mindre der er særlige begrundelser for ikke at tage udgangspunkt i denne. Hvis der er flere data sæt, beregnes T25 for det mest relevante data sæt. Hvis forskellige data sæt giver T25-værdier, som ligger inden for et relativt snævert interval, anvendes gennemsnittet af disse T25-værdier. Hvis sidstnævnte procedure ikke anvendes, skal rationalet for anvendelse af en anden procedure begrundes nøje. T25 kan enten opnås direkte fra et studie, hvor en given dosis netop resulterer i, at 25% af forsøgsdyrene har fået tumorer. Beregning af T25 udfra andre sammenhænge af tumorhyppighed og dosis foretages ved at gange dosis med faktoren 25/p, hvor p er den aktuelle tumorhyppighed. Hvis eksponeringsvarigheden er kortere end standardlevetiden for det pågældende species, hvis studiet afsluttes inden standardlevetiden, og hvis eksponeringen ikke er kontinuert (24 timer per døgn, 7 dage per uge), foretages dosiskorrektion. Er stoffet givet via oral administration eller dermal applikation, foretages der en dosiskorrektion for forskelle i kropsstørrelse mellem dyr og mennesker ved at omregne T25 til den tilsvarende humane dosis HT25 (i mg/kg lgv./dag) ved allometrisk skalering på basis af stofskiftet (afsnit 4.4.1.2), idet HT25 = T25 / W0,25, hvor W0,25 er skaleringsfaktoren med henblik på korrektion af dosis for forskelle i kropsstørrelse mellem dyr og mennesker baseret på stofskiftet, se tabel 4.4.1.2. Den daglige livstidsdosis for udvikling af tumorer kan beregnes for en specifik livstidsrisiko ved at gange livstidsrisikoen med faktoren HT25/0,25. Eller den specifikke livstidsrisiko for udvikling af tumorer kan beregnes for en specifik daglig livstidsdosis ved at dividere dosis med faktoren HT25/0,25. Endelig foretages en overordnet evaluering af, hvorvidt de tilgængelige data indikerer, at den aktuelle risiko må vurderes at være forskellig fra den beregnede risiko. Der indgår 6 forskellige elementer i denne overordnede evaluering: 1) det tilgængelige datamateriale, 2) epidemiologiske studier, 3) dosis-responssammenhæng, 4) tumorsite, species, stamme, køn, 5) mekanistisk relevans for mennesker, og 6) toksikokinetik. På baggrund af denne overordnede evaluering udarbejdes et `commentary statement'. T25-metoden er blevet evalueret ved at sammenligne resultater opnået ved denne metode med resultater opnået ved anvendelse af LMS og LED10 (Sanner et al. 2001). Ved sammenligningerne blev der taget højde for de forskellige forudsætninger, som de enkelte metoder bygger på. Sammenligningen med LMS viste en god korrelation mellem de 2 metoder (korrelationskoefficient på 0,85 i et log-log plot) baseret på data for 33 stoffer. Forholdet (ratio) mellem T25 og LMS (for 10-5 livstidsrisikoen) lå inden for området fra 0,5 til 2,0 for 30 ud af de 33 stoffer. Fordelingen af de beregnede ratio for alle 33 stoffer blev plottet og de følgende parametre for fordelingen blev estimeret: Middelværdi og medianen 1,21; 5 percentil 0,50; 95 percentilen 1,87; minimum 0,45; og maksimum 2,31. For 24 stoffer gav T25 et højere resultat end LMS og for de resterende 9 stoffer et lavere resultat. Sammenligningen med LED10 viste en god korrelation mellem de 2 metoder (korrelationskoefficient på 0,94 i et log-log plot) baseret på data for 68 stoffer. Forholdet (ratio) mellem T1 og LED1 (ratio er uafhængig af hvilken livstidsrisiko, der anvendes i beregningerne) lå inden for området fra 0,5 til 2,0 for 63 ud af de 68 stoffer. Fordelingen af de beregnede ratio for 65 ud af de 68 stoffer blev plottet, og følgende parametre for fordelingen blev estimeret: Middelværdi og median lig med 1,25; 5 percentil lig med 0,54; 95 percentil lig med 1,98; minimum lig med 0,5; og maksimum lig med 2,3. For 44 stoffer gav T25 et højere resultat end LED og for 21 stoffer et lavere resultat. Anvendelsen af T25-metoden ved kvantitativ risikovurdering af genotoksiske carcinogener har for nyligt (november 2000) været til diskussion på en ECETOC workshop (Robert et al. 2001). Det blev konkluderet, at anvendelse af T25-metoden i relation til risikovurdering er problematisk og begrundet i de usikkerheder, der skyldes en falsk forudsætning vedrørende præcision og linearitet af dosis-responskurver for udvikling af tumorer. Dette er imidlertid ikke et argument, rigtigt eller forkert, der kun kan bruges imod T25-metoden, men kan også bruges mod anvendelse af de 3 andre kvantitative risikovurderingsmetoder (one-hit modellen, LMS og LED10) beskrevet i dette kapitel, da alle disse metoder anvender lineær ekstrapolation fra de relativt høje doser, der anvendes i dyreforsøg til lavdosisområdet, der repræsenterer de eksponeringsniveauer, som den generelle befolkning sædvanligvis udsættes for via miljøet. 5.3.5 WHO's retningslinier for kvantitativ risikovurderingVed kvantitativ risikovurdering af genotoksiske carcinogener har WHO (1996) i relation til guidelines for drikkevandskvalitet som standard anvendt den lineære multistadiemodel (LMS). Men andre modeller har også været anvendt i enkelte tilfælde. Ved estimering af TDI med udgangspunkt i dyreeksperimentelle studier med oral administration eller dermal applikation af det givne stof blev der ikke foretaget korrektion af dosis for forskelle i kropsstørrelse mellem dyr og mennesker. Begrundelsen herfor var, at dette medfører et yderst konservativt estimat for TDI. WHO (2000) har i relation til guidelines for luftkvalitet som standard anvendt en kvantitativ risikovurdering for de stoffer, som er placeret i IARC's gruppe 1 eller 2A (se 3.6.3), mens TDI for stoffer placeret i IARC's gruppe 2B, 3 eller 4 estimeres med udgangspunkt i tærskelværdiprincippet (kapitel 4). Det er dog anført, at der i tilfælde af tilstrækkelig evidens kan afviges fra standardreglen. For eksempel kan TDI for stoffer i IARC's gruppe 1 eller 2A estimeres med udgangspunkt i tærskelværdiprincippet, hvis der er tilstrækkelig evidens for, at et givent stof ikke er genotoksisk. Omvendt kan der for stoffer i IARC's gruppe 2B foretages kvantitativ risikovurdering, hvis der er tilstrækkelig evidens for, at et givent stof er genotoksisk. Ved kvantitativ risikovurdering af genotoksiske carcinogener med udgangspunkt i dyreeksperimentelle studier har den lineære multistadiemodel (LMS) været anvendt som standard til beregning af 'unit risk' (UR), det vil sige det risikoestimat, som er associeret med livstidseksponering for en given koncentration (1 µg/m³) af det givne stof i luften. Men andre modeller har også været anvendt i enkelte tilfælde. Ved kvantitativ risikovurdering af stoffer med udgangspunkt i humane data har den såkaldte 'average relative risk method' været anvendt til beregning af 'unit risk' (UR) som følger:
5.4 SammenfatningFor visse typer af effekter findes der muligvis ikke en tærskel. Sådan forholder det sig antageligt for genotoksiske carcinogener, hvor den tilgrundliggende mekanisme for udvikling af tumorer er en beskadigelse af arvematerialet. Dette betyder i teorien, at en hvilken som helst eksponering, hvor lav den end måtte være, vil medføre en risiko for udvikling af tumorer, der er større end nul. Det skal dog understreges, at man internationalt i de senere år har diskuteret og fortsat diskuterer, hvorvidt der foreligger er tærskel for genotoksiske effekter eller ej. Under antagelse af at der for genotoksiske carcinogener ikke findes en tærskel, kan tærskelværdiprincippet (se kapitel 4) således ikke anvendes med henblik på estimering af TDI for sådanne stoffer. I stedet estimeres TDI ved en kvantitativ risikovurdering, og principperne herfor er uddybet i dette kapitel. Ligeledes er fire forskellige kvantitative risikovurderingsmetoder/-modeller beskrevet (se 5.3). I relation til fastsættelse af kvalitetskriterier i luft, jord og drikkevand har Miljøstyrelsen siden 1990 ved estimering af en TDI for genotoksiske carcinogener anvendt den såkaldte one-hit model (se 5.3.1), med mindre der er specifikke forhold, som taler for en anvendelse af andre modeller. Den lineære multistadiemodel (engelsk: Linearised MultiStage (LMS) model – se 5.3.2) har gennem mange år været US-EPA's foretrukne matematiske model til kvantitativ risikovurdering af genotoksiske carcinogener og har også været anvendt af WHO (1996) i relation til guidelines for drikkevandskvalitet. Den simple lineære one-hit model har vist sig at give omtrent samme resultater som LMS i lavdosisområdet. Inden for de generelle usikkerheder på beregningerne, giver det ofte således ikke den store forskel, hvilken af de to modeller der anvendes i den kvantitative risikoberegning. Andre metoder/modeller til kvantitativ risikovurdering af genotoksiske carcinogener er blevet foreslået i de seneste år. For eksempel har US-EPA (1996) fremsat nye retningslinier og anbefaler nu som standard at anvende lineær ekstrapolation fra en benchmark dose (afsnit 3.2.2) med udgangspunkt i dosisdeskriptoren LED10 (den nedre 95% konfidensgrænse for den dosis, der er associeret med 10% ekstra risiko, oftest beregnet ved kurvefitning af tumor- og/eller 'precursor' data) (afsnit 5.3.3) i stedet for den lineære multistadiemodel (LMS). Ulempen ved LED10-metoden er, såvel som ved LMS, at denne kræver anvendelse af computerbaserede programmer, for at beregningerne kan udføres. Inden for EU har der været fremsat forslag om at anvende en mere simplificeret metode baseret på dosisdeskriptoren T25 som basis for den kvantitative risikovurdering (afsnit 5.3.4). T25-metoden kræver således ikke anvendelse af computerbaserede beregningsprogrammer. T25 defineres som den kroniske dosis (enhed: mg/kg legemsvægt per dag), som vil give 25% af forsøgsdyrene tumorer i et specifikt væv, efter korrektion for den spontane hyppighed, indenfor den standardiserede levetid for det pågældende species. T25-metoden er blevet evalueret ved at sammenligne resultater opnået ved denne metode med resultater opnået ved anvendelse af LMS og LED10. Ved sammenligningerne blev der taget højde for de forskellige forudsætninger, som de enkelte metoder bygger på. Sammenligningen med både LMS (for 33 stoffer) og med LED10 (for 68 stoffer) viste en god korrelation mellem de 2 metoder indbyrdes. Dog har T25-metoden en tendens til at give et højere resultat end LMS og LED10, det vil sige, at T25-metoden ikke er ligeså konservativ som LMS og LED10. Oprindeligt er T25-metoden blevet foreslået som en parameter til potensvurdering med henblik på fastsættelse af specifikke koncentrationsgrænser for indhold af kræftfremkaldende stoffer i præparater og produkter i henhold til klassifikation og mærkning af præparater inden for EU. Metoden er siden hen også blevet foreslået anvendt i relation til kvantitativ risikovurdering af genotoksiske carcinogener inden for EU's risikovurderingsprogram for eksisterende stoffer og ventes at blive den anbefalede metode fremover i henhold til den reviderede Technical Guidance Document (TGD) (se 1.2.1). I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand anbefales det enten at anvende one-hit modellen i den kvantitative risikovurdering til beregning af TDI eller TK eller alternativt T25-metoden. En af disse to metoder anbefales frem for LED10-metoden, idet begge metoder vurderes at give resultater, der er sammenlignelige med LED10-metoden, men beregningerne er mere simple og gennemskuelige og kan udføres uden anvendelse af computerbaserede beregningsprogrammer. Et forhold, der taler for anvendelse af T25-metoden frem for one-hit modellen, er, at denne metode er accepteret i relation til kvantitativ risikovurdering af genotoksiske carcinogener inden for EU's risikovurderingsprogram for eksisterende stoffer. Når den kvantitative risikovurdering foretages med udgangspunkt i dyreeksperimentelle studier, hvor teststoffet er givet ved oral administration eller dermal applikation, anbefales det at foretage dosiskorrektion for forskelle i kropsstørrelse mellem dyr og mennesker på basis af stofskiftet (afsnit 4.4.1.2). Dosiskorrektion er ikke relevant i de tilfælde, hvor teststoffer er administreret via inhalation. Der findes hverken nationalt eller internationalt faste regler for, hvilken livstidsrisiko der kan tolereres eller accepteres, da dette i høj grad er et politisk/administrativt spørgsmål. Den tolerable livstidsrisiko kan derfor være forskellig hos forskellige myndigheder. En livstidsrisiko for at udvikle tumorer på 10-6 betyder, at livslang eksponering for den pågældende dosis eller koncentration kan medføre, at én ud af en million eksponerede individer i princippet kan udvikle én tumor, men ikke at det nødvendigvis sker. En livstidsrisiko på mellem 10-6 og 10-7 anses for at være tolerabel, og en livstidsrisiko. Miljøstyrelsens hidtidige administrative praksis i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand har været en livstidsrisiko på 10-6. Inden for EU anvendes et 10-6 livstidsrisikoestimat som udgangspunkt, når der fastsættes drikkevandskvalitetskrav eller grænseværdier for udeluft for genotoksiske carcinogener. WHO angiver i relation til vejledende grænseværdier for drikkevand og luft de koncentrationer, der er associeret med livstidsrisici på 10-4, 10-5 og 10-6, således at den enkelte myndighed selv kan vælge den koncentration, der svarer til den pågældende myndigheds tolerable livstidsrisiko. I US-EPA's database IRIS anføres for carcinogene stoffer et risikoestimat (engelsk: unit risk) for inhalation af luft og for indtagelse via drikkevand. Endvidere anføres de koncentrationer i luft henholdsvis drikkevand, der er associeret med livstidsrisici på 10-4, 10-5 og 10-6. 5.5 ReferencerDragsted LO (1997). Cancer risk assessment and the EPA guidelines. Hum Ecol Risk Assess 3, 501-505. Dybing E, Sanner T, Roelfzema H, Kroese K and Tennant RW (1997). T25: A simplified carcinogenic potency index: Description of the system and study of correlations between carcinogenic potency and species/site specificity and mutagenicity. Pharmacol Toxicol 80, 272-279. Grandjean P (1997). Farlig forurening. Fra risikovurdering til forebyggelse. Sundhedsstyrelsen. Nyt Nordisk Forlag Arnold Busck, København. IARC (1987): Overall evaluations of carcinogenicity: An updating of IARC monographs volumes 1 to 41, Supplement 7. IARC monographs on the evaluation of carcinogenic risks to humans. World Health Organization, International Agency for Research on Cancer. IRIS (2002). http://www.epa.gov/iris/intro.htm. Lovell DP and Thomas G (1996). Quantitative risk assessment and the limitations of the linearized multistage model. Hum Exp Toxicol 15, 87-104. LST (1990). Kvantitativ risikovurdering af kræftfremkaldende stoffer. Levnedsmiddelstyrelsen, Sundhedsministeriet. MEM (1997). Bekendtgørelse om klassificering, emballering, mærkning, salg og opbevaring af kemiske stoffer og produkter. Miljø- og Energiministeriets bekendtgørelse nr. 801 af 23. oktober 1997. MST (2002). Personlig meddelelse. MST (1992). Sundhedsmæssig vurdering af kemiske stoffer i drikkevand. Vejledning fra Miljøstyrelsen Nr. 1 1992. Miljøministeriet, Miljøstyrelsen. MST (1990). Begrænsning af luftforurening fra virksomheder. Vejledning fra Miljøstyrelsen Nr. 6 1990. Miljøministeriet, Miljøstyrelsen. Roberts RA, Crump KS, Lutz WK, Wiegand H-J, Williams GM, Harrison PTC and Purchase IFH (2001). Scientific analysis of the proposed uses of the T25 dose descriptor in chemical carcinogen regulation. An ECETOC Workshop overview. Arch Toxicol 75, 507-512. Sanner T, Dybing E, Willems MI and Kroese ED (2001). A simple method for quantitative risk assessment of non-threshold carcinogens based on the dose descriptor T25. Pharmacol Toxicol 88, 331-341. US-EPA (2001). Cancer risk assessment guidelines. FR notice (November 2001). http://www.epa.gov/NCEA/raf/pdfs/cancer_gls.pdf. US-EPA (1999). Guidelines for carcinogen risk assessment. NCEA-F-0644, July 1999, Reviewed Draft. http://www.epa.gov/NCEA/raf/cancer.htm. US-EPA (1998). Ambient water quality criteria derivation for the protection of human health – Technical Support Document. Final draft. United States Environmental Protection Agency, Office of Water 4304, EPA-822-B-98-005. US-EPA (1996). Proposed guidelines for carcinogen risk assessment (April 23, 1996). Federal Register 61(79):17960-18011. http://www.epa.gov/NCEA/raf/pdfs/propcra_1996.pdf. WHO (2000). Criteria for carcinogenic endpoint. In: Air quality guidelines for Europe, second edition. World Health Organization, Regional Office for Europe, Copenhagen, 20-29. WHO (1999). Principles for the assessment of risks to human health from exposure to chemicals. Environmental Health Criteria 210, International Programme on Chemical Safety, World Health Organization, Geneva. WHO (1996). 12. Chemical and physical aspects: introduction. In: Guidelines for drinking-water quality, second edition. Volume 2 Health criteria and other supporting information. World Health Organization, Geneva, 121-131. 6 Eksponeringsbetragtninger6.1 Humane eksponeringsestimater Den generelle befolkning kan eksponeres for kemiske stoffer ved inhalation af dampe, aerosoler og støv i luften (i indeklima såvel som udendørs), ved indtagelse af levnedsmidler, drikkevand og jord, ved oral kontakt med forbrugerprodukter, samt ved hudkontakt med drikkevand, jord og forbrugerprodukter. Børn vil i mange tilfælde eksponeres for kemiske stoffer i miljøet i højere grad end voksne, fordi børn dagligt indånder større mængder luft, drikker mere vand og spiser mere mad i forhold til voksne, når der sammenlignes på basis af legemsvægt. Endvidere kan der være forskelle i eksponeringsmønstrene mellem børn og voksne, da børn af naturen er nysgerrige og ofte undersøger ukendte ting ved at putte disse i munden. Især kan eksponering for kemiske stoffer via forurenet jord eller støv være markant højere hos børn, da børn oftest leger på jorden og på gulvet. Forskellene mellem børns og voksnes eksponeringer er nærmere belyst i Nielsen et al. (2001). I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand har Miljøstyrelsen siden 1990 anvendt de standardestimater for eksponering, der er angivet i Miljøprojekt nr. 123 ”Risikovurdering af forurenede grunde” (MST 1990) og gengivet i tabel 6. Værdierne bygger dels på litteraturstudier og dels på en høring af en række institutioner i forskellige lande foretaget af Miljøstyrelsen. Det er fremhævet, at værdierne for indtagelse af jord og for hudkontakt med jord er usikre, og at de er det bedste skøn, der kunne foretages på daværende tidspunkt. Tabel 6: Standardestimater for luft, jord og drikkevand (MST 1990)
a Gartnere og lignende særligt jordeksponerede erhvervsgrupper er ikke inkluderet. I det efterfølgende beskrives de forskellige eksponeringsveje, herunder forskelle mellem børns og voksnes eksponeringer (se 6.1). Endvidere beskrives for de mest anvendte forsøgsdyr de standardeksponeringsestimater for foder, drikkevand og luft, der ligger til grund for omregning af koncentrationer af kemiske stoffer i disse medier til indtagen dosis (enhed: mg/kg legemsvægt per dag) (se 6.2). 6.1 Humane eksponeringsestimater6.1.1 LuftDen generelle befolkning kan eksponeres for kemiske stoffer ved inhalation af dampe, aerosoler og støv i luften i indeklima såvel som udendørs. Ved eksponering via inhalation forstås den koncentration af et givent stof, der er i indåndingsluften. Eksponeringen udtrykkes som en gennemsnitlig koncentration over en tidsperiode, for eksempel mg/m³ per dag. Ved risikokarakterisering, det vil sige vurdering af (nul)effektniveauer i forhold til eksponering, kan der ofte være behov for at estimere den gennemsnitlige daglige dosis, som mennesker udsættes for som følge af eksponering for luftbårne stoffer. Eksponeringskoncentrationen i indåndingsluften kan ifølge US-EPA (1997) omregnes til den gennemsnitlige daglige dosis (L)GDD (enhed: mg/kg legemsvægt per dag) på følgende måde:
Luftvejenes anatomi og fysiologi medfører imidlertid, at koncentrationen af det givne stof bliver mindre ved passagen gennem luftvejene til lungerne, således at den mængde af det givne stof, der optages i kroppen fra lungerne (intern dosis), er mindre end eksponeringskoncentrationen (potentiel dosis). Dette forhold er illustreret i figur 6.1.1. Mængden (volumet) af luft indåndet per dag (m³/dag), i det efterfølgende kaldet ventilationen (engelsk: inhalation rate - IR eller ventilation rate - VR), er således en central faktor ved omregning af eksponeringskoncentration til en gennemsnitlig daglig dosis. VR varierer afhængigt af alder, køn, vægt, helbred og fysisk aktivitetsniveau (arbejde, løb, gang, hvile). VR angives typisk som et minutvolumen (liter luft udåndet per minut) og beregnes som respirationsvolumet (det volumen luft der ind-/udåndes i hvert åndedrag - tidal volumen) ganget med antallet af indåndinger per minut. US-EPA (1997) har i relation til udarbejdelse af Exposure Factors Handbook identificeret 5 nøglestudier og 5 andre studier, der belyser ovennævnte forhold. Resultaterne af disse studier, som alle er amerikanske, viste følgende generelle tendenser: 1) børn i hvile indånder relativt større mængder luft sammenlignet med voksne, 2) mænd indånder større mængder luft end kvinder, 3) unge indånder større mængder luft end ældre, 4) fysisk aktivitet medfører øget ventilation (den relative forskel mellem de enkelte grupper (1-3) er også ved fysisk aktivitet som beskrevet under 1-3), 5) ventilationen (VR) er højere udendørs end indendørs, også når der tages højde for forskelle i fysisk aktivitet, og 6) astmatikere (både børn og voksne) har en øget ventilation (VR) i forhold til raske personer inden for de samme aldersgrupper. Figur 6.1.1. Inhalation – sammenhæng mellem eksponering og dosis. Modificeret fra US-EPA (1997).
I tabel 6.1.1A er udvalgte resultater fra et af nøglestudierne (Layton 1993 - citeret fra US-EPA 1997) præsenteret. I dette studie er der anvendt 3 forskellige procedurer til at estimere den daglige gennemsnitlige ventilation over hele levetiden baseret på en meget stor population (ca. 50000 personer). Disse 3 procedurer gav sammenlignelige resultater. US-EPA har generelt i de fleste risikovurderinger anvendt en standardværdi for ventilationen på 20 m³/dag for voksne, og det er også den værdi, der anvendes i IRIS (Integrated Risk Information System). I Exposure Factors Handbook (US-EPA 1997) anbefales for eksponering gennem længere tid en gennemsnitlig ventilation på 11,3 m³/dag for kvinder og 15,2 m³/dag for mænd (alder voksne: 19 år og opefter). Disse værdier er baseret på resultaterne af undersøgelserne foretaget af Layton (1993). For gruppen børn som sådan anbefales ikke en generel standardværdi for eksponering gennem længere tid, men i stedet for anbefales aldersspecifikke standardværdier, også disse værdier er baseret på resultaterne af undersøgelserne foretaget af Layton (1993). For eksponering i kortere tid er der på basis af forskellige aktivitetsmønstre anbefalet en række specifikke standardværdier. På baggrund af det foreliggende datagrundlag blev det vurderet, at det ikke på det tidspunkt var muligt at anbefale øvre grænser (95 percentiler). US-EPA's anbefalinger er samlet i tabel 6.1.1B. Tabel 6.1.1A. Ventilationen (VR) beregnet på basis af energiindtag fra levnedsmidler. Fra Layton (1993 - modificeret fra US-EPA 1997).
a Inaktiv, defineret som søvn, var 8 timer for aldersgruppen fra 15 år og opefter, 9 timer for aldersgruppen fra 9 til 14 år, 10 timer for aldersgruppen fra 3 til 8 år, og 11 timer for aldersgruppen under 3 år. Principperne for risikovurdering af nye og eksisterende stoffer inden for EU er udførligt beskrevet i Technical Guidance Document (TGD) (se 1.2.1), som lige er blevet gennemgående revideret (EU 2002). Der er ikke angivet deciderede anbefalinger til standardværdier for ventilationen (VR), men i et Appendix (Appendix IV C) er samlet de fysiologiske faktorer, som kan anvendes i relation til risikovurderingerne. For ventilationen er angivet de værdier, som US-EPA har anbefalet i deres Exposure Factors Handbook (US-EPA 1997), se tabel 6.1.1B. WHO tager ved fastsættelse af guidelines for luftkvalitet udgangspunkt i de standardværdier for ventilation (VR), der er anbefalet af International Commission on Radiological Protection (ICRP 1974 - citeret i WHO 1994, 1999). Disse værdier er gengivet i tabel 6.1.1C. Det fremgår af Exposure Factors Handbook (US-EPA 1997), at ICRP i 1981 har publiceret reviderede standardværdier. ECETOC (2001) har samlet eksponeringsdata i Exposure Factors Sourcebook for European Populations. For ventilationen (VR) er præsenteret de data, som US-EPA har anbefalet i deres Exposure Factors Handbook (US-EPA 1997), se tabel 6.1.1B. I relation til eksponering gennem kortere tid har ECETOC anført, at disse data formentligt er repræsentative for individer af en hvilken som helst nationalitet, idet det for en given aktivitet forventes, at VR primært er afhængig af køn og størrelse (vægt). I relation til eksponering gennem længere tid har ECETOC anført, at estimaterne for VR kan variere meget afhængigt af forskelle i livsstil og aktivitetsniveau, da disse estimater er baseret på indtagelse af levnedsmidler og energiforbrug over lang tid. ECETOC konkluderede dog, at US-EPA's anbefalede standardværdier også vil være rimelige estimater for den europæiske befolkning. Tabel 6.1.1B. US-EPA's standardværdier for ventilation (VR) som angivet i Exposure Factors Handbook (US-EPA 1997).
a Ventilationen blev beregnet som et gennemsnit af ventilationen for de forskellige aktivitetsniveauer 6.1.1.1 Sammenfatning, daglig ventilationDe fleste undersøgelser af den gennemsnitlige daglige ventilation (VR) over kortere eller længere tid, herunder variationer i relation til alder, køn, vægt, helbred og fysisk aktivitetsniveau, er foretaget i USA. På baggrund af disse undersøgelser, som er præsenteret og evalueret i Exposure Factors Handbook (US-EPA 1997), er det i Exposure Factors Handbook blevet anbefalet for eksponering gennem længere tid at anvende en gennemsnitlig ventilation på 11,3 m³/dag for kvinder og 15,2 m³/dag for mænd (alder voksne: 19 år og opefter). For gruppen børn anbefales en række aldersspecifikke standardværdier. For eksponering i kortere tid er der på basis af forskellige aktivitetsmønstre anbefalet en række specifikke standardværdier. På baggrund af det foreliggende datagrundlag kunne der ikke anbefales øvre grænser (95 percentiler). De anbefalede standardværdier er samlet i tabel 6.1.1B. Tabel 6.1.1C. WHO's standardværdier for ventilation (VR) (WHO 1994, 1999).
a 8 timers hvile, 16 timers let, ikke arbejdsmæssig aktivitet, 20 timer/dag inden døre Inden for EU er der i relation til risikovurdering af nye og eksisterende stoffer ikke angivet deciderede anbefalinger til standardværdier for VR, men i et Appendix (Appendix IV C) til Technical Guidance Document (TGD) er US-EPA's anbefalinger (se tabel 6.1.1B) angivet med henblik på at kunne anvendes i relation til risikovurderingerne. WHO tager ved fastsættelse af guidelines for luftkvalitet udgangspunkt i de standardværdier for VR (gennemsnitsværdier), der er anbefalet af International Commission on Radiological Protection (ICRP 1974 - citeret i WHO 1994, 1999). Disse værdier er gengivet i tabel 6.1.1C. ECETOC (2001) har samlet eksponeringsdata i Exposure Factors Sourcebook for European Populations. For VR er præsenteret de data, som US-EPA har anbefalet i Exposure Factors Handbook, og det er konkluderet, at disse standardværdier også vil være rimelige estimater for den europæiske befolkning. MST har i relation til fastsættelse af kvalitetskriterier for luft samt ved omregning af luftbåren eksponering til en gennemsnitlig daglig dosis som udgangspunkt hidtil anvendt en standardværdi for VR på 20 m³/dag som et dagligt gennemsnit for voksne med mindre, der har været specifikke forhold, der har talt for anvendelse af en anden standardværdi. Undersøgelser, der ligger til grund for US-EPA's anbefalede standardværdier for en gennemsnitlig ventilation (VR) (se tabel 6.1.1B), vurderes at være mere repræsentative med henblik på vurdering af gennemsnitsværdier, også for den generelle danske befolkning, end de undersøgelser, der ligger til grund for ICRP's standardværdier (gennemsnitsværdier), der anvendes af WHO (se tabel 6.1.1C) samt for de standardværdier (gennemsnitsværdier), der hidtil har været anvendt af Miljøstyrelsen (se tabel 6). Dette er begrundet i, at US-EPA's anbefalinger er baseret på nyere studier (1991-1993), som har anvendt nye og veludviklede metoder til estimering af ventilationen, samt at de forskellige undersøgelser har givet sammenlignelige resultater. Standardværdien på 20 m³/dag, som MST hidtil har anvendt i relation til fastsættelse af kvalitetskriterier for udeluft, vurderes i lyset af de nyere undersøgelser som værende en værdi, der ligger over gennemsnittet for befolkningen som helhed. Det skal bemærkes, at US-EPA på baggrund af det foreliggende datagrundlag vurderede, at der ikke kunne fremsættes anbefalinger vedrørende øvre grænser (95 percentiler). Da der ikke siden hen er blevet publiceret yderligere data med henblik på en vurdering af øvre grænser, kan der således heller ikke på nuværende tidspunkt foretages en vurdering heraf, ej heller en vurdering af hvorvidt MST's standardværdi på 20 m³/dag kan betragtes som en repræsentativ værdi for en 95 percentil for voksne eller børn. Inden for EU er US-EPA's anbefalinger (se tabel 6.1.1B) angivet i TGD'en for risikovurdering af nye og eksisterende stoffer (TGD) med henblik på at kunne anvendes i relation til risikovurderingerne. Også ECETOC har konkluderet, at disse standardværdier vil være rimelige estimater for den europæiske befolkning. Det vurderes således, at US-EPA's anbefalinger fremover vil finde bred anvendelse i Europa i relation til risikovurderinger af kemiske stoffer. 6.1.2 JordDen generelle befolkning kan eksponeres for kemiske stoffer i jord via indtagelse af jordpartikler eller ved direkte hudkontakt med jorden. 6.1.2.1 Indtagelse af jordDen generelle befolkning kan eksponeres for kemiske stoffer via indtagelse af jord- og støvpartikler. Eksponeringen udtrykkes som regel som en gennemsnitlig mængde over en tidsperiode, for eksempel mg/dag. Denne eksponeringsvej er især relevant for mindre børn, idet børn af natur er nysgerrige og ofte undersøger ukendte ting ved at putte disse i munden. Der kan således været tale om decideret jordspisning (geophagia), men også indtagelse af jord- eller støvpartikler ved, at børnene sutter på deres hænder, legetøj eller andre genstande. Et øget bidrag hos børn via denne eksponeringsvej skyldes også, at børn oftest leger på jorden og på gulvet. Der kan være en ulige fordeling børn imellem i relation til indtagelse af jord, idet de fleste børn kun indtager relativt små mængder, mens nogle få børn spiser større mængder jord. Det sidste fænomen kaldes pica (pica defineres generelt som en “gentagen indtagelse af non-nutritive substanser”). Voksne kan også eksponeres for kemiske stoffer via indtagelse af jord- og støvpartikler, som sidder på overfladen af for eksempel madvarer, cigaretter, eller deres hænder. Der er publiceret en række undersøgelser med henblik på at estimere børns daglige indtagelse af jord. De fleste af de ældre studier har forsøgt at estimere indtagelsen af jord med udgangspunkt i den mængde snavs, der var på børnenes hænder samt nogle antagelser omkring børns adfærd. I de nyere studier er der anvendt metoder baseret på målinger i fæces og jord af sporelementer (for eksempel aluminium, silicium, og titanium), der findes i relativt høje mængder i jord, men ikke i levnedsmidler, og som stort set ikke absorberes fra mave-tarmkanalen. I de nyeste studier er der foretaget analyser af massebalancen, idet koncentrationen af sporelementerne er målt både i fæces, jord samt levnedsmidler. US-EPA (1997) har i relation til udarbejdelse af Exposure Factors Handbook identificeret 7 nøglestudier, som alle har anvendt metoder baseret på målinger af sporstoffer samt 9 andre studier. Der er i de enkelte nøglestudier anvendt fra 3 til 8 forskellige sporelementer. Ikke alle de anvendte sporelementer viste sig at være velegnede som udgangspunkt for estimering af den daglige indtagelse af jord, og aluminium, silicium og yttrium viste sig at være de mest pålidelige. Resultaterne af disse studier er summeret i tabel 6.1.2.1A. Tabel 6.1.2.1A. Estimater for daglig indtagelse af jord hos børn. Modificeret fra US-EPA (1997). Referencerne er alle beskrevet i US-EPA (1997).
a AIR er syreuopløselig rest Som det fremgår af tabellen, er der en forholdsvis stor spredning på resultaterne. Estimaterne for børns daglige gennemsnitlige indtagelse af jord varierede fra 39 til 271 mg/dag med et gennemsnit på 146 mg/dag for indtagelse af jord alene og på 191 mg/dag for indtagelse af jord og støv. Resultaterne for 95 percentilen varierede fra 106 til 1432 mg/dag med et gennemsnit på 383 mg/dag for indtagelse af jord alene og på 587 mg/dag for indtagelse af jord og støv. Nogle af disse variationer skyldes, 1) at der ganske enkelt er individuelle forskelle mellem børns indtagelse af jord, 2) at indtagelse af de forskellige sporelementer ikke kun forekommer fra jord, men også fra levnedsmidler eller andre ting som børn putter i munden, 3) der kan være usikkerheder med hensyn til opsamling af ekskreter i bleen, primært fæces, 4) ) sporelementerne kan overføres til fæces ved kontakt med bleen, for eksempel kan disse forekomme i visse hudplejemidler, 5) der er forskelle i optagelse af de enkelte sporelementer fra mave-tarmkanalen, og 6) de indsamlede jordprøver er som regel inhomogene og ikke repræsentative for en gennemsnitlig eksponering. Resultaterne som angivet i tabel 6.1.2.1A er ikke nødvendigvis repræsentative som estimat for den gennemsnitlige daglige indtagelse af jord gennem længere tid, da ingen af studierne er foretaget over længere tid (indsamlingsperiode fra 3 til 8 dage). Endelig vurderes det, at estimaterne i tabel 6.1.2.1A ligger i den høje ende, da alle studier, med en enkelt undtagelse (Calabrese et al. 1989), er foretaget om sommeren, hvor direkte kontakt med jorden forekommer hyppigere end om vinteren. I følge US-EPA (1997) findes der meget få oplysninger vedrørende forekomsten af pica adfærd hos børn, men det synes ikke er være et særligt udbredt fænomen. Denne vurdering er baseret på de 7 nøglestudier, som US-EPA har identificeret med henblik på estimering af den daglige indtagelse af jord for børn (se ovenfor), hvor kun et enkelt barn ud af mere end 600 undersøgte børn udviste pica adfærd overfor jord. Resultaterne for dette barn er vist i tabel 6.1.2.1B. Tabel 6.1.2.1B. Daglig indtagelse af jord for et pica-barn. Fra Calabrese et al. (1991) og modificeret fra US-EPA (1997).
US-EPA (1997) har i relation til udarbejdelse af Exposure Factors Handbook kun identificeret 3 studier, hvor der er estimeret en daglig indtagelse af jord hos voksne. I det ene studie, som tog udgangspunkt i antagelser vedrørende jord- og støvmængder på hænderne, såkaldt `mouthing behaviour' samt udendørs og indendørs aktiviteter, blev der estimeret et dagligt gennemsnit over et år på 60,5 mg/dag. I det andet studie, som tog udgangspunkt i målinger af koncentrationen af arsen i urin, `mouthing behaviour' samt informationer om aktivitetsmønstre, blev der estimeret en daglig indtagelse på 50 mg/dag. I det tredje studie, som tog udgangspunkt i målinger af sporelementer, blev der estimeret en daglig indtagelse fra 30 til 100 mg/dag. Det sidste studie blev af US-EPA vurderet som værende det mest pålidelige. US-EPA har generelt i de fleste risikovurderinger anvendt en standardværdi for indtagelse af jord for børn på 200 mg/dag. I Exposure Factors Handbook (US-EPA 1997) anbefales en standardværdi på 100 mg/dag for børn i alderen op til 6 år. Dette er begrundet i, at de høje estimater (se tabel 6.1.2.1A) er baseret på målinger af titanium, som udviste større variabilitet end estimaterne baseret på de øvrige sporelementer, samt at undersøgelserne foretaget af Calabrese et al. (1989) omfattede et pica-barn. Det er dog anført, at 200 mg/dag kan anvendes som et konservativt estimat. Som 95 percentil anbefales en standardværdi på 400 mg/dag. I en risikovurdering af TCDD har US-EPA anvendt en standardværdi på 5 g/dag for pica-børn. I Exposure Factors Handbook (US-EPA 1997) anbefales en standardværdi på 10 g/dag for pica-børn. Det er understreget, at denne standardværdi er baseret på resultater fra kun et enkelt pica-barn (se tabel 6.1.2.1B). Denne værdi anvendes kun i relation til meget kortvarige (akutte) eksponeringer. US-EPA har generelt i de fleste risikovurderinger anvendt en standardværdi for indtagelse af jord for voksne på 50 mg/dag i industriområder og 100 mg/dag i beboelsesområder og i landbrugsområder. I Exposure Factors Handbook (US-EPA 1997) anbefales en standardværdi på 50 mg/dag. Det er understreget, at denne værdi er meget usikker. US-EPA's anbefalinger er samlet i tabel 6.1.2.1C. Tabel 6.1.2.1C. US-EPA's standardværdier for indtagelse af jord som angivet i Exposure Factors Handbook (US-EPA 1997).
a 200 mg/dag kan anvendes som et konservativt estimat WHO (1994) har angivet en daglig indtagelse af jord på 20 mg/dag. Værdien er en medianværdi og stammer fra Health and Welfare Canada. Det er ikke specificeret, hvorvidt standardværdien gælder for voksne eller børn. ECETOC (2001) har samlet eksponeringsdata i Exposure Factors Sourcebook for European Populations. Heri er det angivet, at den estimerede gennemsnitlige indtagelse af jord er 40 mg/dag for børn og 1 mg/dag for voksne. Disse estimater er baseret på to amerikanske undersøgelser (børn: Calabrese et al. 1997; voksne: Stanek et al. 1997), der er publiceret efter, at US-EPA publicerede Exposure Factor Handbook (US-EPA 1997). ECETOC har valgt at basere deres anbefalinger på de 2 nyeste studier, da disse studier vurderes som værende mere pålidelige som følge af forbedrede metoder til estimering af den daglige indtagelse af jord. Vedrørende studiet af børn (Calabrese et al. 1997) er det dog anført, at udendørsområdet var `a grassy Superfund site', og at der således kan have været en nedsat indtagelse af jord som følge af stedets natur og børns ændrede aktivitetsmønstre i forbindelse med et `Superfund site'. Som øvre grænser (95 percentiler) for indtagelse af jord er angivet en værdi på 200 mg/dag for børn og 300 mg/dag for voksne. At estimatet for voksne er højere end for børn, er i følge ECETOC begrundet i datavariabilitet og indikerer usikkerhederne i estimaterne. Der er for nyligt publiceret endnu en amerikansk undersøgelse vedrørende estimater for daglig indtagelse af jord for børn (Stanek & Calabrese 2000). Estimaterne er beregnet med udgangspunkt i resultaterne fra en tidligere undersøgelse (Calabrese et al. 1997 - citeret i ECETOC 2001), men med forbedrede metoder. Der blev estimeret en middelværdi (mean) på 31 mg/dag og en medianværdi (median) på 17 mg/dag. Endvidere blev der estimeret 95 percentiler for jordspisning over 7, 30, 90 samt 365 dage på henholdsvis 133, 112, 108 samt 106 mg/dag. 6.1.2.1.1 Sammenfatning, daglig indtagelse af jord De fleste estimater for daglig indtagelse af jord for børn og voksne er baseret på undersøgelser foretaget i USA. Der er dog 2 hollandske undersøgelser, men begge disse har ikke taget højde for et eventuelt indhold at de målte sporelementer i levnedsmidler, og resultaterne er således behæftet med nogen usikkerhed. På baggrund af 7 nøglestudier (se tabel 6.1.2.1A), som er præsenteret og evalueret i Exposure Factors Handbook (US-EPA 1997), anbefales i denne en standardværdi for gennemsnitlig daglig indtagelse af jord på 100 mg/dag for børn (i alderen op til 6 år) og på 50 mg/dag for voksne. Det er dog anført, at 200 mg/dag kan anvendes som et konservativt estimat. Som 95 percentil for børn anbefales en standardværdi på 400 mg/dag. For pica børn i relation til meget kortvarige (akutte) eksponeringer anbefales en standardværdi på 10 g/dag. De anbefalede standardværdier er samlet i tabel 6.1.2.1C. WHO (1994) har angivet en daglig indtagelse af jord på 20 mg/dag (median). Det er ikke specificeret, hvorvidt standardværdien gælder for voksne eller børn. ECETOC (2001) har samlet eksponeringsdata i Exposure Factors Sourcebook for European Populations. Heri er det angivet, at den estimerede gennemsnitlige indtagelse af jord er 40 mg/dag for børn og 1 mg/dag for voksne. Disse estimater stammer fra to amerikanske undersøgelser, der er publiceret efter, at US-EPA publicerede deres Exposure Factors Handbook. Som øvre grænser (95 percentiler) for indtagelse af jord er angivet en værdi på 200 mg/dag for børn og 300 mg/dag for voksne. MST har i relation til fastsættelse af kvalitetskriterier for jord som udgangspunkt hidtil anvendt en standardværdi på 200 mg/dag som en daglig gennemsnitsværdi for et barn. I relation til meget kortvarig eksponering har der som udgangspunkt været anvendt en værdi på 10 g/dag for at tage højde for eventuelle pica-børn. I den nyeste amerikansk undersøgelse vedrørende estimater for daglig indtagelse af jord for børn (Stanek & Calabrese 2000) blev estimeret en middelværdi på 31 mg/dag og en medianværdi på 17 mg/dag samt 95 percentiler for jordspisning over 7, 30, 90 samt 365 dage på henholdsvis 133, 112, 108 samt 106 mg/dag. I løbet af 1990-erne er publiceret en række studier, der alle peger på, at den gennemsnitlige indtagelse af jord hos mindre børn ligger under eller omkring 100 mg/dag. Der er tale om amerikanske undersøgelser, og det kan således ikke udelukkes, at der kan være en række forskelle, der gør, at resultaterne ikke er repræsentative for børn i Danmark. Der findes imidlertid ingen data, der kan belyse disse forskelle. US-EPA (1997) har anbefalet en standardværdi for gennemsnitlig daglig indtagelse af jord på 100 mg/dag for børn (i alderen op til 6 år) og på 50 mg/dag for voksne, men har dog anført, at 200 mg/dag kan anvendes som et konservativt estimat. ECETOC (2001) har derimod angivet en estimeret gennemsnitlig indtagelse af jord på 40 mg/dag for børn og 1 mg/dag for voksne. Disse estimater er baseret på to amerikanske undersøgelser, der er publiceret efter, at US-EPA publicerede Exposure Factors Handbook, og som af ECETOC vurderes at være mere pålidelige end de ældre metoder på grund af forbedrede metodikker. Standardværdien på 200 mg/dag, som MST hidtil har anvendt som en daglig gennemsnitsværdi for et barns indtagelse af jord i relation til fastsættelse af kvalitetskriterier for jord, vurderes i lyset af de nyere undersøgelser i US-EPA (1997) og ECETOC (2001) som værende en værdi, der ligger væsentligt over gennemsnitsværdien for børn generelt. I den nyeste undersøgelse (Stanek & Calabrese 2000), som er foretaget med udgangspunkt i resultaterne fra en tidligere undersøgelse, men med forbedrede metoder, ligger 95 percentilen for børns indtagelse af jord på lige godt 100 mg/dag. Som 95 percentil for børns indtagelse af jord har US-EPA (1997) anbefalet en standardværdi på 400 mg/dag, mens ECETOC (2001) som øvre grænser (95 percentiler) for indtagelse af jord har angivet en værdi på 200 mg/dag for børn og 300 mg/dag for voksne. Standardværdien på 200 mg/dag, som MST hidtil har anvendt som en daglig gennemsnitsværdi for et barn i relation til fastsættelse af kvalitetskriterier for jord, kan med udgangspunkt i Stanek & Calabrese (2000) således betragtes (forsigtigt skøn) som repræsenterende en 95 percentil for denne aldersgruppe. 6.1.2.2 Hudkontakt med jordDen generelle befolkning kan eksponeres for kemiske stoffer ved direkte hudkontakt med jorden. Eksponeringen udtrykkes i reglen som en gennemsnitlig mængde over en tidsperiode, for eksempel mg/dag. Denne eksponeringsvej er især relevant for mindre børn, idet børn oftest leger på jorden og også mange med jorden. Voksne kan også eksponeres for kemiske stoffer ved direkte hudkontakt, for eksempel ved havearbejde. Der er ikke fundet nogle studier vedrørende den gennemsnitlige daglige hudeksponering, hverken for børn eller voksne. 6.1.3 DrikkevandDen generelle befolkning kan eksponeres for kemiske stoffer, naturligt forekommende såvel som kontaminanter, ved indtagelse af drikkevand. Eksponeringen udtrykkes i reglen som en gennemsnitlig mængde over en tidsperiode, for eksempel liter/dag. Ved risikokarakterisering, det vil sige vurdering af (nul)effektniveauer i forhold til eksponering, kan der ofte være behov for at estimere den gennemsnitlige daglige dosis, som mennesker udsættes for som følge af eksponering for kemiske stoffer i drikkevandet. Hertil kræves viden om, hvor meget drikkevand der indtages. Indtagelsen af drikkevand varierer afhængigt af alder, fysisk aktivitetsniveau (arbejde, løb, gang, hvile), samt omgivelsernes temperatur. US-EPA (1997) har i relation til udarbejdelse af Exposure Factors Handbook identificeret 3 nøglestudier og 9 andre studier, der belyser ovennævnte forhold. Resultaterne af disse studier, som alle på nær en enkelt er amerikanske, viste, at der var en god overensstemmelse på tværs af studierne for estimatet på gennemsnitsværdi og 90 percentilen. Resultaterne viste også, at den gennemsnitlige indtagelse af drikkevand stiger med alderen, med det fysiske aktivitetsniveau samt med stigende temperatur. Det skal bemærkes, at på vægtbasis har børn en højere indtagelse af drikkevand i forhold til voksne og ældre et højere end middelaldrende. I tabel 6.1.3A er resultaterne fra et studie af Ershow & Cantor (1989 - citeret fra US-EPA 1997) præsenteret. I tabel 6.1.3B er resultaterne fra nøglestudierne summeret og i tabel 6.1.3C resultaterne fra de andre studier. US-EPA har generelt i de fleste risikovurderinger anvendt en standardværdi for daglig indtagelse af drikkevand på 2 liter/dag for voksne og 1 liter/dag for spædbørn (op til 10 kg). I Exposure Factors Handbook (US-EPA 1997) anbefales en gennemsnitlig daglig indtagelse på 1,4 liter/dag for voksne (alder: 19 år og opefter). Denne værdi er et gennemsnit af resultaterne af undersøgelserne fra følgende studier: Ershow & Cantor (1989), Roseberry & Burmaster (1992) og Canada Department of Health and Welfare (1981) – alle citeret i US-EPA (1997). Som 90 percentil anbefales for voksne en værdi på 2,3 liter/dag. Det er anført, at en indtagelse på 2 liter/dag svarer til 84 percentilen, hvis der sammenlignes med resultaterne fra undersøgelserne foretaget af Ershow & Cantor (1989 - citeret i US-EPA 1997). Disse standardværdier anbefales som værdier for eksponering gennem hele levetiden på trods af, at undersøgelserne er foretaget for kortere tids eksponering, men det er understreget, at 90 percentilen for langtidseksponering meget vel kan være højere end den anbefalede værdi på 2,3 liter/dag. Tabel 6.1.3A. Gennemsnitlig daglig indtagelse totalta af drikkevand. Fra Ershow & Cantor (1989) og modificeret fra US-EPA (1997).
a Drikkevand, totalt er defineret som hele den mængde drikkevand i husholdningen, der konsumeres direkte i form af drikkevarer eller i forbindelse med tilberedning af mad og drikkevarer Tabel 6.1.3B. Daglig indtagelse af drikkevand. Resultater fra nøglestudierne i Exposure Factors Handbook (US-EPA 1997). Tabellen er modificeret fra US-EPA (1997).
For gruppen børn som sådan anbefales ikke en generel standardværdi, men i stedet for anbefales aldersspecifikke standardværdier (se tabel 6.1.3D), også disse værdier er baseret på resultaterne af de 3 ovennævnte studier. Endelig er der anbefalet specifikke standardværdier for gravide og ammende kvinder, og for aktivitet under forskellige temperaturer (se tabel 6.1.3D). US-EPA's anbefalinger er samlet i tabel 6.1.3D. Tabel 6.1.3C. Daglig indtagelse af drikkevand. Resultater fra andre studier i Exposure Factors Handbook (US-EPA 1997). Tabellen er modificeret fra US-EPA (1997).
WHO anvender som udgangspunkt ved fastsættelse af guidelines for drikkevand en standardværdi for indtagelse af drikkevand for voksne (vægt 60 kg) på 2 liter/dag. Denne standardværdi vurderes som værende på den sikre side i langt de fleste tilfælde. Men det er også anført, at der i visse tilfælde kan være tale om en underestimering af vandindtagelsen, for eksempel for personer der bor under varme klimatiske forhold samt for børn, hvor væskeindtaget per kg legemsvægt er højere end hos voksne. I de tilfælde, hvor det vurderes, at børn udgør en særlig risikogruppe, anvendes der en standardværdi for indtagelse af drikkevand på 1 liter/dag for børn med en vægt på 10 kg og en standardværdi på 0,75 liter/dag for spædbørn med en vægt på 5 kg. (WHO 1996). Tabel 6.1.3D. US-EPA's standardværdier for indtagelse af drikkevand som angivet i Exposure Factors Handbook (US-EPA 1997).
a Fra Ershow & Cantor (1989) ECETOC (2001) har samlet eksponeringsdata i Exposure Factors Sourcebook for European Populations. For daglig indtagelse af drikkevand anbefales en gennemsnitlig værdi på 1,1 liter/dag for voksne og på 0,5 liter/dag for børn i alderen 1 til 11 år. Disse værdier er baseret på en britisk undersøgelse (Hopkins & Ellis 1980 - citeret i ECETOC 2001). Udvalgte resultater fra denne undersøgelse er vist i tabel 6.1.3E. Værdien på 1,1 liter/dag for voksne er i følge ECETOC i overensstemmelse med den værdi på 1 liter/dag, som MAFF har anvendt i en risikovurdering af nitrat og nitrit. Tabel 6.1.3E. Daglig indtagelse af drikkevand. Fra Hopkins & Ellis (1980) og modificeret fra ECETOC (2001).
6.1.3.1 Sammenfatning, daglig indtagelse af drikkevandDe fleste undersøgelser af den daglige indtagelse af drikkevand er foretaget i USA. Derudover er der en britisk undersøgelse og en canadisk undersøgelse. Disse undersøgelser er præsenteret og evalueret i Exposure Factors Handbook (US-EPA 1997). På baggrund af udvalgte undersøgelser (se ovenfor), er det for voksne blevet anbefalet at anvende en gennemsnitlig daglig indtagelse på 1,4 liter/dag og en 90 percentil på 2,3 liter/dag (alder: 19 år og opefter). For gruppen børn som sådan anbefales aldersspecifikke standardværdier (se tabel 6.1.3D). Endelig er der anbefalet specifikke standardværdier for gravide og ammende kvinder, og for aktivitet under forskellige temperaturforhold (se tabel 6.1.3D). WHO anvender ved fastsættelse af guidelines for drikkevand i de fleste tilfælde en standardværdi for voksne på 2 liter/dag. Men i tilfælde af, at børn vurderes at udgøre en særlig risikogruppe, anvendes der en standardværdi på 1 liter/dag for børn med en vægt på 10 kg og en standardværdi på 0,75 liter/dag for spædbørn med en vægt på 5 kg. (WHO 1996). ECETOC (2001) har samlet eksponeringsdata i Exposure Factors Sourcebook for European Populations. For daglig indtagelse af drikkevand anbefales en gennemsnitlig værdi på 1,1 liter/dag for voksne og på 0,5 liter/dag for børn i alderen 1 til 11 år. Disse værdier er baseret på en britisk undersøgelse (Hopkins & Ellis 1980 – citeret i ECETOC 2001 og US-EPA 1997), se tabel 6.1.3E. MST har i relation til fastsættelse af kvalitetskriterier for drikkevand som udgangspunkt hidtil anvendt en standardværdi på 2 liter/dag (gennemsnitsværdi for voksne) med mindre, der har været specifikke forhold, der har talt for anvendelse af en anden standardværdi. Standardværdien, som MST hidtil har anvendt for voksne (2 liter/dag), ligger væsentlig over de værdier, der er foreslået af US-EPA i Exposure Factors Handbook (US-EPA 1997) og af ECETOC (2001) i Exposure Factors Sourcebook. US-EPA (1997) har for voksne anbefalet en gennemsnitlig daglig indtagelse på 1,4 liter/dag (alder: 19 år og opefter). For gruppen børn som sådan er der anbefalet aldersspecifikke standardværdier. Ligeledes er der anbefalet specifikke standardværdier for gravide og ammende kvinder samt for aktivitet under forskellige temperaturforhold. Anbefalingerne er samlet i tabel 6.1.3D. Baseret på en britisk undersøgelse (se tabel 6.1.3E) har ECETOC (2001) for daglig indtagelse af drikkevand anbefalet en gennemsnitlig værdi på 1,1 liter/dag for voksne og på 0,5 liter/dag for børn i alderen 1 til 11 år. Standardværdien på 2 liter/dag, som MST hidtil har anvendt som en gennemsnitsværdi i relation til fastsættelse af kvalitetskriterier for drikkevand, vurderes i lyset af de nyere undersøgelser (US-EPA 1997, ECETOC 2001) som værende en værdi, der ligger noget over den gennemsnitlige indtagelse for befolkningen som helhed. Omvendt vurderes standardværdierne præsenteret af ECETOC (2001) som værende for lave set i lyset af de undersøgelser, der er præsenteret i Exposure Factors Handbook (US-EPA 1997). Det skal dog erkendes, at de højere resultater i de amerikanske undersøgelser kan afspejle forskellene i klimatiske forhold mellem USA og England. US-EPA (1997) har for voksne anbefalet en 90 percentil på 2,3 liter/dag (alder: 19 år og opefter). Der er ligeledes anbefalet 90 percentiler for børn i forskellige aldersgrupper samt for gravide og ammende kvinder. For enkelte af de sidstnævnte grupper er der endvidere anbefalet 95 percentiler. Anbefalingerne er samlet i tabel 6.1.3D. Standardværdien på 2 liter/dag, som MST hidtil har anvendt som en gennemsnitsværdi for indtagelse af drikkevand for voksne i relation til fastsættelse af kvalitetskriterier for drikkevand, er lavere end 90 percentilen for voksne og for gravide kvinder samt lavere end 95 percentilen for gravide og ammende kvinder. Standardværdien på 2 liter/dag for voksne er lig med 95 percentilen for børn i aldersgruppen 11 til 19 år, men højere end 95 percentilen for de yngre aldersgrupper. Standardværdien på 2 liter/dag kan således betragtes som en repræsentativ værdi for en 95 percentil for børn i alderen op til 19 år, men ikke for voksne. 6.1.4 SammenfatningDen generelle befolkning kan eksponeres for kemiske stoffer ved inhalation af dampe og støv i luften (i indeklima såvel som udendørs), indtagelse af levnedsmidler, drikkevand og jord, samt ved hudkontakt med drikkevand og jord. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand har Miljøstyrelsen siden 1990 anvendt de standardestimater for eksponering, der er angivet i Miljøprojekt nr. 123 ”Risikovurdering af forurenede grunde” (MST 1990a) og gengivet i tabel 6. Værdierne bygger dels på litteraturstudier og dels på en høring af en række institutioner i forskellige lande foretaget af Miljøstyrelsen. Det er fremhævet, at værdierne for indtagelse af jord og for hudkontakt med jord er usikre, og at de er det bedste skøn, der kunne foretages på daværende tidspunkt. Gennemgangen af den nyere viden om, hvor meget luft, jord/støv og drikkevand børn og voksne indtager, indikerer, at en revision af de hidtil anvendte gennemsnitsværdier bør overvejes. Ved revurderingen heraf kunne der tages udgangspunkt i de gennemsnitlige værdier for daglig indånding af luft samt daglig indtagelse af jord/støv og drikkevand, der er angivet i tabel 6.1.4., som primært er de anbefalede gennemsnitsværdier i Exposure Factors Handbook (US-EPA 1997). For daglig indtagelse af drikkevand er der i tabel 6.1.4. ligeledes medtaget øvre grænser (90 og 95 percentiler) baseret på anbefalingerne i Exposure Factors Handbook (US-EPA 1997). Datagrundlaget i relation til daglig indånding af luft samt daglig indtagelse af jord/støv er ikke tilstrækkeligt med henblik på en vurdering af øvre grænser. Der er ikke fundet nogle studier vedrørende den gennemsnitlige daglige hudeksponering eller øvre grænser herfor, hverken for børn eller voksne. Tabel 6.1.4. Gennemsnitlige værdier for daglig indånding af luft samt daglig indtagelse af jord/støv og drikkevand. Baseret primært på US-EPA (1997).
6.1.4.1 LuftStandardværdien på 20 m³/dag, som MST hidtil har anvendt i relation til fastsættelse af kvalitetskriterier for udeluft, vurderes i lyset af de nyere undersøgelser som værende en værdi, der ligger over gennemsnittet for befolkningen som helhed. Således har US-EPA (1997) anbefalet for eksponering gennem længere tid at anvende en gennemsnitlig ventilation på 11,3 m³/dag for kvinder og 15,2 m³/dag for mænd (alder voksne: 19 år og opefter). For gruppen børn anbefales en række aldersspecifikke standardværdier. For eksponering i kortere tid er der på basis af forskellige aktivitetsmønstre anbefalet en række specifikke standardværdier. De anbefalede standardværdier er samlet i tabel 6.1.1B. Inden for EU er US-EPA's anbefalinger (se tabel 6.1.1B) angivet i TGD'en for risikovurdering af nye og eksisterende stoffer (TGD) med henblik på at kunne anvendes i relation til risikovurderingerne. Også ECETOC har konkluderet, at disse standardværdier vil være rimelige estimater for den europæiske befolkning. Det vurderes således, at US-EPA's anbefalinger fremover vil finde bred anvendelse i Europa i relation til risikovurderinger af kemiske stoffer. På baggrund af det foreliggende datagrundlag vurderede US-EPA (1997), at der ikke kunne fremsættes anbefalinger vedrørende øvre grænser (95 percentiler). Da der ikke siden hen er blevet publiceret yderligere data med henblik på en vurdering af øvre grænser, kan der således heller ikke på nuværende tidspunkt foretages en vurdering heraf, ej heller en vurdering af hvorvidt MST's standardværdi på 20 m³/dag kan betragtes som en repræsentativ værdi for en 95 percentil for voksne eller børn. 6.1.4.2 JordStandardværdien på 200 mg/dag, som MST hidtil har anvendt som en daglig gennemsnitsværdi for et barns indtagelse af jord i relation til fastsættelse af kvalitetskriterier for jord, vurderes i lyset af de nyere undersøgelser som værende en værdi, der ligger væsentligt over gennemsnitsværdien for børn generelt. I løbet af 1990-erne er publiceret en række studier, der alle peger på, at den gennemsnitlige indtagelse af jord hos mindre børn ligger under eller omkring 100 mg/dag. Der er tale om amerikanske undersøgelser, og det kan således ikke udelukkes, at der kan være en række forskelle, der gør, at resultaterne ikke er repræsentative for børn i Danmark. Der findes imidlertid ingen data, der kan belyse disse forskelle. US-EPA (1997) har anbefalet en gennemsnitlig daglig indtagelse af jord på 100 mg/dag for børn (i alderen op til 6 år) og 50 mg/dag for voksne, men har dog anført, at 200 mg/dag kan anvendes som et konservativt estimat. ECETOC (2001) har derimod angivet en estimeret gennemsnitlig indtagelse af jord på 40 mg/dag for børn og 1 mg/dag for voksne. Disse estimater er baseret på to amerikanske undersøgelser, der er publiceret efter, at US-EPA publicerede Exposure Factors Handbook, og som af ECETOC vurderes at være mere pålidelige end de ældre metoder på grund af forbedrede metodikker. I den nyeste undersøgelse (Stanek & Calabrese 2000), som er foretaget med udgangspunkt i resultaterne fra en tidligere undersøgelse, men med forbedrede metoder, ligger 95 percentilen for børns indtagelse af jord på lige godt 100 mg/dag. Som 95 percentil for børns indtagelse af jord har US-EPA (1997) anbefalet en standardværdi på 400 mg/dag, mens ECETOC (2001) som øvre grænser (95 percentiler) for indtagelse af jord har angivet en værdi på 200 mg/dag for børn og 300 mg/dag for voksne. Standardværdien på 200 mg/dag, som MST hidtil har anvendt som en daglig gennemsnitsværdi for et barns indtagelse af jord i relation til fastsættelse af kvalitetskriterier for jord, kan således (forsigtigt skøn) betragtes som repræsenterende en 95 percentil for denne aldersgruppe. Der er ikke fundet nogle studier vedrørende den gennemsnitlige daglige hudeksponering, hverken for børn eller voksne. 6.1.4.3 DrikkevandStandardværdien på 2 liter/dag, som MST hidtil har anvendt som en gennemsnitsværdi i relation til fastsættelse af kvalitetskriterier for drikkevand, vurderes i lyset af de nyere undersøgelser som værende en værdi, der ligger noget over den gennemsnitlige indtagelse for befolkningen som helhed. US-EPA (1997) har for voksne anbefalet en gennemsnitlig daglig indtagelse på 1,4 liter/dag (alder: 19 år og opefter). For gruppen børn som sådan er der anbefalet aldersspecifikke standardværdier. Ligeledes er der anbefalet specifikke standardværdier for gravide og ammende kvinder samt for aktivitet under forskellige temperaturforhold. Anbefalingerne er samlet i tabel 6.1.3D. Baseret på en britisk undersøgelse (se tabel 6.1.3E) har ECETOC (2001) for daglig indtagelse af drikkevand anbefalet en gennemsnitlig værdi på 1,1 liter/dag for voksne og på 0,5 liter/dag for børn i alderen 1 til 11 år. Disse standardværdier vurderes imidlertid som værende for lave set i lyset af de undersøgelser, der er præsenteret i Exposure Factors Handbook (US-EPA 1997). Det skal dog erkendes, at de højere resultater i de amerikanske undersøgelser kan afspejle forskellene i klimatiske forhold mellem USA og England. US-EPA (1997) har for voksne anbefalet en 90 percentil på 2,3 liter/dag (alder: 19 år og opefter). Der er ligeledes anbefalet 90 percentiler for børn i forskellige aldersgrupper samt for gravide og ammende kvinder. For enkelte af de sidstnævnte grupper er der endvidere anbefalet 95 percentiler. Anbefalingerne er samlet i tabel 6.1.3D. Standardværdien på 2 liter/dag, som MST hidtil har anvendt som en gennemsnitsværdi for indtagelse af drikkevand i relation til fastsættelse af kvalitetskriterier for drikkevand, er lavere end 90 percentilen for voksne og for gravide kvinder samt 95 percentilen for gravide og ammende kvinder. Værdien 2 liter/dag er lig med 95 percentilen for børn i aldersgruppen 11 til 19 år, men højere end 95 percentilen for de yngre aldersgrupper. Værdien 2 liter/dag kan således betragtes som en repræsentativ værdi for en 95 percentil for børn i alderen op til 19 år, men ikke for voksne. For børn i alderen op til 10 år er 95 percentilen lig med 1,5 liter/dag. 6.2 Standardindtagelser (foder, drikkevand, luft) for forsøgsdyrI de fleste dyreforsøg er eksponeringen for et givent stof oftest angivet som en koncentration af stoffet i foder, drikkevand, eller luft. For at kunne foretage sammenligninger af (nul)effektniveauer på tværs af forskellige studier i en dyreart eller på tværs af studier for forskellige dyrearter samt ved risikokarakterisering (vurdering af (nul)effektniveauer i forhold til eksponering) kan der ofte være behov for at estimere den gennemsnitlige daglige dosis (enhed: mg/kg legemsvægt/dag), som dyrene eksponeres for. Til brug herfor er det nødvendigt at anvende nogle standardværdier for legemsvægt, foderindtagelse, indtagelse af drikkevand samt for ventilationen (indåndingsvolumen over tid) for de mest anvendte dyrearter. Der er ikke internationalt fastsatte standardværdier for disse parametre, men forskellige myndigheder og organisationer har fastsat standardværdier for enkelte af disse parametre, som er gengivet i det efterfølgende. 6.2.1 EU risikovurdering af nye og eksisterende kemiske stofferPrincipperne for risikovurdering af nye og eksisterende stoffer er udførligt beskrevet i Technical Guidance Document (TGD) (se 1.2.1), som lige er blevet gennemgående revideret (EU 2002). I TGD'en er der anbefalet standardværdier for forskellige parametre, som anvendes i de tilfælde, hvor der i de enkelte risikovurderingsrapporter ikke er beskrevet målte værdier. De anbefalede værdier i TGD er baseret på en rapport udarbejdet af TNO i Holland (Paulussen et al. 1998). Nedenfor er gengivet tabellerne vedrørende standardværdier for legemsvægt (tabel 6.2.1 og 6.2.2) og ventilation (tabel 6.2.3). Tabel 6.2.1.1. Standardværdier for legemsvægte i orale og inhalationsstudier. Modificeret fra TGD (EU 2002).
a Legemsvægte er givet i gram (g) for rotter og mus og i kilogram (kg) for hunde. Der er i TGD'en (EU 2002) ikke gengivet standardværdier for indtagelse af foder eller drikkevand med den begrundelse, at eksperimentelle forhold kan influere på disse standardværdier. I stedet er der gengivet allometriske ligninger, således at det er muligt at estimere standardværdier case-by-case. De allometriske ligninger er inddelt i species-specifikke ligninger (tabel 6.2.4) så vel som generelle ligninger (tabel 6.2.5). TGD'en anbefaler, at de species-specifikke ligninger anvendes i det omfang, som det er muligt. De allometriske ligninger stammer oprindeligt fra en rapport udgivet af US-EPA i 1988 (Blackburn K: Recommendations for and documentation of biological values for use in risk assessment, EPA/600/6-87/008, February 1988). Tabel 6.2.1.2. Standardværdier for legemsvægte i dermale studier. Modificeret fra TGD (EU 2002).
a Legemsvægte er givet i gram (g) for rotter og marsvin og i kilogram (kg) for kaniner. Tabel 6.2.1.3. Standardværdier for ventilation (indåndingsvolumen over tid). Modificeret fra TGD (EU 2002).
a Værdierne er baseret på species-specifikke allometriske sammenhænge. Tabel 6.2.1.4. Species-specifikke ligninger for indtagelse af foder. Modificeret fra TGD (EU 2002).
F = indtagelse af foder i kg/dag; W = legemsvægt i kg Tabel 6.2.1.5. Generelle ligninger for indtagelse af foder og drikkevand. Modificeret fra TGD (EU 2002).
a F = indtagelse af foder i kg/dag; L = indtagelse af væske (vand) i liter/dag; W = legemsvægt i kg. Ligningerne, der beskriver indtagelse af foder og væske, kan ikke anvendes på drægtige dyr, som anvendes i multi-generationsstudier. Ligningerne 1-4 anvendes, når enten indtagelse af vand eller af foder kendes, og den manglede værdi ønskes estimeret. Hvis foderet er specificeret nærmere, eller med rimelig sikkerhed kan formodes at være enten tørt eller vådt, anbefales henholdsvis ligning 1 eller 2. Ligninger 5-8 anvendes til estimering af indtagelse af foder eller vand, når legemsvægten er kendt eller kan estimeres. 6.2.2 OECDOECD (2000) har udgivet en rapport 'Guidance Notes for Analysis and Evaluation of Repeat-Dose Toxicity Studies'. I denne rapport er der en tabel, som angiver de fleste laboratoriedyrs gennemsnitsvægt, daglige foderindtagelse samt omregningsfaktorer mellem koncentrationen af et givent stof i foderet (enhed: ppm = mg/kg) til dosis udtrykt i mg/kg legemsvægt per dag. Tabellen, som er gengivet nedenfor (tabel 6.2.2.1) stammer oprindeligt fra en publikation af Lehman (1954) og er tidligere publiceret i WHO (1987). Disse omregningsfaktorer anvendes af Scientific Committee for Food (SCF) i EU, the Joint FAO/WHO Expert Committee on Food Additives (JECFA), the Joint FAO/WHO Meeting on Pesticide Residues (JMPR), OECD og Institut for Fødevaresikkerhed og Ernæring (IFSE) ved vurderinger af tilsætningsstoffer og kontaminanter i levnedsmidler. I OECD rapporten er det endvidere anført, at i de tilfælde hvor den daglige indtagelse er målt, så anføres ppm i foderet som værende lig med (engelsk: equal) dosis udtrykt i mg/kg legemsvægt per dag. I de tilfælde hvor omregningsfaktorerne anvendes, så anføres ppm i foderet som værende ækvivalent med (engelsk: equivalent) dosis udtrykt i mg/kg legemsvægt per dag. Tabel 6.2.2.1. Relation mellem koncentration i foder og dosis udtrykt i mg/kg legemsvægt. Modificeret fra OECD (2000).
a Væske undtaget. Med hensyn til vandforbruget er der ingen generelle tal. I en tabel i OECD rapporten (OECD 2000) er det angivet, at en rotte (Rattus Norvegicus) indtager 24 til 35 ml vand per dag. En gennemsnitlig daglig indtagelse af vand på 30 ml per dag svarer til 75 ml/kg legemsvægt for en voksen rotte (vægt: 400 gram). Med henblik på omregning af koncentrationer i drikkevand (enhed: ppm = mg/liter) til dosis udtrykt i mg/kg legemsvægt per dag i længerevarende studier (90 dage og derover), estimeres en omregningsfaktor på 0,075. Det vil sige, at 1 ppm i drikkevandet svarer til 0,075 mg/kg legemsvægt per dag. 6.2.3 Standardværdier i relation til kvalitetskriterierI relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand har der ikke hidtil været fastlagt specifikke standardværdier for legemsvægt, foderindtagelse, indtagelse af drikkevand samt for ventilationen (indåndingsvolumen over tid). Dog har omregningsfaktorerne angivet i tabel 6.2.2.1 mellem koncentration af stof i foder (ppm) og dosis udtrykt i mg/kg legemsvægt per dag været anvendt i en lang række vurderinger. Tabel 6.2.3.1. Standardværdier for legemsvægt, indtagelse af foder og drikkevand samt ventilation for forskellige forsøgsdyr.
Med baggrund i de data, der er præsenteret i afsnit 6.2.1 og 6.2.2, foreslås de standardværdier for legemsvægt, foderindtagelse, samt for indtagelse af drikkevand, der er angivet i tabel 6.2.3.1. For ventilationen foreslås de værdier, der er angivet i tabel 6.2.1.3. 6.3 ReferencerECETOC (2001). Exposure Factors Sourcebook for European populations (with focus on UK data). Technical Report No. 79. European Centre for Ecotoxicology and Toxicology of Chemicals, Brussels. EU (2002). Technical Guidance Document on risk assessment in support of Commission Directive 93/67/EEC on risk assessment for new notified substances and Commission Regulation (EC) No 1488/94 on risk assessment for existing substances and Directive 98/8/EC of the European Parliament and of the Council concerning the placing of biocidal products on the market. 2. Udgave. European Commission. Endnu ikke publiceret. MST (1990). Risikovurdering af forurenede grunde. Miljøprojekt nr. 123. Miljøministeriet, Miljøstyrelsen. Nielsen E, Thorup I, Schnipper A, Hass U, Meyer O, Ladefoged O, Larsen JC, Østergaard G, Sørensen TL og Larsen PB (2001). Children and the unborn child. Exposure and susceptibility to chemical substances – an evaluation. Miljøstyrelsen, Miljø- og Energiministeriet. http://www.mst.dk/udgiv/publications/2001/87-7909-574-7/html/ OECD (2000). Guidance notes for analysis and evaluation of repeat-dose toxicity studies. OECD Environment, Health and Safety Publications. Series on Testing and Assessment No. 32 and Series on Pesticides No. 10. Environment Directorate, Organisation for Economic Co-operation and Development, Paris 2000. Paulussen JJC, Mahieu CM og Bos PMJ (1998). Default values in occupational risk assessment. TNO report V98.380. TNO Nutrition and Food Research Institute, Zeist, Holland. Stanek III EJ and Calabrese EJ (2000). Daily soil ingestion estimates for children at a Superfund site. Risk Anal 20, 627-635. US-EPA (1997). Exposure Factors Handbook EPA/600/P-95/002Fa. Update to Exposure Factors Handbook EPA/600/8-89/043 - May 1989. http://www.epa.gov/nceawww1/pdfs/efh/front.pdf WHO (1999). Principles for the assessment of risks to human health from exposure to chemicals. Environmental Health Criteria 210. International Programme on Chemical Safety, World Health Organization. WHO (1996). 12. Chemical and physical aspects: introduction. In: Guidelines for drinking-water quality, second edition. Volume 2 Health criteria and other supporting information. World Health Organization, Geneva, 121-131. WHO (1994). Assessing human health risks of chemicals: derivation of guidance values for health-based exposure limits. Environmental Health Criteria 170. International Programme on Chemical Safety, World Health Organization. WHO (1987). Principles for the safety assessment of food additives and contaminants in food. Environmental Health Criteria 70. International Programme on Chemical Safety, World Health Organization. 7 Kvalitetskriterier7.1 Overskridelse af kvalitetskriterier eller TDI Et sundhedsmæssigt baseret kvalitetskriterie for luft, jord og drikkevand beregnes ud fra den estimerede tolerable daglige indtagelse (TDI - se kapitel 4 og 5) ved at dividere med den gennemsnitlige daglige standardeksponering for det relevante medie (se kapitel 6). Det vil sige, at selve fastsættelsen af det enkelte kvalitetskriterie ud fra TDI er ens for stoffer, hvor der findes en tærskelværdi for de(n) kritiske effekt)er) og for genotoksiske carcinogener. De sundhedsmæssigt baserede kvalitetskriterier skal betragtes som sikkerhedsgrænser, der beskytter befolkningens sundhed, idet kvalitetskriterierne er lagt på et niveau, hvor der ikke forventes at optræde sundhedsskadelige effekter. På trods af de procedurer, der anvendes ved fastsættelse af kvalitetskriterierne, kan man ikke sikre sig, at der ved anvendelse af kvalitetskriterier kan være en vis, ikke nærmere defineret, risiko, da fuldstændig sikkerhed aldrig kan garanteres. I de tilfælde, hvor der ikke er en tærskelværdi for stoffets kritiske effekt, kan denne risiko være udtrykt kvantitativt, da det tolerable risikoniveau indgår i selve beregningen af TDI (se kapitel 5). Men selv om det sundhedsmæssigt baserede kvalitetskriterie fastsættes med udgangspunkt i tærskelværdiprincippet, er kvalitetskriteriet således et udtryk for, at en vis, ikke nærmere defineret, risiko tolereres. Dette gælder for eksempel risikoen for, at datagrundlaget ikke har været tilstrækkeligt, at de tilgængelige undersøgelser ikke har været udført tilstrækkeligt omhyggeligt, at de anvendte undersøgelsesmetoder ikke har kunnet afsløre alle skadelige effekter, eller at de undersøgte individer (mennesker eller dyr) ikke i tilstrækkelig grad har været repræsentative for de udsatte befolkningsgrupper. (MST 1990, MST 1992). 7.1 Overskridelse af kvalitetskriterier eller TDIAnvendelse af de principper og den praksis, der er indarbejdet i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand, repræsenterer efter Miljøstyrelsens opfattelse en forsigtighedstilgang, der har medført, at de fastsatte kvalitetskriterier opfylder målsætningen om et højt beskyttelsesniveau for befolkningen. Kvalitetskriterierne skal, som nævnt, opfattes som sikkerhedsgrænser og ikke faregrænser, det vil sige, at overskridelse af kvalitetskriterierne ikke er ensbetydende med fare, men mere et udtryk for at det beskyttelsesniveau, der sædvanligvis tilstræbes ved en konkret overskridelse, formindskes. Begrebet acceptabel daglige indtagelse (ADI), som anvendes ved vurdering af for eksempel tilsætningsstoffer, pesticidrester samt rester af veterinære lægemidler i levnedsmidler, blev oprindeligt foreslået af the Joint FAO/WHO Expert Committee on Food Additives (JECFA) og defineret som den estimerede mængde af et tilsætningsstof (enhed: mg/kg legemsvægt per dag), der kan indtages dagligt gennem et helt livsforløb uden væsentlige sundhedsmæssige risici. Den tolerable daglige/ugentlige indtagelse (TDI / PTWI), som JECFA anvender i relation til kontaminanter i levnedsmidler, defineres på tilsvarende måde. (Larsen & Richold 1999). JECFA har imidlertid ikke givet retningslinier for, hvordan overskridelser af ADI / TDI skal vurderes. WHO (1987 – citeret fra Larsen & Richold 1999) har anført, at da data i de fleste tilfælde ekstrapoleres fra dyreeksperimentelle studier, hvor dyrene er eksponeret gennem hele levetiden, så relaterer ADI sig til en livstidsanvendelse og giver dermed en tilstrækkelig sikkerhedsmargen, så overskridelse af ADI i kortere tid ikke anses for kritisk. JECFA (1989 – citeret fra Larsen & Richold 1999) har konkluderet, at eksponering i kortere tid der medfører en overskridelse af PTWI for en kontaminant ikke giver anledning til bekymring, forudsat at den gennemsnitlige indtagelse over længere tid ikke medfører en overskridelse af PTWI. JECFA har også anført, at det ikke er muligt at foretage en generalisering i relation til det tidsrum, gennem hvilket en overskridelse af ADI / TDI / PTWI vil give anledning til forekomst af skadelige effekter, da en hvilken som helst skadelig effekt afhænger af 'nature of toxicity' og den biologiske halveringstid for det givne kemiske stof. I 1998 afholdt ILSI Europe Acceptable Daily Intake Task Force i samarbejde med the Food Chemical Intake Task Force en workshop med henblik på at belyse problematikken i relation til overskridelse af ADI / TDI / PTWI. Det specifikke formål med workshoppen var at diskutere følgende spørgsmål (Larsen & Richold 1999):
Nedenfor opsummeres konklusionerne fra workshoppen, som beskrevet af Larsen & Richold (1999). For tilsætningsstoffer, pesticidrester samt rester af veterinære lægemidler blev det vurderet, at overskridelse af ADI ikke forekommer særligt hyppigt, mens det vil forekomme jævnligt for kontaminanter. Baseret på erfaringer mente toksikologerne ikke, at der er grund til bekymring i relation til alvorlige sundhedsmæssige risici som følge af overskridelse af ADI en gang imellem, da der er en stor sikkerhedsmargen indbygget ved estimeringen af ADI og forudsat, at indtagelse over en længere periode gennemsnitligt ikke ligger over ADI. Det blev dog understreget, at overskridelse af ADI ikke er ønskelig. Det blev også erkendt, at der ikke er nogle principper, der kan tage højde for såkaldte ekstreme indtagelsesmønstre. Der var også generel enighed om, at en vurdering af betydningen af en overskridelse af ADI skal foretages case-by-case. I relation til hvilke metoder, der bør anvendes ved estimering af daglig indtagelse, er det som et første trin anbefalet at anvende en konservativ, teoretisk/hypotetisk tilgang. Hvis denne metode ikke giver et estimat for daglig indtagelse, der medfører en overskridelse af ADI, er der ikke behov for yderligere vurdering af daglig indtagelse. I tilfælde af overskridelse af ADI er den næste anbefalede tilgang at estimere en daglig indtagelse ud fra en 3-dages kostundersøgelse suppleret med et spørgeskema med en minimumpopulation på 200 individer. Hvis den estimerede daglige indtagelse stadig er højere end ADI, anbefales det at foretage en decideret risikovurdering. I relation til hvor meget ADI kan overskrides og i hvor lang tid, blev det konkluderet, at der ikke kan gives nogle generelle retningslinier herfor, og at en case-by-case vurdering altid bør foretages. Dog er der givet nogle fingerpeg, der kan være til hjælp i denne case-by-case vurdering. I de tilfælde, hvor ADI er baseret på et kronisk studie, er en overskridelse af ADI ikke acceptabel, hvis denne overskridelse forekommer gennem størstedelen af menneskets levetid. Hvis den indtagelse, der medfører en overskridelse af ADI, er for en kortere tidsperioden end den, der er anvendt i det studie, hvorpå ADI er baseret, kan man foretage en vurdering af betydningen af denne overskridelse udfra et nul-effektniveau fra et studie af kortere varighed. For tilsætningsstoffer, pesticidrester samt rester af veterinære lægemidler i levnedsmidler er den biologiske halveringstid i reglen kort, og de observerede effekter i kroniske studier skyldes muligvis gentagen påvirkning (kronisk stress) og ikke effekter som følge af akkumulering af det givne stof i organismen. For stoffer, hvor effekterne ikke er fuldstændigt reversible, bør der også foretages en vurdering af betydningen af kortvarige overskridelse af ADI med udgangspunkt i (nul)effektniveauer fra korttidsstudier. I relation til kontaminanter, hvor der er fastsat en TDI eller PTWI, blev det anbefalet at anvende de samme principper som for vurdering af overskridelse af ADI. Det blev understreget, at kontaminanter med meget lang biologisk halveringstid akkumulerer i organismen, og at de skadelige effekter som følge af eksponering først optræder, når den kritiske koncentration er opnået i målorganet. Endvidere er det anført, at der sædvanligvis er store forskelle mellem de doser, der giver anledning til effekter efter forholdsvis kort tids eksponering og de doser, der giver anledning til effekter efter lang tids eksponering. I disse tilfælde blev det vurderet, at overskridelse af TDI / PTWI for kort tids eksponering (dage, uger, måneder) eller en mindre overskridelse for lidt længere tidsperioder (måneder, år) næppe vil have nogen betydning forudsat, at den kumulerede eksponering gennem længere tid ikke medfører en overskridelse af den kritiske koncentration i målorganet. De overordnede synspunkter fra workshoppen var således, at den gennemsnitlige daglige indtagelse af tilsætningsstoffer og kontaminanter ikke bør medføre en overskridelse af ADI / TDI / PTWI gennem længere perioder. Dette princip gør sig gældende for den generelle befolkning som sådan, og hvis en overskridelse observeres for en undergruppe af den generelle befolkning bør betydningen heraf vurderes case-by-case, inklusive vurdering af indtagelsesmønstre, det toksikologiske datasæt, præcisionen af estimaterne for daglig indtagelse, samt den eksisterende lovgivning. I relation til overskridelse af kvalitetskriterier har Miljøstyrelsen i forbindelse med overskridelse af kvalitetskriterierne for drikkevand anvendt følgende retningslinier (MST 1992): For kemiske stoffer, hvor der findes en tærskelværdi for den kritiske effekt, kan overskridelse af kvalitetskriteriet med indtil en faktor 10 tolereres i en kortere periode, mens overskridelse med mere end en faktor 10 ikke kan tolereres. For stoffer, hvor ikke findes en tærskelværdi for den kritiske effekt (genotoksiske carcinogener), kan påvisning af stoffet i koncentrationer, der indebærer en livstidsrisiko for udvikling af tumorer på fra 10-6 op til 10-5 tolereres i en kortere periode, hvis det ikke er muligt straks at forbedre vandkvaliteten, indtil der kan indføres permanente afhjælpende foranstaltninger. Påvises stoffet i en koncentration, der indebærer en beregnet livstidsrisiko for udvikling af tumorer, der er større end 10-5, bør den pågældende vandforsyning lukkes. En livstidsrisiko for udvikling af tumorer på 10-6 eller mindre anses af Miljøstyrelsen for at være tolerabel, når den efter omstændighederne i øvrigt ikke kan undgås. 7.2 AllokeringNår der fastsættes et kvalitetskriterie for et kemisk stof i luft, jord eller drikkevand, bør der tages hensyn til alle de eksponeringsveje, ad hvilke den generelle befolkning kan blive eksponeret for stoffet, det vil sige via luft, levnedsmidler, og drikkevand. Dette betyder, at TDI før beregningen af det enkelte kvalitetskriterie skal fordeles på disse forskellige medier. (MST 1990, MST 1992). Såfremt det vides, at der forekommer en væsentlig eksponering for et givent kemisk stof fra andre medier end det medie, der skal fastsættes et kvalitetskriterie for, tildeles (allokeres) kun en vis procentdel af TDI til det pågældende medie, således at man sikrer sig, at bidragene fra de forskellige medier samlet ikke overskrider TDI. Denne procedure kaldes allokering. Som udgangspunkt allokeres 10% af TDI til det pågældende medie. For visse kritiske stoffer, hvor hovedbidrag kommer fra øvrige kilder kan der dog allokeres helt ned til kun 1% af TDI (for eksempel for visse pesticider), men også højere fordelingstal kan anvendes i forbindelse med konkrete vurderinger. I de tilfælde hvor bidrag fra andre medier ikke er kendt, fastsættes kvalitetskriterierne som udgangspunkt ved, at hele TDI allokeres til det pågældende medie. (Nielsen et al. 1995). I WHO (1994) er det vedrørende allokering anbefalet, at generelt bør mindre end 100% af TDI anvendes ved fastsættelse af guideline-værdier for at tage højde for, at i langt de fleste tilfælde vil eksponeringen for alle medier ikke være kendt. Endvidere er det anbefalet, at i de tilfælde hvor eksponeringen via et givent medie er meget lille (mindre end et par procent) i forhold til den totale eksponering fra alle medier, bør der ved fastsættelse af en guideline-værdi for det pågældende medie ikke anvendes allokering. WHO (1996) har i relation til fastsættelse af guidelines for drikkevand anført, at eksponeringen for kemiske stoffer via drikkevand ofte er lav i forhold til andre kilder som for eksempel levnedsmidler og luft. Derfor er der ved fastsættelse af guidelines baseret på tærskelværdiprincippet taget højde for eksponering fra andre kilder ved at allokere en vis procentdel af TDI til indtagelse via drikkevandet. I de tilfælde hvor det har været muligt, er der ved fastsættelse af guideline taget udgangspunkt i data vedrørende indtagelse af det givne stof fra de forskellige kilder, eller indtagelsen er estimeret udfra vurderinger af fysisk-kemiske egenskaber. I øvrige tilfælde er der som udgangspunkt anvendt en arbitrær faktor på 10% for indtagelse via drikkevandet. Denne faktor vurderes i de fleste tilfælde som værende tilstrækkelig til at tage højde for indtagelse af det givne stof fra andre kilder. CEN har en generel holdning til allokering i forbindelse med legetøj, idet det er hovedsynspunktet, at der kun skal allokeres 10% af TDI til legetøj. Da CEN p.t. forbereder et forslag til EU-kommissionen om en ny standard for visse organiske forbindelser i legetøj, vil dette princip blive foreslået for nogle grupper af kemikalier, men ikke for andre. 7.3 Andre forholdI relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand kan der i visse tilfælde være behov for at justere disse yderligere. Et eksempel herpå er fastsættelse af et kvalitetskriterie for en gruppe af nærtbeslægtede stoffer (for eksempel phthalater), som ikke alle er lige godt undersøgt, og hvor kvalitetskriteriet fastsættes på baggrund af den viden, der foreligger for enkelte af de stoffer, der er inkluderet i gruppen. Her kan der således anvendes en reduktionsfaktor til at tage højde for usikkerhederne i relation til den manglende viden for det enkelte stof i gruppen. Andre forhold, som også indgår ved fastsættelse af kvalitetskriterier for luft, jord og drikkevand, er stoffets fysisk-kemiske egenskaber (for eksempel vandopløselighed, flygtighed og persistens i miljøet), dannelse af aerosoler, binding af stoffet til for eksempel partikler, baggrundsniveauer i luft, jord og drikkevand, samt udseende, lugt og smag. 7.4 Fastsættelse af sundhedsmæssigt baseret luftkvalitetskriterium og B-værdiDet sundhedsmæssigt baserede kvalitetskriterium i luft (QCluft) for et konkret stof beregnes ud fra den estimerede tolerable daglige indtagelse (TDI – se kapitel 4 og 5) eller et NOAEC/LOAEC fra et inhalationsstudie ved at dividere med den gennemsnitlige daglige ventilation (VR) (se 7.4.1). QCluft danner udgangspunkt for fastsættelse af en B-værdi (se 7.4.2), som er den enkelte virksomheds samlede maksimalt tilladelige bidrag til tilstedeværelsen af et forurenende stof i luften i omgivelserne udenfor virksomheden, det vil sige immissionen. 7.4.1 Sundhedsmæssigt baseret luftkvalitetskriteriumFølgende fremgangsmåde anvendes til beregning af det sundhedsmæssigt baserede luftkvalitetskriterium (QCluft), når TDI for de(n) kritiske effekt(er) er fra orale studier og angivet i enheden mg/kg legemsvægt/dag:
I relation til fastsættelse af kvalitetskriterier for udeluft har MST hidtil anvendt en standardværdi for ventilation (VR) på 20 m³/dag og en standardværdi for legemsvægt (w) på 70 kg. Endvidere forudsættes 100% absorption efter inhalation med mindre datagrundlaget for et konkret stof er tilstrækkeligt til at foretage en konkret vurdering af absorptionsfraktionen. Følgende fremgangsmåde anvendes, når (nul)effektniveauer (NO(A)EC eller LO(A)EC) for de(n) kritiske effekt(er) er fra inhalationsstudier og angivet i enheden mg/m³:
For langt de fleste typer af systemiske effekter anses det at være den samlede dosis og ikke stoffets koncentration i luften som sådan, der er af betydning for udvikling af disse effekter. Ved fastsættelse af luftkvalitetskriteriet i disse tilfælde foretages en omregning af det fastlagte (nul)effektniveau til et gennemsnitligt døgnniveau (kontinuert eksponering) ud fra de i studiets aktuelle eksponeringsbetingelser. Det vil sige, at der kompenseres for, at eksponeringen ikke har foregået i alle døgnets timer over en fuld uge. Hvis eksponeringen for eksempel er foretaget 6 timer per dag i 5 dage per uge, korrigeres der med en faktor 6/24 til kontinuer eksponering gennem et helt døgn og en faktor 5/7 til kontinuer eksponering gennem hele ugen. For lokale effekter (effekter, der optræder lokalt i luftvejene samt direkte effekter på hud og øjne) anses det sædvanligvis at være stoffets koncentration i luften og ikke den samlede dosis som sådan, der er af betydning for udvikling af disse effekter, hvorfor der i disse tilfælde ikke omregnes til en kontinuert eksponering. Det skal bemærkes, at i nogle tilfælde vil det være stoffets lugt og ikke dets sundhedsmæssige effekter, der er bestemmende for luftkvalitetskriteriet og dermed B-værdien. Hvis der for det givne kemiske stof er foretaget en bestemmelse af lugttærsklen, danner denne udgangspunkt for fastsættelse af B-værdien. Miljøstyrelsen er mere og mere gået over til den praksis selv at få foretaget bestemmelse af lugtgrænse, idet metoder og værdier angivet i litteraturen i mange tilfælde ikke vurderes at være pålidelige. Kun litteraturværdier, hvor metoden ud fra en grundig beskrivelse vurderes for pålidelig, anvendes i relation til fastsættelse af lugtbaserede B-værdier. 7.4.2 B-værdiI henhold til Luftvejledningen (MST 2001) defineres B-værdien som den enkelte virksomheds samlede maksimalt tilladelige bidrag til tilstedeværelsen af et forurenende stof i luften som immission. Ved immission forstås forekomst i udendørs luft af forurenende stoffer i fast, flydende eller gasformig tilstand – normalt i ca. 1½ meters højde – over jordoverfladen. B-værdien anvendes ved den OML-beregning (OML: Operationel Meteorologisk Luftkvalitetsmodel) af skorstenshøjden, der skal foreligge for ethvert afkast, som udsender forurenende stoffer til luften. Fastsættelsen af luftkvalitetskriteriet, som det er beskrevet ovenfor, er baseret på en konstant udsættelse for et givent kemisk stof. Selv om emissionen fra en virksomhed er konstant, vil koncentrationen i udeluften i nedslagsområderne (immissionskoncentrationsbidraget) svinge på grund af skiftende meteorologiske forhold. I praksis vil den enkelte persons udsættelse derfor altid være stærkt varierende. Ifølge Luftvejledningen (MST 2001) er B-værdien ved OML-beregningerne en middelværdi over en time, der ikke må overskrides mere end 1% af tiden, det vil sige højst 7 timer af en måneds samlede timer. B-værdien er således ikke alene en koncentrationsangivelse, men tillige en oplysning om den tid, stoffet må virke i. Der skelnes mellem stoffer, der alene har en langtidseffekt, og hvor det er den akkumulerede dosis, der er afgørende for risikoen, og stoffer hvor virkningen er akut. Ved implementeringen af luftkvalitetskriteriet til en B-værdi er spørgsmålet om karakteren af stoffets skadelige virkninger, og den tid de virker i, afgørende for fastsættelse af B-værdien. Der skelnes mellem 4 forskellige tilfælde (MST 1990):
7.4.3 Luftkvalitetskriteriet anvendt i forbindelse med afdampning fra forurenet jordSom en generel regel må afdampning af kemiske stoffer fra en forurenet grund ikke medføre et højere bidrag til den ovenstående luft end luftkvalitetskriteriet. Et sådant kriterium for afdampning bliver især anvendt i forbindelse med afdampning af flygtige stoffer fra jorden og deres indsivning i indeklima. Luftkvalitetskriteriet anvendes alene til bidraget fra forureningerne til indeklimaet og kan således ikke anvendes som et overordnet kvalitetskriterium for det samlede indhold af den pågældende komponent i indreluften, da andre myndigheder (Arbejdstilsynet og Erhvervs- og Boligstyrelsen) har ansvaret for den overordnede kvalitet i indeklimaet. Tilsvarende anvendes luftkvalitetskriteriet som udgangspunkt til fastsættelse af tolerabelt bidrag fra forurenende stoffer, der fra en erhvervsvirksomhed siver ind i boliger i samme bygning, for eksempel indsivning af dampe fra renserivirksomhed til nabolejligheder. 7.5 Fastsættelse af sundhedsmæssigt baseret jordkvalitetskriteriumFølgende fremgangsmåde anvendes ved beregning af det sundhedsmæssigt baserede jordkvalitetskriterium, når de(n) kritiske effekt(er) ikke er akut toksicitet:
I relation til fastsættelse af kvalitetskriterier for jord har MST hidtil anvendt en standardværdi for daglig indtagelse af jord (EI) på 0,2 g/dag, en standardværdi for daglig hudkontakt med jord (EH) på 1 g/dag, og en standardværdi for legemsvægt (w) på 10 kg. Følgende fremgangsmåde anvendes ved beregning af jordkvalitetskriteriet, når den kritiske effekt er akut toksicitet:
Fastsættelsen af jordkvalitetskriteriet, som det er beskrevet ovenfor, foretages for den mest følsomme anvendelse af jordområdet. Der er således taget hensyn til, at små børn, der vurderes som den mest udsatte gruppe, kommer i maksimal kontakt med jorden og bliver eksponeret i forbindelse med indtagelse af jord og ved hudkontakt med jord. 7.6 Afskæringskriterier i relation til jordSom nævnt tidligere fastsættes jordkvalitetskriteriet for den mest følsomme anvendelse af jordområdet (parcelhuse og børneinstitutioner), det vil sige, at der tages hensyn til, at børn kommer i maksimal kontakt med jorden og bliver eksponeret i forbindelse med indtagelse af jord og ved hudkontakt med jord (se 7.4). Der findes imidlertid lettere forurenede områder, primært i byer, som er forurenet i et omfang, der overstiger de fastsatte jordkvalitetskriterier. Ofte drejer det sig om bly og PAH forureninger. Områderne har en geografisk udbredelse, der ikke gør det muligt at fjerne forureningen. For sådanne områder kan der fastsættes såkaldte afskæringskriterier, hvor afskæringskriteriet angiver et niveau, der kræver fuldstændig afskæring fra forureningen. Intervallet mellem jordkvalitetskriteriet og afskæringskriteriet benævnes rådgivningsintervallet. Til dette interval, hvor jorden betragtes som lettere forurenet, er der knyttet visse forholdsregler og restriktioner til anvendelse af arealerne, når disse anvendes til boliger, børneinstitutioner og offentlige legepladser. Disse forholdsregler tilsigter at begrænse den direkte eksponering for jord ved hudkontakt og indtagelse. Miljøstyrelsen har udgivet en vejledning vedrørende rådgivning af beboere i lettere forurenede områder (MST 2000). Principperne for fastsættelse af afskæringskriterier, som er beskrevet i vejledningens bilag C (MST 2000), gengives kort her. Afskæringskriteriet fastsættes med udgangspunkt i de samme vurderinger af TDI, som har være anvendt ved fastsættelse af jordkvalitetskriteriet for det pågældende stof. En gennemgang (Larsen 1998) af international praksis har vist, at man i flere lande anvender differentierede jordkvalitetskriterier afhængig af arealanvendelsen, og at den accepterede stofkoncentration i jord stiger med en faktor 5 til 10, når anvendelsen af arealet medfører mindre adgang til frie jordoverflader, for eksempel legeplads i forhold til villahave eller park. Som udgangspunkt for fastsættelse af afskæringskriteriet er der for stoffer, hvis skadelige effekter er en følge af lang tids udsættelse, anvendt en faktor på op til 10 i forhold til jordkvalitetskriteriet, idet det skønnes at efterlevelse af rådgivning og iværksættelse af eksponeringsreducerende foranstaltninger vil kunne reducere eksponeringen med forureningskomponenterne med en faktor 10. For de stoffer, hvor jordkvalitetskriteriet er fastsat på grundlag af akut toksicitet, fastsættes afskæringskriteriet til det samme som jordkvalitetskriteriet, da det ikke anses for muligt, med tilstrækkelig sikkerhed, gennem rådgivning af forebygge, at børn i enkeltstående tilfælde indtager større mængder jord. Endvidere bør der også i forbindelse med afskæringskriteriet gælde det almene æstetiske og hygiejniske krav om, at jorden ikke må have afvigende lugt eller udseende. 7.7 Fastsættelse af sundhedsmæssigt baseret drikkevandkvalitetskriteriumFølgende fremgangsmåde anvendes ved beregning af det sundhedsmæssigt baserede drikkevandkvalitetskriterium:
I relation til fastsættelse af kvalitetskriterier for drikkevand har MST hidtil anvendt en standardværdi for daglig indtagelse af vand (EI) på 2 liter/dag og en standardværdi for legemsvægt (w) på 70 kg. Kemiske stoffer, der kan trænge gennem huden, kan optages i en ikke uvæsentlig mængde i forbindelse med karbadning og brusebadning. Stoffer, der let fordamper (for eksempel mange opløsningsmidler), kan endvidere frigives fra vandet ved brusebadning og dermed indåndes og således også give et bidrag til den samlede optagelse af det pågældende stof. Omfanget af optagelsen via karbadning og brusebadning afhænger af det enkelte stofs hudgennemtrængelighed, og i hvor stor udstrækning stoffet frigives ved fordampning fra vandet. Der bør derfor i konkrete tilfælde, hvor disse bidrag vurderes særligt relevante (meget hudgennemtrængelige stoffer og/eller stoffer med høj flygtighed fra vandfasen), også tages højde for disse bidrag ved fastsættelse af drikkevandkvalitetskriteriet, således at den samlede optagelse via drikkevand og badevand ikke overskrider den del at TDI, som er allokeret til drikkevand. (MST 1992). Smag, lugt og udseende af drikkevandet har en væsentlig betydning, også selv om det ikke udgør en sundhedsfare i relation til indtagelse af drikkevand. Det vil således i nogle tilfælde være lugt, smag, eller udseende, og ikke stoffets sundhedsmæssige effekter, der er bestemmende for værdien af drikkevandkvalitetskriteriet. (MST 1992). Det er hovedprincippet, at forurening med kemiske stoffer i drikkevand ikke bør forekomme, og at man bør minimere eventuelle forureninger mest muligt, også selv om drikkevandkvalitetskriteriet ikke er overskredet (MST 1992). 7.8 ReferencerLarsen JC and Richold M (1999). Report of workshop on the significance of excursions of intake above the ADI. Regul Toxicol Pharmacol 30, S2-S12. Larsen PB (1998). Afskæringskriterier for forurenet jord. Baggrundsrapport for fastsættelse af afskæringskriterier. Miljøprojekt Nr. 425 1998. Miljø- og Energiministeriet, Miljøstyrelsen. MST (2001). Luftvejledningen. Vejledning Nr. 2 2001. Miljøstyrelsen, Miljø- og Energiministeriet. MST (2000). Rådgivning af beboere i lettere forurenede områder. Vejledning fra Miljøstyrelsen Nr. 7 2000. Miljøstyrelsen, Miljø- og Energiministeriet. MST (1992). Sundhedsmæssig vurdering af kemiske stoffer i drikkevand. Vejledning fra Miljøstyrelsen Nr. 1 1992. Miljøministeriet, Miljøstyrelsen. MST (1990). Begrænsning af luftforurening fra virksomheder. Vejledning fra Miljøstyrelsen Nr. 6 1990. Miljøministeriet, Miljøstyrelsen. Nielsen E, Larsen PB, Hansen E, Ladefoged O, Mortensen I, Strube M og Poulsen M (1995). Toksikologiske kvalitetskriterier for jord og drikkevand. Projekt om jord og grundvand fra Miljøstyrelsen Nr. 12 1995. Miljø- og Energiministeriet, Miljøstyrelsen. WHO (1996). 12. Chemical and physical aspects: introduction. In: Guidelines for drinking-water quality. Volume 2 Health criteria and other supporting information. World Health Organization, Geneva, 121-131. WHO (1994). Assessing human health risks of chemicals: derivation of guidance values for health-based exposure limits. Environmental Health Criteria 170. International Programme on Chemical Safety, World Health Organization. 8 Kombinationseffekter8.1 Rapport vedrørende kombinationseffekter af kemiske stoffer i blanding Risikovurderinger af kemiske stoffer og efterfølgende reguleringsmæssige tiltag, som for eksempel fastsættelse af kvalitetskriterier for kemiske stoffer i luft, jord og drikkevand, baseres generelt på data fra undersøgelser af de enkelte stoffer. Mennesker udsættes imidlertid for en lang række kemiske stoffer samtidigt, og disse stoffer kan potentielt have både de samme og forskellige effekter. Som følge heraf er myndighederne nødt til at tage stilling til sådanne kemiske “coctails” for at sikre, at disse ikke har uforudsete helbredseffekter. Institut for Fødevaresikkerhed og Ernæring har derfor for Miljøstyrelsen og Fødevaredirektoratet udarbejdet en rapport over den nuværende viden om kombinationseffekter af kemiske stoffer i blanding (MST 2002). Nedenfor (se 8.1) er et kort uddrag fra denne rapport, inklusive rapportens hovedkonklusioner. Som udgangspunkt fastsættes kvalitetskriterier for kemiske stoffer i luft, jord og drikkevand for det enkelte stof på baggrund af en vurdering af det pågældende stofs toksikologiske egenskaber. Såfremt der foreligger toksikologiske data for blandinger af flere stoffer, bør disse selvsagt benyttes i relation til fastsættelse af kvalitetskriterier. Men oftest foreligger denne form for data ikke. MST har for kombinationseffekter i relation til B-værdier og drikkevand fremsat administrative principper. Disse er kort gengivet i 8.2 og 8.3 respektivt. 8.1 Rapport vedrørende kombinationseffekter af kemiske stoffer i blandingInstitut for Fødevaresikkerhed og Ernæring har, som nævnt ovenfor, udarbejdet en rapport over den nuværende viden om kombinationseffekter af kemiske stoffer i blanding (MST 2002). Her gives er et kort uddrag fra denne rapport, inklusive rapportens hovedkonklusioner. Kombinationseffekter og interaktioner mellem kemiske stoffer, for eksempel lægemidler, som gives til mennesker i store doser, har været kendt i mange år inden for farmakologien. Erfaringerne herfra er dog ikke direkte anvendelige til forudsigelse af toksiske effekter af blandinger af kemiske stoffer i miljøet og fødevarer, hvor eksponeringsniveauerne for den generelle befolkning er relativt lave, og interaktioner, som forekommer ved høje doser, er ikke nødvendigvis repræsentative for eksponering i lavdosis niveauet. Det er imidlertid et faktum, at hovedparten af vores viden om kombinationseffekter af kemiske stoffer stammer fra undersøgelser, som har anvendt doser, der er langt højere end dem, der forekommer i miljøet. Ofte har doserne overskredet tærsklerne for effekter af de enkelte stoffer i blandingen. Den umiddelbare forventning er, at eksponering for meget lavere doser sandsynligvis vil resultere i mindre grad af interaktion, som primært skyldes forskellige tærskelværdier og mætningsfænomener, for eksempel med hensyn til metabolismen (aktivering/deaktivering) af stofferne i organismen. Forudsigelse af de toksikologiske egenskaber af en blanding af kemiske stoffer kræver ideelt detaljeret information om blandingens sammensætning og om virkningsmekanismerne for hvert enkelt stof i blandingen. Som regel er sådanne detaljerede oplysninger ikke til stede. Et af de vigtigste punkter, som indledningsvis skal afklares, er hvorvidt der vil forekomme interaktioner eller ej. Interaktioner i form af synergistiske eller antagonistiske effekter. Synergistiske effekter optræder, når den samlede effekt af to kemiske stoffer er større end summen af effekterne af hvert enkelt stof alene, mens en antagonistisk effekt optræder, når den samlede effekt af to kemiske stoffer er mindre en summen af effekterne af hvert enkelt stof alene. Selv om stofferne ikke påvirker hinanden (ingen interaktion), er der alligevel grund til at overveje, om de skal vurderes ud fra principper om dosis addition eller effekt/respons addition. Dosis addition anvendes, når stofferne har helt samme virkningsmåde og danner baggrund for toksicitetsækvivalenter, mens effekt/respons addition (hvor den teoretiske baggrund er kompliceret) typisk finder anvendelse, når stofferne har enten samme type effekt (f.eks. kræftfremkaldende effekt) eller respons (f.eks. samme målorgan, men forskellig virkningsmåde). Disse tre basale principper (ingen interaktion, synergi, antagonisme) for kombinationseffekter af kemiske stoffer er helt teoretiske, og ofte er man nødt til at tage hensyn til to eller alle tre koncepter samtidig, når blandinger består af mere end to stoffer, og toksicitetsmålene er komplekse. En Hollandsk forskergruppe har gennemført et forskningsprogram for at afprøve en hypotese om, at eksponering for blandinger af kemiske stoffer i (lave) ikke-toksiske doser af de individuelle stoffer, som en regel ikke vil være sundhedsmæssigt betænkeligt. En af grundene var, at de fleste retningslinier fra nationale og internationale myndighed ofte foreslår at anvende modeller baseret på simpel ”dosis addition” eller ”respons addition” til vurdering af stofblandinger, og herved totalt ignorerer ethvert kendskab til stoffernes virkningsmekanismer. Det er klart, at sådanne fremgangsmåder i høj grad vil overestimere risikoen, når der er tale om stoffer med virkningsmekanismer, hvor antagelsen af additive effekter ikke gælder. For blandinger af stoffer, som vides at have samme virkningsmekanisme og derfor ikke vil udvise interaktioner, er en kumulativ fremgangsmåde baseret på enten dosis- eller respons addition det rette valg. Ud fra resultaterne af eksperimentelle korttids studier konkluderede den Hollandske gruppe, at eksponering for blandinger af arbitrært udvalgte kemiske stoffer klart viste fravær af fuld additiv effekt og gav en vis evidens for delvis additivitet, når alle stofferne i blandingen blev indgivet i doser svarende til deres egne nul-effektniveauer (NOAELs). Ved en smule lavere dosisniveauer, blev der ikke fundet tegn på toksiske effekter. Denne konklusion gjaldt kombinationer af stoffer, som enten havde forskellige målorganer og/eller forskellige mål i det samme organ, det vil sige, at de havde forskellige virkningsmekanismer. Derfor udgør eksponering for sådanne blandinger ikke en større risiko end eksponering for det enkelte stof under forudsætning af, at eksponeringsniveauerne er enten omkring eller lavere end de individuelle stoffers NOAELs. Ved eksponeringsniveauer, som er højere end NOAELs, kan der ses både synergistiske og antagonistiske effekter, afhængigt af stofferne. Gruppen konkluderer samlet, at når eksponeringsniveauerne for de enkelte stoffer i en blanding er på ADI/TDI-niveau, så kan der ikke forventes nogen øget risiko. Anvendelse af “dosis addition” metoder i risikovurderingen af kemiske stoffer i blandinger er kun videnskabeligt forsvarlig, når alle kemiske stoffer i blandingen virker på samme måde, med den samme mekanisme, og kun er forskellige med hensyn til deres potens. Samme virkningsmekanisme kan antages at være til stede hvis to stoffer:
Risikovurderingen af eksponering for blandinger af veldefinerede kemiske stoffer bør udnytte de toksikologiske databaser optimalt. Ideelt bør udgangspunktet for vurderingen være en dosis af hver enkelt stof, der er associeret med et bestemt biologisk respons (f.eks. ED10, ED20), da dette tager hensyn til alle tilgængelige dosis-respons data. Et udgangspunkt baseret på doser, som giver et bestemt respons, bør altid foretrækkes frem for at anvende NOAEL, fordi NOAEL er en enkelt punktværdi og ikke et mål for et biologisk respons. Udgangspunktet for de individuelle stoffer bør ideelt også baseres på studier med samme dyreart og med samme administrationsvej. De data, der er tilgængelige for de fleste kemiske stoffer, tillader imidlertid ikke en estimering af for eksempel ED10 og relative potenser må derfor ofte baseres på NOAEL som udgangspunkt. 8.1.1 Forslag til generel fremgangsmådeDet er vigtigt først at skelne mellem simple blandinger og komplekse blandinger. En simpel blanding består af et relativt lille antal stoffer (f.eks. 10 eller færre), og blandingens sammensætning er kendt, både kvalitativt og kvantitativt. En kompleks blanding indeholder derimod mere end 10, måske hundreder eller tusindvis af stoffer, og den kvalitative og kvantitative sammensætning kendes ikke fuldt ud. Der bør også skelnes mellem analyse af hele blandinger (top-down approach) og analyse af de enkelte komponenters interaktioner (bottom-up approach). Den sidstnævnte fremgangsmåde kræver forståelse af de grundlæggende koncepter vedrørende kombinationseffekter af kemiske stoffer. Groten et al. (2001) har foreslået generelle fremgangsmåder til sikkerhedsvurdering af simple og komplekse blandinger. For simple blandinger er den mest pragmatiske og måske enkleste måde at teste blandingens toksicitet uden at identificere hvilke typer af interaktioner, der optræder mellem blandingens komponenter. Resultaterne af en sådan testning kan imidlertid kun anvendes til fareidentifikation og risikovurdering af eksponering for denne specielle blanding. En mere detaljeret fremgangsmåde er at undersøge kombinationseffekterne mellem blandingens individuelle komponenter. Her kan der anvendes forskellige undersøgelsesdesign, afhængig af blandingens kompleksitet og antallet af stoffer i blandingen. Det største problem ved vurderingen af resultaterne er at afgøre, hvorvidt komponenterne påvirker de samme toksikologiske processer, gennem den samme virkemåde, eller om deres virkemåder er funktionelt uafhængige. Med hensyn til komplekse blandinger anbefales det at anvende en to-trins fremgangsmåde, dersom blandingen ikke kan testes direkte. Først identificeres de ”n” (f.eks. 10) mest risikofyldte stoffer i blandingen. Dernæst gennemføres fareidentifikation og risikovurdering af de (10) prioriterede stoffer under anvendelse af procedurerne beregnet til vurdering af simple, veldefinerede blandinger: Udvælgelse af “top 10” stofferne under første trin bør baseres på eksponeringsniveauerne og graden af toksicitet af de individuelle stoffer. Jo større risikokvotienten (RQ) er, jo højere er sandsynligheden for skadelige effekter i mennesker (dvs. højere risiko), og des højere skal stoffet rangeres på listen over de prioriterede stoffer. Fareidentifikation og risikovurdering af den udvalgte blanding af stoffer (“top 10” stofferne) bør baseres på toksicitetsdata, på kendskab til virkningsmekanismerne for de individuelle stoffer, og på forudsigelse af tilstedeværelse eller fravær af additive eller potentierende interaktive effekter mellem de udvalgte stoffer. For at kunne forudsige kumulativ effekt (additivitet) eller interaktion er det nødvendigt med kendskab til den formodede virkningsmekanisme. Udarbejdelse af et system til klassifikation af kemiske stoffer på basis af deres virkningsmekanismer vil derfor være ekstremt nyttigt. Trin 1: Identifikation af prioriterede stoffer Udvælg et begrænset antal stoffer (f.eks. 10) med det største potentiale for risiko, ved at anvende risikokvotienten (RQ):
Med andre ord, udvælg blandingens “top 10” stoffer Trin 2: Farekarakterisering og risikovurdering Identificer fare og vurder risiko for den udvalgte blanding af de (“10”) prioriterede stoffer, under anvendelse af principperne for vurdering af simple blandinger af kemiske stoffer. En pragmatisk fremgangsmåde: Foretag begrænsede toksikologiske studier f.eks. et 4-ugers rotteforsøg og en screeningstest for genotoksicitet med den udvalgte blanding af de (“10”) prioriterede stoffer, under anvendelse af eksponeringskoncentrationer, der er f.eks. 3-10 gange højere end i den komplekse blanding. Vurderingen antager implicit, at fare og risiko ved eksponering for den definerede (“top 10”) blanding af stoffer er repræsentativ for fare og risiko ved eksponering for hele den komplekse blanding. 8.1.2 Simple blandinger af pesticider i fødevarerInstituttet for Fødevaresikkerhed og Ernæring har udarbejdet en rapport om kombinationseffekter af pesticider i fødevarer for Fødevaredirektoratet (Reffstrup 2002). I rapporten foreslås det, at risikovurderingen af pesticidrester i fødevarer foretages på en “case-by-case” basis, hvor de kemiske og toksikologiske data vurderes i en proces, der lægger vægt på den foreliggende evidens. Det foreslås generelt at anvende hazard index metoden med ADI (hvor de individuelle sikkerhedsfaktorer er inkluderet) og ikke NOAEL (hvor sikkerhedsfaktorer ikke er inkluderet) som den acceptable nævner ved beregning af de individuelle hazardkvotienter. Dog bør toksicitets ækvivalentfaktor metoden (TEF) anvendes i de tilfælde, hvor vægten af evidens peger på, at stofferne i blandingen har samme virkningsmåde (som f.eks. organophosphaterne). 8.2 Kombinationseffekter i relation til B-værdierMST (2001) har for kombinationseffekter i relation til B-værdier og samtidig emission af flere stoffer fremsat følgende administrative principper: Når stofferne virker uafhængigt af hinanden, det vil sige ikke påvirker hinandens effekt, skal B-værdierne for stofferne hver for sig være overholdt. Afkasthøjden skal fastsættes på grundlag af det stof, der giver den største spredningsfaktor. Hvis stofferne påvirker hinandens effekter og virkemåde, vil man for visse stofblandinger kunne risikere, at stofferne kan forstærke hinandens effekter, det vil sige have en synergistisk virkning. Erfaringerne fra dyreforsøg tyder på, at sådanne effekter først gør sig gældende, når stofferne hver især er til stede i en koncentration i luften, der i sig selv vil medføre effekter, det vil sige er til stede over deres individuelle nul-effektniveau. Da B-værdien for det enkelte stof typisk er fastsat ud fra data vedrørende nul-effektniveauet og under anvendelse af en usikkerhedsfaktor, vil der ikke forventes effekter ved B-værdien. Overholdelse af stoffernes individuelle B-værdier vurderes derfor umiddelbart at sikre, at interaktion mellem stofferne, herunder forstærkende effekter, ikke kan forekomme. Afkasthøjden skal derfor fastsættes på grundlag af det stof, der giver den største spredningsfaktor. Når stofferne har samme effekter og virkemåde, er der grundlag for at summere eksponeringsbidraget for de enkelte stoffer. Der bør i praksis ske addition i B-værdisammenhæng for ensvirkende stoffer, når 1) stofferne er homologe stoffer (stoffer fra samme kemiske stofgruppe som for eksempel alkoholer, ketoner eller ethere) og 2) stofferne tilhører samme stofgruppe i Luftvejledningen og 3) stofferne har sundhedsrelaterede B-værdier (det vil sige, at de ikke er mærket med et L). Hvis alle 3 punkter er opfyldt, bør afkastberegningen foretages på grundlag af den samlede emission af stofferne. Det kan ske ve at fastlægge den resulterende Br-værdi (se formlen nedenfor). Denne Br-værdi er udtryk for en samlet B-værdi for blandingen, beregnet på grundlag af de enkelte stoffers kildestyrke og B-værdier.
8.3 Kombinationseffekter i relation til kvalitetskriterier for drikkevandSom udgangspunkt fastsættes kvalitetskriterier for kemiske stoffer i drikkevand for det enkelte stof på baggrund af en vurdering af det pågældende stofs toksikologiske egenskaber. Dog er der i henhold til bekendtgørelse om vandkvalitet og tilsyn med vandforsyningsanlæg (MEM 2001) fastsat kvalitetskrav for en række grupper af stoffer i drikkevand, for eksempel sum af trihalomethaner, sum af øvrige phthalater, anioniske detergenter. MST (1992) har for kombinationseffekter i relation til forekomst af nærtbeslægtede stoffer hidtil anvendt additionsformlen:
Det er anført, at additionsformlen kun kan benyttes for stoffer, der medfører beslægtede effekter, det vil sige stoffer, der for eksempel alle er neurotoksiske. Ved anvendelsen af additionsformlen tages der således ikke hensyn til en eventuel synergistisk eller antagonistisk effekt. Såfremt der er kendskab til størrelsen af synergistiske eller antagonistiske effekter, bør denne viden selvfølgelig indgå i vurderingen. 8.4 ReferencerGroten JP, Feron VJ and Sühnel J (2001). Toxicology of simple and complex mixtures. Trends in Pharmacological Sciences 22, 316-322. MEM (2001). Bekendtgørelse om vandkvalitet og tilsyn med vandforsyningsanlæg. BEK nr. 871 af 21. september 2001. Miljø- og Energiministeriet. MST (2002). Combined actions and interactions of chemicals in mixtures. The toxicological effects of exposure to mixtures of industrial and environmental chemicals. Miljøprojekt …… Endnu ikke publiceret. MST (2001). Luftvejledningen. Vejledning Nr. 2 2001. Miljøstyrelsen, Miljø- og Energiministeriet. MST (1992). Sundhedsmæssig vurdering af kemiske stoffer i drikkevand. Vejledning fra Miljøstyrelsen Nr. 1 1992. Miljøministeriet, Miljøstyrelsen. Reffstrup TK (2002). Combined actions of pesticides in food. Ministeriet for Fødevarer, Landbrug og Fiskeri, Fødevaredirektoratet. FødevareRapport 2002:19. Bilag A TerminologiBenchmark dose - BMD: Den nedre konfidensgrænse for en koncentration eller dosis, der fører til en given kritiske effektstørrelse, det vil sige den incidens (f.eks. 1, 5 eller 10%), der estimeres for en given effekt. B-værdi: Bidragsværdi, som er den enkelte virksomheds samlede maksimalt tilladelige bidrag til tilstedeværelsen af et forurenende stof i luften i omgivelserne udenfor virksomheden, det vil sige immissionen. Default værdi: Pragmatisk fastlagt værdi som anvendes når datagrundlaget ikke er tilstrækkeligt med henblik på fastlæggelse af en eksakt værdi. Anvendes blandt andet i relation til usikkerhedsfaktorer (UF) og standardeksponeringer. På dansk: udgangsværdi eller standardværdi. Dosis: Den mængde af et stof (angives sædvanligvis i enheden mg eller mg/kg legemsvægt) som er tilgængelig for optagelse i organismen, f.eks. er det ved indånding volumet af indåndet luft (angives sædvanligvis i enheden m³) ganget med stofkoncentrationen (angives sædvanligvis i enheden mg/m³). Dosis (koncentration) – effekt vurdering: Karakterisering af sammenhængen mellem dosis (eller koncentration) af stoffet og de observerede effekter. Dosis (koncentration) – respons vurdering: Karakterisering af sammenhængen mellem dosis (eller koncentration) af stoffet og forekomst/hyppighed (engelsk: incidence) og alvorlighed (engelsk: severity) af en effekt. Eksponeringsvurdering: Vurdering af de koncentrationer (eller doser) af et stof i forskellige medier (f.eks. luft, jord, vand, levnedsmidler, forbrugerprodukter, indeklima), som et individ udsættes for. Angives for luft sædvanligvis i mg/m³, for jord i mg/kg jord, for drikkevand i mg/liter, og for levnedsmidler i mg/kg levnedsmiddel. Andre angivelser kan være mg/dag og mg/kg legemsvægt per dag. Farlighedsvurdering: Vurdering af et kemisk stofs forskellige iboende toksikologiske (sundhedsskadelige) effekter. Grænseværdi: En administrativt fastsat værdi, typisk for det maksimale indhold i et givent medie, eventuelt i et produkt. Er som regel et lovbundet reguleringsinstrument fastsat gennem udstedelse af en bekendtgørelse. Tidligere anvendtes termen i forbindelse med en sundhedsmæssigt baseret grænseværdi, hvor der i dag i stedet anvendes betegnelsen kvalitetskriterie. Hormesis: En dosis-responssammenhæng, hvor der ved lave doser ses en stimulation og ved høje doser en inhibition og giver sig udtryk ved et U-formet eller omvendt U-formet kurveforløb. Kravværdi: Anvendes på tilsvarende vis som grænseværdi, se ovenfor. Kritisk effekt: Den toksikologiske effekt, der anses for at være afgørende, det vil sige danner udgangspunkt, ved fastsættelsen af kvalitetskriterier i luft, jord og vand. Kvalitetskriterium: Vejledende værdi, det vil sige ikke en lovbundet værdi, der fastsættes af Miljøstyrelsen i relation til for eksempel luft, jord, drikkevand og grundvand. Der eksisterer både sundhedsmæssigt baserede og økotoksikologiske kvalitetskriterier. Denne rapport omhandler udelukkende de sundhedsmæssigt baserede kvalitetskriterier. LOEL/C (Lowest Observed Effect Level / Concentration) – laveste observerede effektniveau: Den laveste dosis / koncentration af et stof, som medfører en ændring i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer, og som kan påvises i forhold til en sammenlignelig kontrolgruppe. De ændringer, der ses ved LOEL/C, vurderes ikke at være af skadelig karakter. LOAEL/C (Lowest Observed Adverse Effect Level / Concentration): Den laveste dosis / koncentration af et stof, som medfører en skadelig ændring i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer, og som kan påvises i forhold til en sammenlignelig kontrolgruppe. Lokale effekter: Effekter, der optræder lokalt i luftvejene eller i mave-tarmkanalen samt direkte effekter på hud og øjne ved direkte kontakt med stoffet. NOEL/C (No Observed Effect Level / Concentration) – observeret nul-effektniveau: Den højeste dosis / koncentration af et stof, som ikke medfører ændringer i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer i forhold til en sammenlignelig kontrolgruppe. NOAEL/C (No Observed Adverse Effect Level / Concentration): Den højeste dosis / koncentration af et stof, som ikke medfører påviselige skadelige ændringer i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer. Der kan ved NOAEL/C ses ændringer i ovennævnte parametre, som ikke vurderes at være af skadelig karakter. Provisorisk tolerabel ugentlig indtagelse (PTWI): En foreløbig beregnet indtagelse som mennesker vurderes at kunne indtage ugentligt (tolerere) gennem et helt livsforløb uden at der optræder sundhedsskadelige effekter. PTWI angives sædvanligvis i enheden mg/kg legemsvægt per uge. Skadelig effekt (adverse effect): Ændring i morfologi, fysiologi, funktion, vækst, udvikling, eller levetid hos eksponerede individer, som resulterer i svækkelse af kapaciteten til at kompensere for yderligere stress eller øget følsomhed for andre miljøbetingede effekter. Systemiske effekter: Effekter der optræder i organismen efter optagelse af stoffet for eksempel via luftvejene, mave-tarmkanalen, og/eller huden. Toksikodynamik: Omhandler kemiske stoffers virkning på organismen (på cellulært og molekylært plan) og som fører til observerbare effekter. Toksikokinetik: Omhandler kemiske stoffers skæbne i organismen, dvs. optagelse, fordeling, metabolisme samt udskillelse, og beskriver både kvalitative og kvantitative aspekter. Tolerabel daglig indtagelse (TDI): En beregnet indtagelse som mennesker vurderes at kunne udsættes for (tolerere) gennem et helt livsforløb uden at der optræder sundhedsskadelige effekter. TDI angives sædvanligvis i enheden mg/kg legemsvægt per dag. Tolerabel koncentration (TK): Den koncentration af et stof i luft, jord eller drikkevand som mennesker vurderes at kunne udsættes for (tolerere) gennem et helt livsforløb uden at der optræder sundhedsskadelige effekter. TK angives i enheden mg/m³ (luft), mg/l (drikkevand), og i mg/kg (jord). Tærskelværdi: Den koncentration eller dosis hvorunder der ikke observeres effekter, se også NOEL / NOAEL. Usikkerhedsfaktorer (UF) – tidligere sikkerhedsfaktorer: Anvendes ved beregning af TDI og kvalitetskriterier for stoffer hvor der anses at være en tærskelværdi for den kritiske effekt.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||