|
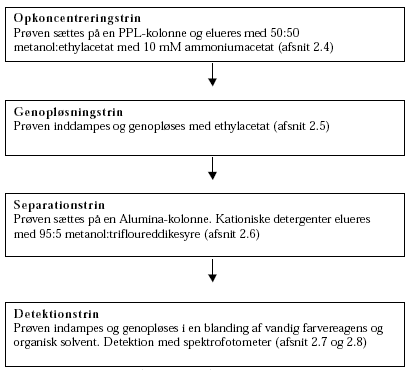
Metodeudvikling og -validering kationiske detergenter 2 Metodeudvikling2.1 Den anvendte metodeMetoden i denne metodeundersøgelse bygger på en adskillelse i 3 detergenttyper. Princippet er beskrevet af Kloster et al (1994) /15/ og N. Buschmann et al (1992) /14/. K.-H. Theil (2002) /8/ har på baggrund heraf undersøgt forskellige kolonnetyper og eluenter, og det er dette arbejde, denne metodeudvikling bygger videre på. De enkelte trin og principperne for denne metode er vist i nedenstående figur.
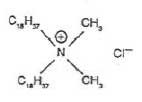
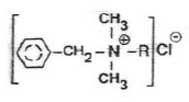
Figur 2.1-1 Principperne og grundtrinnene i metoden. Metodeundersøgelsen blev gennemført således, at de enkelte deltrin blev undersøgt og optimeret enkeltvis (jf. Figur 2.1-1 og afsnit 2.3 til 2.8). Når der for enkelttrin blev fundet acceptable genfindinger og repeterbarheder, blev hele metoden undersøgt samlet (afsnit 2.9). I metodeundersøgelsen blev alkyldimethylbenzylammoniumchlorid (ADMBAC) benyttet som modelstof. Denne kationiske detergent bliver benyttet som standardstof i den danske metode (Østergaard et al, 1999) /9/. Disteryldimethylammoniumchlorid (DSDMAC) blev benyttet som standardstof. DSDMAC er standardstoffet, der benyttes i den tyske standard (DIN 38409 Teil 20, 1989) /5/. I Tabel 2.1-1 er strukturformlerne for de to kationiske detergenter gengivet.
Tabel 2.1-1 Strukturformler for de anvendte kationiske detergenter (Merry et al, 1999) /1/. 2.2 Generelle betingelser og apparatur i undersøgelsenFor at forenkle beskrivelsen af forsøgsbetingelserne ved de enkelte delundersøgelser i metodeudviklingen er de generelle betingelser og anvendt apparatur beskrevet her. Hvis intet andet er anført i teksten nedenfor, er undersøgelserne udført under disse betingelser. De anvendte kemikalier og reagenser er svarende til dem, der er beskrevet i metodeforskriften i Bilag A. Alt glasudstyr var detergentvasket inden brug efter metoden, der ligeledes er beskrevet i Bilag A. Syntetiske prøver blev fremstillet med en stofmængde svarende til 50 μg ADMBAC i 500 mL Milli-Q vand. Alle genfindinger blev udregnet på baggrund af den tilsatte stofmængde og ikke koncentrationerne. Grunden til denne fremgangsmetode var, at prøvevolumenet varierede i mange af de undersøgte deltrin. Stofmængderne blev sammenlignet for at tage højde for disse volumenændringer. Det anvendte udstyr til fastfaseekstraktionen var en VAC ELUT SPS 24 vacuum manifold fra Varian. Herpå blev Bond Elut PPL-kolonner (3 mL, 200 mg) benyttet til opkoncentreringstrinnet og Mega BE-AL-B alumina-kolonner (6 mL, 1g) til separationstrinnet. Detektionstrinnet i metodeudviklingen blev gennemført efter følgende princip: Prøven blev udrystet med bromthymolblåt i 250 mL skilletragt med chloroform. Et gulfarvet kompleks blev derved ekstraheret over i chloroformfasen, der blev bestemt med spektrofotometri ved 416 nm. Metoden er beskrevet af Østergaard et al (1999) /9/. I afsnit 2.7 undersøges en alternativ detektionsmetode svarende til den tyske standard (DIN 38409 Teil 20, 1989) /5/. I undersøgelserne efter afsnit 2.7 blev denne metode benyttet. 2.3 Indledende undersøgelserFor at kunne undersøge de enkelte trin i analysen blev det først undersøgt, om modelstoffet (ADMBAC) og standardstoffet (DSDMAC) gav identiske molære absorptionskoefficienter i forskellige matricer efter udfarvningen med bromthymolblåt. Resultaterne i matricerne blev undersøgt over for den molære absorptionskoefficient udregnet fra en 4-punkts kalibreringskurve for både ADMBAC og DSDMAC i methanol. Methanol svarer til matricen i den eksterne kalibreringskurve, når den fremstilles efter tysk /5/. Matricerne blev valgt svarende til prøvematricen i de forskellige trin i den planlagte procedure for opkoncentrering og separation. Dette var henholdsvis 50:50 methanol/ethylacetat med 10 mM ammoniumacetat svarende til trinnet for opkoncentrering og 95:5 methanol/trifloureddikesyre svarende til trinnet for separation i proceduren.
Tabel 2.3-1 Sammenligning af molære absorptionskoefficienter for bromthymolblåt i forskellige matricer. Resultaterne for methanol var udregnet på baggrund af en 4-punkts kalibreringskurve. Andre matricer var undersøgt med 3-dobbelte bestemmelser på en 50 μg prøve. A) Variationskoefficienten (CV) er givet som standardafvigelsen/gennemsnittet*100%. Originaldata findes i Bilag B. Undersøgelsen viste, at den molære absorptionskoefficient varierede med matricen. Sammenlignes i stedet de molære absorptionskoefficienter for ADMBAC og DSDMAC inden for den samme matrice, blev der fundet en genfinding på 96% (1,59*106 /1,65*106) af DSDMAC relativt til ADMBAC i 50:50 methanol/ethylacetat med 10 mM ammoniumacetat og 105% (1,25*106/1,20*106) i 95:5 methanol/trifloureddikesyre. Dette viste, at ADMBAC og DSDMAC havde tilnærmelsesvis ens molære absorptionskoefficienter inden for de forskellige matricer. I undersøgelsen af de enkelte deltrin blev samme matrice derfor benyttet for den eksterne kalibreringskurve som prøvematricen efter det undersøgte deltrin. I den endelige metode er det nødvendigt, at den eksterne kalibreringskurve fremstilles og måles i samme matrice som prøverne. 2.4 Opkoncentreringstrin (PPL-kolonne)I dette trin foregik en applikation af prøven på en PPL-kolonne, efter at kolonnen var blevet konditioneret. Ved applikation menes, at detergenterne i prøven tilbageholdes på kolonnen. Prøven blev fremstillet direkte i 500 mL målekolbe og blev overført til PPL-kolonnen med et slangesystem i teflon fra Supelco. PPL-kolonnen blev herefter tørret med luft. Elueringen skete med 50:50 methanol/ethylacetat med 10 mM ammoniumacetat. I de første undersøgelser blev elueringen som udgangspunkt foretaget med 5 mL eluent over i 10 mL spidsglas. Opkoncentreringstrinnet blev undersøgt for blindniveau, optimeret med hensyn til elueringsvolumen, eluering med stop, applikationshastighed og applikationsvolumen. 2.4.1 BlindI indledende forsøg (ikke medtaget her) blev der observeret en stigende blindværdi igennem en række forsøgsserier. Proceduren for vask af glasudstyr blev gennemgået og endte svarende til forskriften angivet i Bilag A. For at sikre, at problemet var løst, og eventuelt finde andre kilder til blindværdier blev følgende forsøg udført.
Tabel 2.4-1 Absorbanser ved 416 nm for blindforsøg på PPL-kolonne. Resultaterne bygger på 4 bestemmelser for blindforsøgene og dobbeltbestemmelser for den eksterne blind og kontrol. A) Angiver den benyttede mængde af 50:50 methanol/ethylacetat med 10 mM ammoniumacetat i konditioneringstrinnet. Som det fremgår af Tabel 2.4-1, var blindniveauerne lave. Idet en svag reduktion i blindniveau blev observeret ved stigende volumen anvendt til konditionering, blev 15 mL eluent benyttet i det efterfølgende. 2.4.2 Applikationshastighed og elueringsvolumenFor at fastlægge applikationshastigheden blev to forskellige hastigheder på PPL-kolonnen undersøgt. Disse var henholdsvis 25 og 45 minutter til applikation af 500 mL prøve. Samtidig blev elueringsvolumenet undersøgt. Det anvendte udstyr gjorde, at der maksimalt kunne benyttes 10 mL eluent.
Tabel 2.4-2 Genfinding af eksterne standarder ved forskellige kombinationer af applikationshastighed og elueringsvolumen. De to forskellige genfindingsresultater henfører til to forskellige analysedage. Originaldata findes i Bilag C og D. A) Denne kombination blev ikke medtaget på forsøgsdag 2. Det fremgår af Tabel 2.4-2, at en langsom applikation (45 minutter eller 30 dråber på 10 sekunder) sammen med det maksimale elueringsvolumen (10 mL) gav den bedste genfinding. Genfindingen var dog ikke tilfredsstillende på begge forsøgsdage, og derfor blev det forsøgt at opdele elueringen i 4 del-elueringer med 3 pauser på hver 5 minutter. De opnåede resultater er opstillet i Tabel 2.4-3.
Tabel 2.4-3 Genfinding af ekstern standard med og uden pauser i elueringen. Originaldata findes i Bilag E. Som det fremgår af Tabel 2.4-3, påvirkede indlagte pauser (og dermed længere tid til at opnå ligevægt imellem kolonnematerialet og eluat) ikke genfindingsresultatet. Derfor blev eluering med 10 mL uden pauser på 45 minutter bibeholdt. Genfindingen er stadig uacceptabelt lav. 2.4.3 ApplikationsvolumenetFor at undersøge, om den relativt lave genfinding skyldtes, at ADMBAC blev vasket ud af PPL-kolonnen på grund af det høje prøvevolumen (500 mL), blev prøver med forskellige prøvevolumener, men med konstant stofmængde undersøgt.
Tabel 2.4-4 Genfinding af 50 μg ADMBAC-prøver med variabel prøvevolumen fremstillet direkte i målekolber. I undersøgelsen blev der sammenlignet med A) Ekstern standard fremstillet i 50:50 methanol/ethylacetat med 10 mM ammoniumacetat. B) Applikation direkte på kolonne, først 5 mL standard efterfulgt af 10 mL Milli-Q vand. C) 25 μg ADMBAC prøve i 500 mL. Originaldata findes i Bilag F. Tabel 2.4-4 viser, at ADMBAC ikke blev vasket ud af kolonnen ved store prøvevolumener. Da disse genfindingsprocenter var acceptable i forhold til kravene i Forordet, blev de andre trin i metoden undersøgt. De forbedrede genfindingsprocenter i forhold til Tabel 2.4-3 blev tillagt øget rutine med metoden hos teknikeren, der udførte forsøgene. Endvidere blev det fundet vigtigt at undersøge, hvorvidt de efterfølgende trin gav anvendelige resultater, før dette trin eventuelt blev endeligt optimeret. 2.4.4 DelkonklusionUndersøgelserne viste, at en lang applikationstid (45 minutter) sammen med et højt elueringsvolumen (10 mL) gav de bedste genfindinger af den eksterne standard. Endvidere blev det vist, at pauser i elueringen ikke gav målelig forbedring af genfinding, og at et øget prøvevolumen ikke bidrog negativt til resultaterne. En øget mængde af 50:50 mehanol/ethylacetat med 10 mM ammoniumacetat i konditionneringstrinnet af PPL-kolonnen reducerede antydningsvis blindværdien. Derfor konditioneres i metodeforskriften i Bilag A med 15 mL. En acceptabel genfinding blev fundet for dette deltrin i de afsluttende forsøgsserier. 2.5 Genopløsningstrin mellem PPL- og alumina-kolonneNår prøven var elueret fra PPL-kolonnen, var den opløst i en 50:50 methanol/ethylacetat med 10 mM ammoniumacetat opløsning. Den blev inddampet til tørhed og genopløst i 5 mL ethylacetat på ultralydsbad i 5 minutter. Herefter kunne detergenterne i prøven sættes på en alumina-kolonne efter opvarmning. Til undersøgelse af dette trin blev prøver i en 50:50 mehanol/ethylacetat med 10 mM ammoniumacetat fremstillet direkte i spidsglassene. Disse blev inddampet til tørhed og genopløst. Genfindingsprocenterne over for eksterne standarder er vist i nedenstående tabel.
Tabel 2.5-1 Genfinding af 50 μg og 100 μg ADMBAC prøver efter inddampning fra 50:50 methanol/ethylacetat med 10 mM ammoniumacetat og genopløsning i ethylacetat. I undersøgelsen blev der sammenlignet med en ekstern standard fremstillet i ethylacetat. Originaldata findes i Bilag G. Som det fremgår af Tabel 2.5-1, var genfinding for høj i forhold til kravet i Forordet. I undersøgelsen fandt vi en gennemsnitlig blindabsorbans på 0,005 svarende til 3% af absorbansen i en 50 μg prøve. Altså var der intet betydeligt blindbidrag fra dette trin. Genfindingsprocenter større end 100 skyldtes usikkerheden i fremstillingen af standarderne. Konklusionen var, at der kunne opnås en acceptabel genfinding sammen med en lav blindabsorbans. Resultaterne viste, at dette trin kunne benyttes i analysemetoden. Her blev kun undersøgt syntetiske prøver. I afsnit 2.9 blev problemer i dette trin for spildevandsprøver observeret. Derfor blev dette trin efterfølgende modificeret, se afsnit 2.9. 2.6 Separationstrinnet (Mega BE-AL-B alumina-kolonne)Separationen af nonioniske, kationiske og anioniske detergenter foregik i dette trin på Mega BE-AL-B alumina-kolonner. Efter applikation af prøven på kolonnen kunne de forskellige detergentfraktioner isoleres i forskellige eluenter:
Der blev elueret med 6 mL af hver eluent. I det følgende undersøges, om isoleringen af kationiske detergenter fra anioniske detergenter er effektiv. Endvidere undersøges om anioniske detergenter kan genfindes efter eluering med 75:25 methanol/2 M HCl. 2.6.1 Isolering af kationiske detergenterDe tre eluentfraktioner blev undersøgt for kationiske detergenter.
Tabel 2.6-1 Genfinding af kationiske detergenter i de tre fraktioner i separationstrinnet over for en ekstern standard af 95:5 methanol/trifloureddikesyre. Originaldata findes i Bilag H. A) Enkeltbestemmelse. Som det fremgår af Tabel 2.6-1, blev hovedparten af de kationiske detergenter genfundet i den forventede fraktion. I de to andre fraktioner var absorbansniveauet af samme størrelsesorden som blindbiddraget. 2.6.2 Interferensundersøgelse fra anioniske detergenter (LAS)Genfindingen af kationiske detergenter i prøver tilsat forskellige niveauer af anioniske detergenter (LAS) er gengivet i Tabel 2.6-2.
Tabel 2.6-2 Genfinding af kationiske detergenter i separationstrinnet efter tilsætningen af anioniske detergenter over for en ekstern standard i 95:5 methanol/trifloureddikesyre. Originaldata findes i Bilag I. A) På baggrund af en 50 μg ADMBAC opløsning. Undersøgelsen viste, at genfindingen ikke blev systematisk påvirket af tilstedeværelsen af anioniske detergenter, og at den overholdt kravene fra Forordet. 2.6.3 Genfinding af anioniske detergenter (LAS) i 3. fraktionFor at dokumentere, at de anioniske detergenter blev separeret fra de kationiske detergenter i fraktion 2 og opsamlet i 3. fraktion, blev LAS undersøgt i de forskellige fraktioner. Resultaterne af denne undersøgelse vises i Tabel 2.6-3. Til analyse af LAS blev en specifik analyse anvendt (HPLC med UV- og fluorescensdetektion (UV-FLD) /32/).
Tabel 2.6-3 Fordeling af LAS i de forskellige fraktioner efter seperationstrinnet. 1. fraktion er nonioniske detergenter, 2. trin er kationiske detergenter og 3. fraktion er anioniske detergenter. Resultaterne er i procent af den total målte mængde fra den specifikke analyse af LAS målt med HPLC med UV-FLD /15/. A) målingen af
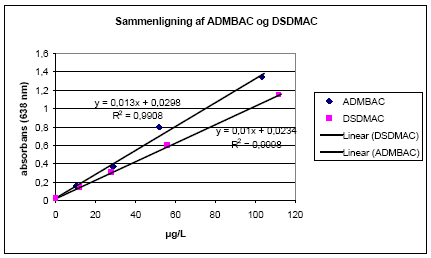
LAS i denne prøve gav intet resultat. Undersøgelsen viste, at hovedparten af LAS blev elueret i 3. fraktion. Den del, der blev elueret med i 2. fraktion, udgjorde mindre end 3%. Derfor blev det konkluderet, at separationen af kationiske og anioniske detergenter var tilfredsstillende. 2.6.4 DelkonklusionUndersøgelserne dokumenterede, at hovedparten (97%-108%) af de kationiske detergenter blev elueret i 2. fraktion (95:5 methanol/trifloureddikesyre), og at metoden fjernede interferensen fra anioniske detergenter i tilstrækkeligt omfang. Endvidere blev det vist, at anioniske detergenter blev elueret i 3. fraktion (>97%). En acceptabel genfinding af eksten standard blev fundet i dette deltrin. 2.7 DetektionstrinnetEt detektionsprincip svarende til den tyske standard (DIN 38409 Teil 20, 1989) /5/ blev undersøgt. Prøven blev først inddampet i et 50 mL bægerglas. Herefter blev 10 mL disulfinblåtbuffer og 25 mL 95:5 chloroform/butanol tilsat, og der blev omrørt kraftigt i 5 minutter på magnetomrører. Det blå kompleks i den organiske fase blev bestemt spektrofotometrisk ved 628 nm. Detektionstrinnet blev først undersøgt for linearitet, følsomhed og præcision. Dernæst blev de molære absorbanser for ADMBAC og DSDMAC sammenlignet. Effekten af ufuldstændig inddampning blev også undersøgt. 2.7.1 Linearitet, følsomhed og præcisionDet sås ud fra 4-punkts kalibreringskurven givet i Bilag J, at den testede detektionsmetode gav en god lineær sammenhæng (R2=0,9999). Endvidere gav metoden en god følsomhed. Sammenlignede man følsomheden i absorbansenheder med den eksisterende VKI-metode (Østergaard et al, 1999 /9/), sås en forbedring med ca. en faktor 10. Variationskoefficienten (CV) på de enkelte punkter i kalibreringskurven var maksimalt 2%, hvilket var tilfredsstillende. 2.7.2 Sammenligning af ADMBAC og DSDMACAbsorptionskoefficienterne for ADMBAC og DSDMAC blev sammenlignet.
Figur 2.7-1. Kalibreringskurver med ADMBAC og DSDMAC. Originaldata er gengivet i Bilag K. I Figur 2.7-1 ses det, at kalibreringskurven for ADMBAC gav en højere absorbans og dermed en bedre følsomhed end kalibreringskurven for DSDMAC. Absorptionskoefficienten for DSDMAC udgjorde kun ca. 80% af den for ADMBAC. Det fremgår af kalibreringskurven for ADMBAC, at R2 var dårligere end resultatet fundet i afsnit 2.7.1. Dette skyldtes det næsthøjeste punkt. Ved at udelade dette punkt kunne samme linearitet (R2=0,9993) findes. Denne udeladelse ændrer ikke på andre af konklusionerne. Da det var ønskeligt, at resultaterne med denne metode kunne sammenlignes med den tyske standard (DIN 38409 Teil 20, 1989) /5/, skulle standardkurven med DSDMAC benyttes i den endelige metode. 2.7.3 Effekten af ufuldstændig inddampningEffekten af en ufuldstændig inddampning af 95:5 methanol/trifloureddikesyre (solvent fra eluering af alumina-kolonne) inden tilsætningen af disulfinblå og 95:5 chloroform/butanol til bæreglasset er vist i Tabel 2.7-1.
Tabel 2.7-1 Absorbanser for 3-dobbeltbestemmelse af 50 μg ADMBAC. Det fremgår af Tabel 2.7-1, at følsomheden faldt til 72%, hvis der ikke blev inddampet fuldstændigt. Endvidere blev præcisionen dårligere. Dette skyldtes formentlig, at remanensen var trifloureddikesyre, som ændrede pH i farvereaktionen. Derfor var en fuldstændig inddampning nødvendig. 2.7.4 DelkonklusionUdfarvningsmetoden svarende til den tyske standard /5/ med disulfinblåt blev undersøgt. Undersøgelsen viste en forbedret følsomhed (over 10 gange højere målt i absorbansenheder i forhold til den nuværende VKI-metode /9/. Ligeledes blev der fundet en god præcision (CV på de enkelte punkter i kalibreringskurven på 2%). I undersøgelsen af kalibreringskurver for ADMBAC og DSDMAC viste det sig, at følsomheden for DSDMAC kun er ca. 80% af den for ADMBAC. I denne metode blev en magnetomrører benyttet til væske-væske ekstraktionen, og derved blev den manuelle udrystning af en 250 mL skilletragt i tre gange 30 sekunder undgået. Dette gjorde metoden betydeligt mere arbejdsmiljøvenlig og mindre arbejdsintensiv end den eksisterende metode. 2.8 Alternative solventerDet er ønskeligt at undgå chloroform i laboratoriet. Derfor blev det undersøgt, om det var muligt at substituere den benyttede 95:5 chloroform/butanol-blanding med et andet organisk solvent med lignende egenskaber (specielt polaritet). Substitutionen blev forsøgt med butylacetat, ethylacetat, methylisobutylketon (MIBK) og N-butanol. Undersøgelsen blev udført kvalitativt, ved at 5 mL standard (50 μg ADMBAC) blev overført til et 50 mL bægerglas. Efterfølgende blev 10 mL disulfinblåtbufferopløsning og 25 mL organisk solvent overført. Herefter blev der omrørt kraftigt i 5 minutter på magnetomrøreren. De organiske faser blev vurderet visuelt og på UV-spektrofotometret. Samme procedure blev fulgt for alle de organiske solventer for blindprøver. De kvalitative resultater er gengivet i Tabel 2.8-1.
Tabel 2.8-1 Observationer ved undersøgelse af alternative solventer til 95:5 chloroform/butanol-blanding i detektionstrinnet med disulfinblåt som farvekompleks. I Tabel 2.8-1 ses en ubetydelig ekstraktion af farvekompleks fra den vandige fase til den organiske fase. Kun for N-butanol sås en farveændring. Da denne farveændring var ligesom farveændringen observeret for blindprøven, kunne dette solvent ikke benyttes. Generelt blev det også observeret, at faseadskillelse mellem den organiske fase og vandfasen var både hurtigere og mere fuldstændig for 95:5 chloroform/butanol-blanding end for de andre undersøgte solventer. Samme forsøgsdesign blev gennemført for bromthymolblåt. Her blev der observeret en betydelig ekstraktion af gul farvekompleks over i den organiske fase, når der blev anvendt butylacetat, ethylacetat og MIBK. Desværre var det også tilfældet for blindprøverne. Efter ekstraktionen indeholdt N-butanol-fasen samme blå farve både for 50 μg standarden og for blindprøverne. Konklusionen var, at det ikke umiddelbart vil være muligt at erstatte chloroform med andre solventer. Derfor blev chloroform ikke erstattet. Da udrystningen foregik på en magnetomrører, kunne en reduktion af chloroform fra 50 mL i den eksisterende VKI-metode (Østergaard et al, 1999 /9/) til 25 mL i den nye udformning opnås. Yderligere undersøgelser kunne klarlægge, om det ville være muligt at ændre forholdet mellem chloroform og butanol fra 95%-chloroform til en mindre andel. 2.9 MetodeafprøvningDa de enkelte deltrin i metoden nu viste acceptable genfindinger og en acceptabel repeterbarhed, blev en afprøvning af hele metoden gennemført. Til metodeafprøvningen blev der benyttet renset spildevand fra Sjælsø Renseanlæg udtaget den 5. november 2001. Inden analyse blev spildevandet filtreret gennem et 0,45 μm filter. Indledende forsøg med denne metode (ikke medtaget her) viste, at indholdet af kationiske detergenter var mindre end 5 μg pr. 500 mL spildevand. I nedenstående undersøgelser blev spildevandet derfor spiket. Hele metoden blev testet med dobbeltbestemmelser af blindprøve, syntetiske prøver (7,5 μg og 80 μg ADMBAC), naturlige prøver (spildevand spiket med 20 μg og 60 μg ADMBAC) og en kontrolprøve (syntetisk prøve 40 μg ADMBAC). Disse prøver vil svare til dem, der skal medtages i en endelig metodevalidering (U. Lund et al, 1994 /13/).
Tabel 2.9-1 Genfinding af spikede naturlige og syntetiske prøver efter hele analysemetoden. Originaldata findes i Bilag L. A) Farvebuffer var grøn efter udrystning Det fremgår af Tabel 2.9-1, at genfindingen af tilsatte ADMBAC-standarder ikke var acceptabel efter analyse efter hele proceduren. I inddampningstrinnet efter PPL-kolonnen (beskrevet i afsnit 2.5) blev der for de naturlige prøver observeret brune/sorte udfældninger i spidsglasset efter inddampningen. Dette kunne være en medvirkende årsag til den dårlige genfinding. Dette bliver undersøgt i afsnit 2.9.1. For at undersøge, hvorvidt PPL-kolonnens volumen kunne være for lille og dermed bidrage til den lave genfinding, er der i 2.9.2 undersøgt to forskellige kolonnestørrelser. 2.9.1 InddampningsvolumenFor at undersøge, om den dårlige genfinding kunne skyldes udfældninger af kationiske detergenter, der ikke kunne genopløses, blev følgende undersøgelse udført. Både spiket spildevand og syntetiske prøver blev inddampet i spidsglas til henholdsvis tørhed, ca. 100 μL restvolumen og ca. 200 μL restvolumen. Derudover var forsøgsbetingelserne identiske med dem beskrevet i afsnit 2.5 på nær udfarvningsmetoden, hvor princippet i afsnit 2.7 blev benyttet. Resultaterne fra disse undersøgelser er gengivet i Tabel 2.9-2.
Tabel 2.9-2 Genfinding af spike til naturlige og syntetiske prøver ved varierende grad af inddampning inden genopløsning. Forsøgsbetingelserne er beskrevet i afsnit 2.5, dog med detektionsmetoden beskrevet i afsnit 2.7. Originaldata findes i Bilag M og N. Tabel 2.9-2 viser, at genfindingen af spiket til den naturlige prøve blev forbedret fra ca. 40% til ca. 75%, når inddampning ikke var fuldstændig. Genfindingsprocenterne af den syntetiske prøve blev ikke påvirket af den ufuldstændige inddampning. Genfindingen af den syntetiske prøve viste stor spredning og en lidt mindre generel genfinding end forventet. Denne spredning kunne ikke umiddelbart forklares. Der blev i afsnit 2.5 fundet en genfinding på ca. 100% på syntetiske prøver. 2.9.2 Kolonnestørrelse af PPL-kolonnenI trinnet til opkoncentrering blev der benyttet Bond Elut PPL-kolonner (3 mL, 200 mg). I et forsøg på at øge genfindingen blev Bond Elut PPL-kolonner (3 mL, 500 mg) undersøgt over for denne.
Tabel 2.9-3 Genfinding af spike til naturlige og syntetiske prøver ved varierende kolonnestørrelse for PPL-kolonnen. Forsøgsbetingelserne er beskrevet i afsnit 2.4, dog med detektionsmetoden beskrevet i afsnit 2.7. Originaldata findes i Bilag O. Det fremgår af Tabel 2.9-3, at genfindingen af den syntetiske prøve ikke blev påvirket af ændringen i kolonnestørrelsen. For spildevandsprøven sås en klar forbedring fra en gennemsnitlig genfindingsprocent på 60% til 78%. Da blindbiddraget ikke blev påvirket af den øgede kolonnestørrelse, blev det konkluderet, at der skulle skiftes kolonnestørrelse. 2.9.3 DelkonklusionFor naturlige prøver viste resultaterne, at en inddampning til total tørhed i trinnet efter PPL-kolonnen gav dårlig genfinding. Dette blev i 2.9.1 undersøgt, og konklusionen var, at der skulle inddampes, til et restvolumen på 200 μL. Endvidere blev det i 2.9.2 vist, at en PPL-kolonne med 500 mg kolonnemateriale gav bedre genfinding af spike til spildevand end ved 200 mg. 500 mg PPL-kolonne blev derfor benyttet i det efterfølgende. 2.10 Endelig metodeudformningI det sidste forsøg i metodeafprøvningen blev i begge genopløsningstrin i proceduren indført et grundigere skylletrin end tidligere. Først blev henholdsvis 2 mL ethylacetat/4 mL MeOH anbragt i spidsglasset. Bund og sider blev skyllet grundigt ved at trække solventet op i en pasteurepipette 10 gange og mellem hver gang at lade det løbe ned langs siderne på spidsglasset.
Tabel 2.10-1 Genfinding af spike til naturlige og syntetiske prøver. Metoden beskrevet i Bilag A blev benyttet. Resultaterne blev baseret på 4 replikater for de syntetiske prøver og 3 replikater for det spikede spildevand. Originaldata findes i Bilag P. Resultaterne i Tabel 2.10-1viste, at metoden gav samme genfinding for syntetiske (87%) og naturlige prøver (89%). Genfindingen af syntetiske prøver opfylder ikke det ønskede krav på ±5% svarende til kvalitetsklasse 3 i bekendtgørelse 637 /2/. Formentligt vil teknikerens øgede rutine kunne forbedre genfindingen. Hvis det i den efterfølgende metodevalidering viser sig, at metoden generelt giver en lavere genfinding end krævet for kvalitetsklasse 3, kan der korrigeres for genfinding af syntetisk standard. Repeterbarheden for metoden var henholdsvis 2,5% for syntetiske prøver og 6,2% for spike til naturlige prøver. Kravet fra bekendtgørelse 637 /2/ er ±7%, hvilket derfor blev opfyldt. Den foreløbige detektionsgrænse (endelig detektionsgrænse fastlægges i metodevalideringen) var henholdsvis 12 μg/L for syntetiske prøver og 40 μg/L for spike til naturlige prøver. Den foreløbige detektionsgrænse for de syntetiske prøver var meget tæt på kravet på 10 μg/L, mens resultaterne for de naturlige prøver viser et højere resultat. Der er relativ stor usikkerhed på disse resultater, da der kun indgår 4 og 3 replikater i beregningerne. Normalt skal der benyttes mindst 6 replikater i udregningen af en foreløbig detektionsgrænse /13/. Samtidigt var koncentrationerne i prøverne 10 gange den forventede detektionsgrænse. Normalt skal der benyttes prøver med en koncentration 5 gange den forventede detektionsgrænse /13/. Det kan derfor konkluderes, at metoden er klar til en metodevalidering /13/.
|