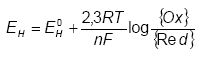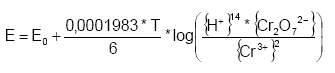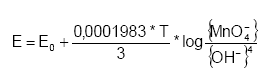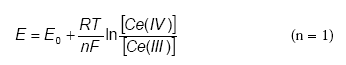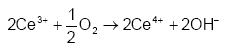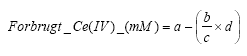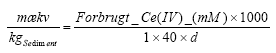|
Miljøprojekt nr. 1024, 2005 Metode til analyse af reducerende stoffer i sedimenterIndholdsfortegnelse2 Reducerede forbindelser i danske sedimenter
4 Lokaliteter - geologi og redoxforhold 5 Udvalgte oxidationsmidler til analyse af reducerende forbindelser i sedimenter
6 Målte og beregnede indhold af reducerende forbindelser i sedimenter
7 Metodebeskrivelse til analyse af reducerende forbindelser i sedimenter ved brug af Ce(SO4)2 ForordDer findes i dag ingen standardiserede analysemetoder til bestemmelse af reducerende forbindelser i sedimenter. På baggrund af rapporten "Principper for beregning af nitratreduktion i jordlagene under rodzonen" (Ernstsen et al., 2001) har der imidlertid vist sig et behov for en metode, der beskriver indholdet af reducerede forbindelser i sedimenter. Formålet med denne rapport der derfor ved gennemgang af relevant litteratur at identificere egnede oxidationsmidler og analysemetoder til bestemmelse af reducerede forbindelser. Dernæst at optimere samt afprøve et antal af disse oxidationsmidler ved forsøg i laboratoriet og sluttelig at udarbejde en hurtig metode til brug for bestemmelse af sedimenters samlede indhold af reducerede forbindelser. Undersøgelsen er gennemført af Danmarks og Grønlands Geologiske Undersøgelse (GEUS) for midler fra Miljøstyrelsen. Desuden har Fyns Amt, Nordjyllands Amt, Ringkøbing Amt og Vejle Amt stillet sedimentprøver samt analyseresultater og geologiske oplysninger til rådighed for projektet. En tak til amterne for deres bidrag til projektet. Sammenfatning og konklusionerIndledende litteraturstudier identificerede kemisk oxidation som den bedst egnede metode ved bestemmelse af indholdet af reducerende forbindelser i sedimenter. Kemisk oxidation har tidligere været anvendt til bestemmelse af chemical oxygen demand (COD) i for eksempel spildevand og til bestemmelse af nitratreduktionskapacitet i sedimenter. Gennem litteraturen fremkom en række mulige oxidationsmidler og efter en yderligere vurdering af fordele og ulemper i forbindelse med brugen af disse oxidationsmidler, blev det besluttet at afprøve i alt tre, der alle forekom egnede for de påtænkte undersøgelser af danske sedimenter. Det drejede sig om kaliumdicromat (K2Cr2O7), kaliumpermanganat (KMnO4) og en cerium-forbindelse med Ce(IV). Disse udvalgte oxidationsmidler blev herefter undersøgt i laboratoriet, hvor der for hver oxidationsmiddel blev udviklet en modificeret og reproducerbar metode, gennem en forsøgsrække hvor bl.a. temperatur, koncentration syre og reaktionstider blev optimeret. Disse modificerede metoder blev afprøvet på sedimentprøver fra forskellige danske lokaliteter, for at undersøge metodernes effektivitet på forskellige geologiske udgangsmaterialer, der ligeledes repræsenterede forskellige redoxforhold. Undersøgelserne viste at alle tre metoder var i stand til at skelne mellem indholdet af reducerende forbindelser i oxiderede og reducerede sedimenter. Således viste alle 3 metoder et væsentligt højere forbrug af oxidationsmidlet i reducerede sedimentprøver end i oxiderede sedimentprøver. Desuden viste det sig muligt at skelne mellem reducerede lerede og sandede sedimenter, idet lerede sedimenter generelt havde et højere indhold af reducerede forbindelser end de sandede lokaliteter. Dog viste sandede sedimenter med et forholdsvis høje indhold af organisk stof et indhold af reducerende forbindelser på sammen niveau eller højere end for typiske reducerede lerholdige sedimenter. En sammenligning af de tre oxidationsmidler viste store forskelle i forbruget af oxidationsmiddel beregnet i iltningsækvivalenter. Således er KMnO4 i neutral opløsning et væsentligt dårligere oxidationsmiddel end K2Cr2O7 og Ce(SO4)2, hvilket betyder at indholdet af reducerede forbindelser målt ved førstnævnte metode er langt mindre end for de to sidstnævnte. Sammenlignes resultaterne fremkommet ved den modificerede KMnO4-metode endvidere med en beregnet potentiel sumkapacitet, baseret på målte indhold af TOC, pyrit og ferrojern, er det tydeligt at KMnO4-metoden ikke kan præstere værdier der af samme størrelsesorden som den beregnede værdi, hvorfor metoden ikke vurderes egnet til analyse af reducerende forbindelser i sedimenter. For de resterende to oxidationsmidler, K2Cr2O7 og Ce(SO4)2 gav den modificerede metode med Ce(SO4)2 generelt højere indhold af reducerende forbindelser i de undersøgte sedimenter end tilsvarende fremkommet ved den modificerede metode med K2Cr2O7. Mængden af reducerende forbindelser bestemt ved brug af K2Cr2O7 udgjorde mindre end den mængde der blev bestemt ved brug af Ce(SO4)2. Indholdet bestemt ved Ce(SO4)2-metoden viste sig ofte lavere end det beregnet ved den potentielle sumkapacitet men ofte i god overensstemmelse med den beregnede reducerede sumkapacitet, når der tages hensyn til den formodede tilgængelighed af reducerende forbindelser i sedimenterne. En beskrivelse af metoden til bestemmelse af reducerede forbindelser under brug af cerium findes i afsnit 7 i nærværende rapport. Summary and conclusionsA review of relevant literature identified a chemical oxidation process as the most appropriate for the measurement of reduced compounds in sediments and a number of oxidizing agents were listed. An examine of these oxidizing agents eliminated potassium-dichromate (K2Cr2O7), potassium-permanganate (KMnO4), and a cerium compound with Ce(IV) as the most useful in the development of a new methods. Each of these three oxidizing agents was studied in lab experiments under different conditions, e.g., temperatures, concentrations of oxidizing agent, concentrations of acid, and reaction times to optimize the analysis methods. Sediment samples from localities within Denmark of different geological and redox properties were analyzed using the three optimized methods. The results obtained for these sediments showed for all three oxidizing agents greater contents of reduced compounds in sediments samples from the reduced zones than for samples from the oxidized geochemical environments. Also the content of reduced compounds was greater in clayey sediments than in sandy sediments of comparable redox status, except for the sandy samples very rich in organic matter which had total amounts of reduced compounds like, or even higher than, than for the reduced clayey sediments. In general, the potassium-permanganate (KMnO4) method gave only low amounts of reduced compounds comparable to the amount that was calculated based on the measured contents of total organic matter, pyrite and ferrous iron. These low contents exclude the potassium-permanganate method as a useful method in this study. The concentrations of reduced compounds obtained by the use of potassium-dichromate (K2Cr2O7) were lower than the amounts obtained by the cerium sulfate Ce(SO4)2 method. The latter method was identified as the one that came up with contents of reduced compound in the sediments close to the total amount calculated on the basis of the estimated available contents of organic matter, pyrite, and ferrous. The total content of reduced compounds measured with the Ce(SO4)2-method was always lower than the calculated total amount based on the total amounts of organic matter, pyrite, and ferrous iron. The method to measure the amount of reduced compounds in sediments by Ce(SO4)2 is described (in Danish) in section 7 in the present report. 1 IndledningMed reference til bl.a. rapporten om principper for beregning af nitratreduktionskapaciteten i jordlagene under rodzonen (Ernstsen et al., 2001), har der vist sig et behov for at udvikle en metode til bestemmelse af reducerende forbindelser i sedimenter. På trods af et meget stort antal forskellige analysemetoder til jord og sedimenter findes der ingen standardiseret metode til bestemmelse af det summerede indhold af reducerende forbindelser i sedimenter. Nogle få danske og udenlandske undersøgelser er gennemført af sedimenters totale nitratreduktionskapacitet (TRC), men generelt for de anvendte metoder er at de medtager langt flere og mere af de reducerende forbindelser end der normalt vil være aktive hvorved der sker en overestimering af sedimenters totale nitratreduktionskapacitet (Ernstsen et al., 2001). Mangel på en enkelt analyse metode til bestemmelse af sedimenters indhold af reducerende forbindelser betyder, at jordlagenes indhold af reducerende forbindelser p.t. ofte baseres på beregninger af differencen mellem indholdet i henholdsvis den reducerede og den oxiderede zone, hvor forskellen i indhold tages som udtryk for den aktuelt mængde, der er tilgængelig for oxidationsprocesser. Formålet med projektet er gennem et studium af eksisterende litteratur at finde egnede metoder til bestemmelse af reducerede forbindelser i jord og sedimenter. På baggrund af de fundne kemiske oxidationsmetoder ønskes der efterfølgende at udvikle en metode til bestemme af sedimenternes samlede indhold af reducerede forbindelser. Resultaterne for det summerede indhold af reducerende forbindelser sammenholdes med fordelingen af det beregnede indhold baseret på fordelingen af TOC, pyrit samt ferrojern. Selv om der forventes at forekomme en sammenhæng mellem sedimenternes indhold af reducerende forbindelser og jordlagenes evne til at reducere nitrat så forsøges der ikke i rapporten opstillet en sådanne sammenhæng. Dette skyldes bl.a. manglende viden om tilgængeligheden af reducerende forbindelser samt en for indeværende ufuldstændig forståelse af specielt den abiotiske nitratreduktionsproces. Analysemetoden er udviklet med henblik på at kunne beskrive variationen i det summerede indhold af reducerende forbindelser, hvor supplerende analyser vil kunne belyse den aktuelle sammensætning af de reducerende forbindelser. 2 Reducerede forbindelser i danske sedimenterEfter aflejringen af de kvartære sedimenter, har der siden sidste istid (gennem mere end 12000 år) kunne foregå en iltning ved tilførsel af iltningsmidler, bl.a. opløst ilt i det nedsivende vand. Den fremad skridende iltning af sedimenterne kan i dag iagttages ved brunlige, gullige og/eller orange farvenuancer. Udbredelsen af den oxiderede zone vil være afhænge af bl.a. det oprindelige indhold af reducerende forbindelser og tilførselen af oxiderende forbindelser. Således vil dybden af den oxiderede zone - alt andet lige - fordobles hvis mængden af nedsivende vand fordobles eller hvis indholdet af reducerende forbindelser halveres (Ernstsen et al., 2001) I områder med ringe nedsivning eller for sedimenter med høje indhold af reducerende forbindelser vil udbredelsen af den oxiderede zone normalt være forholdsvis begrænset. Forbruget af reducerende forbindelser vil ikke nødvendigvis forekomme som en jævnt front, der udbreder sig til stadig dybere jordlag. Således vil der i områder med geologiske vinduer eller i områder med meget heterogene geologiske forhold kunne forekomme flere redox sekvenser med skiftende oxiderede og reducerede forhold. Denne vekslen og gentagelse i redoxforhold afspejler vandets foretrukne strømningsveje. I ældre dele af det glaciale landskab (eksempelvis på bakkeøerne) kan den længere periode efter aflejring have resulteret i dannelse af endog meget dybe oxiderede zoner. Lokale forhold kan ved eksempelvis periglaciale processer (inkl. jordflydning og erosion) have bevirket en gradvis udglatning af landskabet og dermed også en nedslidning og dermed følgende formindskelse af den oxiderede zone. For ældre tertiære aflejringer kan iltningen have foregået på et langt tidligere tidspunkt og forbruget af reducerende forbindelser kan ikke umiddelbart relateres til de nuværende overfladegeologiske forhold. Sedimenternes indhold af reducerende forbindelser kan som nævnt variere meget. Ligeledes kan sammensætningen af de reducerende forbindelser variere fra sted til sted - fra en type til anden. Ofte henregnes organisk stof samt reducerede former af svovl, jern og mangan til gruppen af reducerende forbindelser. Til gruppen af reducerede svovlformer indgår bl.a. pyrit og til gruppen af reduceret jern høre såvel lettilgængelig (ombytteligt) ferrojern samt strukturelt bundet ferrojern i jernholdige lermineraler og andre jernholdige mineraler. Reducerede former af mangan kan ligeledes bidrage. Desuden kan andre reducerende forbindelser som bl.a. methan også lokalt have en stor betydning. Når puljen af reducerende forbindelser gøres op kan bl.a. sammensætning og bindingsforhold være afgørende for i hvor høj grad de enkelte former af reducerende forbindelser bidrager. Når den totale pulje af reducerende forbindelser beregnes for et givent sediment vil denne ofte være forskellig fra den aktuelle pulje af reducerende forbindelser idet ikke hele puljen er reaktiv. Sammensætningen af det oprindelige organiske stof, der blev indlejret i sedimenterne på aflejringstidspunktet, er til tider af en sådanne karakter at det ikke umiddelbart kan anvendes af mikroorganismer ligesom binding til eksempelvis leroverflader nedsætte tilgængeligheden i forbindelse med iltningsprocessen. Når bidraget fra organisk stof skal beregnes, udtrykt i iltningsækvivalenter, kan det derfor være vanskeligt at fastlægge den tilgængelige mængde. Usikkerheden knyttet til beregningen af organisk stofs bidrag knytter sig ikke blot til den tilgængelige mængde men er ligeledes knyttet til i hvor høj grad det organiske stof er reduceret og dermed hvor mange iltninsgækvivalenter bidraget pr. mol forventes at være. Barcelona og Holm (1991) anfører således i deres beregninger for sedimenter fra nogle grundvandsmagasiner at hver mol organisk bundet kulstof bidrager med 2 iltningsækvivalenter idet der drages en sammenligning til phthalsyre For typiske danske sedimenter tyder nærværende undersøgelse på at det indlejrede organiske stof måske bidrager med færre iltningsækvivalenter de her nævnte 2 (Ernstsen et. al, 1998). Sammensætningen af det organiske stof der løbende tilføres fra overfladen under dyrkede arealer adskille sig normalt fra det ældre og indlejrede organiske stof ved en større biotilgængeligt andel. Forekomsten af jern i dets forskellige former vil ligeledes påvirke udnyttelsesgraden og dermed bidraget til puljen af reducerende forbindelser. Ombytteligt ferrojern, der normalt forekommer under reducerende forhold, betragtes normalt som en lettilgængelig fase, hvorimod strukturelt bundet ferrojern i bl.a. lermineraler antagelig repræsenterer en mindre aktiv fase, der normalt ikke kan udnyttes fuldt ud. Således vil det tilbageblevne indhold i oxiderede lerholdige sedimenter være omkring 10 % af det totale jernindhold i ler. Ferrojern bundet i mørke mineraler, som eksempelvis pyrozener og amfiboler, antages at repræsentere meget vanskelig tilgængelig pulje og tænkes således ikke at bidrage i noget væsentlig omfang. Tilgængeligheden af mangan i en ombyttelig fase regnes analogt til ferrojern for en tilgængelig fase mens mineralbundet mangan formodes i langt mindre grad at bidrage til puljen af reducerende forbindelser. Indholdet af mangan i sedimenter er normalt lavere end for jern. 3 Kemiske oxidationsmetoder
3.1 Generel introduktion til redoxkemiI dette afsnit beskrives begreberne oxidationsmiddel, reduktionsmiddel, oxidation, reduktion, oxiderende- og reducerende forbindelser, samt oxidations- og reduktionskapacitet. Et oxidationsmiddel defineres som et stof, der er i stand til at optage elektroner, mens et reduktionsmiddel er et stof der kan afgive elektroner ved reaktion (Bjerrum, 1932). Oxidationsmidler og reduktionsmidler korresponderer til hinanden på lignende måde som syre og baser. Det vil sige at et stof, der reduceres, og har virket oxiderende kaldes et oxidationsmiddel, mens et stof, der oxideres har virket som reduktionsmiddet (eller reduktant) (Bjerrum, 1932). Forløbet kan beskrives som: Ox + ne-→ Red, hvor Ox er oxidationsmidlet, Red er reduktionsmidlet, n er antal og e- er elektroner. Denne ligning kaldes en halvcelle reaktion og skrives for nemheds skyld med den oxiderede forbindelse på venstre side i reaktionsligningen. Da der ikke forekommer frie elektroner kan den ovenstående reaktion kun forløbe, hvis en anden forbindelse stiller elektroner til rådighed for reaktionen, dvs. et reduktionsmiddel skal være tilstede. På denne baggrund kan en halvcelle reaktion ikke eksistere alene. En proces, hvor der foregår både en reduktion og en oxidation, kaldes en redoxproces. Et eksempel på en redoxproces (3) er reaktionen mellem ferrojern (Fe2+) og ilt (O2), der består af to halvreaktioner (1 + 2): Fe2+ + 3H2O →Fe(OH)3 + 3H+ + e- (1) 2H2O →O2 + 4H+ + 4e- (2) 4Fe2+ + O2 + 10H2O →4Fe(OH)3 + 8H+ (3) Processen i ligning (3) kan både benævnes oxidation og reduktion, da begge dele forekommer i ligningen (Schachtschabel et al., 1989). Om reaktionen benævnes som oxidation eller reduktion er derfor afhængig af formålet med reaktionen (Bjerrum, 1932). En forbindelses evne til at oxidere eller reducere er defineret udfra redoxpotentialet EH.
hvor EH0 er redoxpotentialet [V] (i forhold til en standard hydrogen elektrode) under standard betingelser (alle aktiviteter = 1, PH2 = 1 atm, {H+} = 1 M). F er Faradays konstant (96490 C/mol), n er antallet af ombyttede elektroner, R er gaskonstanten (8,314 J/mol*K) og T er temperaturen målt i kelvingrader (°K). Ovenstående ligning kaldes Nernsts ligning og udtrykker den mængde energi, der frigives, når et oxidationsmiddel bliver reduceret i en halvcelle reaktion (Smith, 1995). Måles redoxpotentialet til en positiv værdi er der et overskud af oxiderende forbindelser, mens negative værdier betyder at der er overskud af reducerende forbindelser, jævnfør figur 3.1. Forbindelser med høje positive redoxpotentialer er på denne baggrund gode oxidationsmidler, mens forbindelser med høje negative værdier er gode reduktionsmidler.
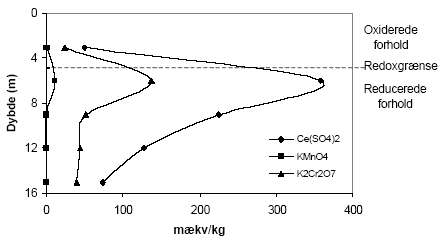
Figur 3.1.Typiske redoxpotentialer (mV) i naturlige systemer ved neutrale pH værdier. Redoxpotentialer i den positive retning (op til ca. +800 mV ) indikerer tilstedeværelse af stærke oxidanter, mens negative værdier indikerer tilstedeværelsen af stærke reduktanter (modificeret efter Sigg, 2000). Oxidationskapacitet defineres som det antal iltningsækvivalenter som et molekyle kan afgive når det virker oxiderende. Et ilt-atom repræsenterer 2 iltningsækvivalenter, mens et optag af et brintatom eller en elektron svarer til 1 iltningsækvivalent. Et reduktionsmiddels reduktionskapacitet er på tilsvarende vis bestemt ved antallet af iltningsækvivalenter som reduktionsmidlet optager under reduktionen (Bjerrum, 1932). Oxidations- eller reduktionskapaciteten for en given forbindelse kan variere med ydre omstændigheder, idet redoxreaktionen generelt er påvirket af pH, temperatur, og mængden af den korresponderede reducerede/oxiderede forbindelse. Disse udefra kommende påvirkningers indflydelse på oxidations- eller reduktionspotentialet kan beregnes gennem Nernsts-ligning (Smith, 1995). Tilgængeligheden af reduktanter (reducerende forbindelser) har ligeledes betydning for hvor hurtigt og effektiv den kemiske oxidationproces kan forløbe. Således vil frie reducerede forbindelser hurtigt reagere i oxidationsprocessen, mens høj ionstyrke eller kompleksdannelse betyder, at det bliver vanskeligere for oxidationsmidlet at komme i kontakt med elektrondonorer (Smith, 1995). Tilstedeværelse af reducerede forbindelser bl.a. ferrojern, der findes bygget ind i og beskyttet i lermineralstrukturen kan således have indflydelse på oxidationsprocessen, idet sandsynligheden for at kollidere med det anvendte oxidationsmiddel bliver formindsket. Sedimenter kan således være vanskelige at oxidere fuldstændigt. 3.2 Redoxkemi i sedimenterDe vigtigste redox-aktive forbindelser i forskellige sedimenter omfatter foruden O2 også nitrat, sulfat, organisk stof, forskellige reducerede former af svovl (inkl. pyrit), jern og mangan. Sammensætningen af disse forbindelser afspejles i sedimentets redoxpotentiale. I figur 3.2. er de vigtigste redoxpotentialer for ovennævnte forbindelser vist. Det ses på figuren at redoxpotentialerne er meget afhængig af pH. Samtidig ses det, at de reducerede forbindelser generelt kun har lave redoxpotentialer (0 til ca. – 300 mV), hvilket betyder der skal anvendes et stærkt oxidationsmiddel, hvis der skal ske en redoxreaktion med disse forholdsvise svage reducerende forbindelser.
Figur 3.2. Redoxpotentialer for nogle af de vigtigste redoxaktive forbindelser i sedimenter for henholdsvis pH 7 (mørke pile) og pH 8 (lyse pile). Redoxpotentialerne er beregnet for aktiviteterne {red} = 1 og {Ox} = 1; og i forbindelse med Fe(II) og Mn(II), er {Mn2+} = 1x10-6 og {Fe2+} = 1x10-6 (Sigg, 2000). 3.3 Metode til bestemmelse af reducerende forbindelser i sedimenterIndholdet af reducerende forbindelser i jord og sedimenter bestemmes i dag ofte ved enkeltanalyser af udvalgte forbindelser, som eksempelvis indholdet af ferrojern, pyrit og organisk materiale (TOC). De enkelte resultater bliver efterfølgende anvendt til at bestemme sedimenternes samlede pulje af reducerende forbindelser og den aktive pulje af de reducerende forbindelser bestemmes ved forskellen mellem indholdet i den reducerede zone og den oxiderede zone (Ernstsen et al., 2001). Der findes for indeværende kun ganske få analysemetoder, der udviklet med henblik på at undersøge den samlede pulje af reducerende forbindelser i sedimenter (Ernstsen, 2001). Den eneste metode, der så vidt vides er afprøvet på danske sedimenter er udviklet af Pedersen (1992) til bestemmelse af den totale nitratreduktionskapacitet (TRC-værdi) i jord og grundvand. Denne metode er baseret på vådkemisk oxidation af reducerende forbindelser med en blanding af kaliumdichromat (K2Cr2O7) og svovlsyre. Metoden indebærer ifølge Pedersen (1992) en total nedbrydning af organiske forbindelser, samt reducerende forbindelser, heriblandt Fe(II), pyrit og Mn(II). Resultaterne af ovennævnte metode for både sandede og lerede sedimenter viste ifølge Pedersen (1992) et sammenhæng imellem de bestemte TRC-værdier og og sedimentprøvernes geokemiske miljøer, således havde reducerede sedimentprøver højere TRC-værdier end oxiderede sedimentprøver. På baggrund af TRC-værdier og totale indhold af carbon (TOC) konkluderede Pedersen (1992) således lerholdige sedimenter hovedsageligt kan tilskrives organisk materiale udfældet på eller adsorberet til lerpartiklerne. De højere TRC-værdier der generelt findes for lerede sedimenter i forhold til sandede sedimenter tilskriver Pedersen (1992) således lerpartiklernes høje specifikke overfladeareal og øgede sorptionsegenskaber. Graversen et al. (1990) vurderede imidlertid, at metoden gav realistiske TRC-værdier for sandede sedimenter med organisk stof, men at metoden for øvrigte sedimenter gav urealistiske høje værdier. I begrænset omfang har kaliumpermanganat (Siegrist et al., 2002, Chambers et al., 2000) og forskellige cerium(IV)-holdige forbindelser (Kammer et al., 2000) været anvendt til bestemmelse af sedimenters indhold af reducerende forbindelser. Ligeledes har persulfat været anvendt ved analyse af sedimenters indhold af reducerende forbindelser - og dermed mulige reduktionskapacitet over for nitrat (Pedersen, 1992). 3.4 Analysemetoder til bestemmelse af reducerende forbindelser i sedimentprøver3.4.1 Oxygen (O2)Ilt (O2) er fra et termodynamisk synspunkt en stærk oxidant i en vandig opløsning: O2 + 4H+ +4e- →2H2O (1,229 V) Reaktionshastigheden af reaktionerne mellem O2 og redoxaktive metaller som Fe(II), og Mn(II) er derimod langsom, da reaktionshastigheden hovedsagelig afhænger af O2's partialtryk i den vandige opløsning, hvilket som udgangspunkt er lavt under normale omstændigheder. Ilt er på denne baggrund ikke anvendelig som oxidationsmiddel, når en hurtig metode ønskes og dette oxidationsmiddel blev derfor ikke yderligere i dette projekt. 3.4.2 Hydrogenperoxid (H2O2)Anvendelse af hydrogenperoxid (H2O2) sker bl.a. ved in situ oxidation af især forurenede grunde med forbindelser efter oliespild, hvor bl.a. napthalen, phenantren, pyren og phenoler er almindeligt forekommende. Hydrogenperoxid har ligeledes forsøgsvis været anvendt ved in situ remediering af chlorerede forbindelser som trichlorethylen og perchlorethylenb (Siegrist et al. 2001; Watts og Smith, 1991; Murphy et al.,1989; Barcelona og Holm, 1991). Undersøgelsen er udført med henblik på at undersøge dels sedimenters forbrug af H2O2 ved in situ oxidation af forurenende organiske forbindelser og dels kinetikken af H2O2 reduktionen. Resultaterne viste at nettoreduktionen af H2O2 er på samme niveau eller svagt overstiger den målte reduktionskapacitet bestemt ved oxidation med kaliumdichromat (K2Cr2O7) i en stærk sur opløsning. Oxidationen af organiske forbindelser under forbrug af H2O2 forløber let i sedimenter ved tilstedeværelsen af jernholdige forbindelser. Hydrogenperoxid danner ved tilstedeværelsen af jern det såkaldte Fenton reagens, der frigiver frie hydroxyl radikaler, som er stærke men ikke specifikt virkende oxidanter, som hurtigt nedbryder mange forskellige organiske forbindelser. Generelt deltager Fe2+/Fe3+ eller andre redoxaktive metaller som henholdsvis elektrondonorer og elektronacceptorer i mange af reaktionerne. Det vil derfor være vanskeligt at anvende H2O2 ved bestemmelse af sedimenters indhold af reducerende forbindelser, da disse redoxaktive metaller vil kunne udgøre en del af de reducerende forbindelser. Herved vil brugen af den metode være begrænset til analyse af organisk stof, som eneste komponent i beregningerne, dersom andre redoxaktive metaller med stor sandsynlighed både vil kunne oxideres og reduceres. Hydrogenperoxid vil under tilsætning af ferrojern oxidere bedst under meget sure forhold (pH 2 – 4). Reagenset bliver ineffektiv under neutrale til basiske forhold og/eller under forhold hvor H2O2 er i stand til at autonedbryde (til H2O og O2) ved kontakt med mineraloverflader, bl.a. karbonater (Siegrist et al., 2001). Brugen af oxidationsmidlet H2O2 under samtidig tilsætning af ferrojern blev ikke undersøgt yderligere dersom ferrojern indgår i puljen af potentielle reducerende forbindelser i sedimenter. 3.4.3 Ozon (O3)Ozon (O3) har ligesom H2O2 været forsøgt anvendt ved in situ remediering og dermed iltning af flere forskellige organiske forureninger. Her knyttes brugen af O3 gerne til teknikker med gennemblæsning af luft i forbindelse med grundvandsforureninger af f.eks. chlorerede forbindelser (Siegrist et al. 2001). Ozon er almindeligt anvendt ved rensning af drikkevand. Da O3 er svagt opløseligt i vand kan denne oxidere forurenende forbindelser direkte eller gennem dannelse af hydroxyl radikaler. Nedbrydningen af O3 er stærkt påvirket af pH-forhold, koncentrationen af O3 og tilstedeværelsen af forbindelser der destruere frie radikaler, primært bikarbonat. I dybereliggende jordlag kan uorganiske forbindelser som OH- og Fe2+ starte eller fremskynde nedbrydningen af O3. For at sikre en effektiv dannelse af radikaler bliver H2O2 ofte tilsat ved anvendelse af O3 (Siegrist et al. 2001). Dersom O3 er meget reaktivt og ustabilt, skal produktionen af O3 helst foregå tæt på brugsstedet hvilket gør det uhensigtsmæssig at anvende i laboratoriet. Dertil kommer, at O3 er giftigt i selv små mængder (Siegrist et al. 2001). På denne baggrund blev det besluttet at brugen af O3 ikke skulle undersøges yderligere som aktiv komponent i den ønskede metode. 3.4.4 Kaliumdichromat (K2Cr2O7)Kaliumdichromat (K2Cr2O7) har tidligere været et alment benyttet oxidatinsmiddel men anvendes nu i langt mindre omfang dersom K2Cr2O7 er giftig og kræftfremkaldende, selv i små doser. Meget forskning vedrørende kemisk oxidation har dog været udført med K2Cr2O7, da denne forbindelse i kemiske sammenhænge er et bedre oxidationsmiddel end for eksempel kaliumpermanganat (KMnO4). Oxidation med dichromat (Cr2O72-) i sur opløsning kan beskrives udfra følgende halvreaktion: Cr2O7- + 14H+ + 6e- ? 2Cr3+ + 7H2O Oxidationspotentialet E (V) kan ved ligevægt beregnes udfra Nernsts ligning:
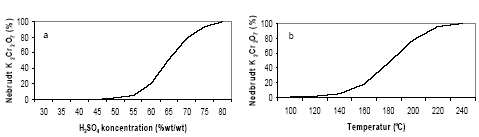
Hvor E0 er standardelektrodepotentialet (V) og T er temperaturen i kelvingrader (°K) (Kolhoff og Steger, 1942). Det fremgår af Nernstes ligning, at ligevægten afhænger af temperatur, pH, samt aktiviteterne af dichromat ionen (Cr2O72-) og chrom-ionen (Cr3+). Temperaturens indflydelse på reaktionen med Cr2O72- er stor og der vil ske en ufuldstændig oxidation af de reducerede forbindelser i prøven ved lave temperaturer, mens der ved høje temperature ske en fuldstændig oxidation. Når K2Cr2O7 udsættes for stærk opvarmning vil der ske en dekomponering af molekylet efter følgende: 4K2Cr2O7 ? 4K2CrO4 + 2Cr2O3 + 3O2 Udfra reaktionsligningen ses det, en opvarmning af K2Cr2O7 i sur opløsning kan producere O2, der forlader opløsningen uden at medvirke til oxidationen af reducerende forbindelser (Pedersen, 1992) og risikoen for dekomponering stiger med stigende temperatur. Således fandt Pedersen (1992), at dekomponering af K2Cr2O7 fandt sted i begrænset omfang ved temperaturer under 140 °C, hvorefter dekomponeringen af K2Cr2O7 stiger eksponentielt til ca. 95% ved 220 C. Ligeledes Stones (1978) har beskrevet denne sammenhæng, figur 3.3. Det vil derfor ikke være hensigtsmæssigt at opvarme K2Cr2O7 til temperaturer højere end 140 °C.
Figur 3.3. Dekomponering af K2Cr2O7 i forbindelse med syrekoncentration og temperatur. a) K2Cr2O7 nedbrydning som funktion af H2SO4 koncentrationen. b) Nedbrydning af K2Cr2O7 i 78,3% H2SO4 som funktion af temperatur (Stones, 1978). Foruden temperaturen har koncentrationen af syre ligeledes stor betydning for dekomponering af K2Cr2O7. Som det fremgår af figur 3.3 stiger dekomponeringen med stigende syrekoncentration. Endvidere er dekomponeringen, som allerede nævnt, også afhængig af Cr2O72- koncentrationen. Således vil koncentrationen af Cr2O72- falde, efterhånden som de reducerede forbindelser oxideres. Hermed bliver dekomponeringen af K2Cr2O7 også afhængig af prøvens indhold af reducerede forbindelser. Da resultatet af analysen således afhænger af forbruget at oxidationsmiddel vil det være hensigtsmæssigt at anvende en temperatur og en syrekoncentration, der begrænser dekomponeringen af Cr2O72- samtidig med at oxidationsforholdene tilgodeses. Under hensyntagen til de nævnte forhold omkring dekomponering findes der i litteraturen flere metoder til oxidation af organisk materiale med K2Cr2O7 som afviger fra hinanden ved forskellige reaktionstider og –temperaturer samt forskelle i syre- og Cr2O72- koncentrationer (Eaton et al., 1995; Pedersen, 1992, Barcelona og Holm, 1992). Således variere de typiske koncentrationer af Cr2O72- mellem 0,05 og 0,2 M og for (svovl)syre mellem på 5– 10 M. Kaliumdichromats evne til at oxidere forskellige organiske forbindelser afhænger meget af de organiske forbindelsers opbygning, idet ikke alle organiske molekyler vil opnå samme grad af oxidation under ens forsøgsbetingelser. Således vil reducerede sukker forbindelser, som eksempelvis glykose, kunne oxideres af K2Cr2O7 uden opvarmning, mens visse heterocycliske forbindelser, som eksempelvis pyridin, kun i ringe grad vil bliver oxideret efter kogning i 2 timer ved 140 °C (Dasgupta og Petersen, 1990). Endvidere er oxidationshastigheden af reducerede forbindelser af første orden med hensyn til både Cr2O72- og den reducerede forbindelse. Dette betyder, at medmindre Cr2O72- er tilstede i et meget stor overskud under en reaktion, vil den mængde reducerede forbindelser, der vil blive oxideret over et givent tidsinterval afhænge af begyndelseskoncentrationen af K2Cr2O7 (Dasgupta og Petersen, 1990). Ved oxidation af sedimenter vil der derfor kunne optræde betydelige forskelle i graden af oxidation bestemt ved især sammensætningen af det organiske materiale og samt begyndelseskoncentrationen af Cr2O72-. 3.4.5 Kaliumpermanganat (KMnO4)Kaliumpermanganat (KMnO4) er et yndet oxidationsmiddel i industrien, idet permanganat ikke bryder carbon skelettet i mange organiske forbindelser under oxidationsprocessen. Når KMnO4 kan anvendes til denne form for oxidation skyldes det at KMnO4 ikke danner frie radikaler. I stedet sker oxidationsprocessen med KMnO4 ved direkte elektron overførsel til reducerede forbindelser. Dette betyder at der skal skabes en direkte kontakt mellem MnO4- og den reducerede forbindelse før reaktionen kan finde sted. Udover de mange anvendelsesformer i industrien, bliver KMnO4 også brugt til in situ remediering af forurende grunde med organiske forbindelser som olie og chlorerede forbindelser (Siegrist et al. 2001). Kaliumpermanganat anvendes desuden også ved måling eksempelvis spildevands kemiske oxygen behov (Chemical Oxgen Demand eller COD) (Eaton et al., 1995, Bigham, 1996, Fresenius et al., 1988). Kaliumpermanganat har potentiale til at oxidere både organiske og uorganiske forbindelser, såsom ferrojern (Fe2+) og reduceret managan (Mn2+) samt naturligt forekommende organisk materiale. Oxidationsprocessen med KMnO4 variere med pH-forholdene og kan beskrives udfra følgende halvcelle reaktioner: MnO4- + 8H + 5e- ? Mn2+ + 4H2O (pH < 3,5) MnO4- + 2H2O + 3e- ? MnO2(s) + 4OH- (pH 3,5 – 12) MnO4- + e- ? MnO42- (pH > 12) Under naturlige forhold kan oxidationspotentialet for KMnO4 beregnes udfra Nernsts ligning:
Hvor E0 er standardelektrodepotentialet (V) og T er temperaturen i kelvingrader (°K) (Kolhoff og Steger, 1942). Det fremgår af Nernsts ligning, at ligevægten afhænger af temperaturen, samt aktiviteterne af MnO4- og den dannede OH-. MnO4--ionen har under naturlige forhold et redoxpotentiale på 0,58 V og et redoxpotentiale på 1,68 V under sure betingelser (Weast,1985), hvilket betyder at KMnO4 er et bedre oxidationsmiddel under sure betingelser end under neutrale forhold Reaktionen mellem MnO4- og reaktanten er meget temperatur afhængige. Forsøg med trichlorethylen viser, at reaktionshastigheden for KMnO4 tredobles når temperaturen øges fra 10 °C til 20 °C (Siegrist et al. 2001). Temperaturafhængigheden skyldes en høj aktiveringsenergi ved redoxreaktioner med KMnO4. Udover temperaturen har især koncentrationen af MnO4- stor betydningen for oxidationen af de reducerede forbindelser. Således viser forsøg at en øget koncentration af MnO4- øger oxidationen af reducerede organiske forbindelser. Årsagen til et øget forbrug af KMnO4 ved 3000 mg/L er ifølge Siegrist et al. (2001) en øget chance for kollision mellem MnO4- og de reducerede forbindelser. Permanganats nedbrydning er relativt upåvirket af pH indenfor intervallet 4 til 8. Ved værdier over 8 har produktionen af OH- en stigende betydning for redoxpotentialet, idet jorden over denne pH-værdi selv indeholder så mange OH- ioner, at reaktionen mellem KMnO4 og reducerede forbindelser begynder at aftage. Derimod vil der ofte i forbindelsen med redoxprocesser dannes brint-ioner (H+), som i ikke bufferede systemer med lav pH, kan medføre en yderligere sænkning af pH, til værdier på 2 til 3 (Siegrist et al., 2001). Dette drastiske fald i pH kan påvirke redoxreaktionerne for en række forbindelser, der findes naturligt i jorden. Permanganat findes i to forskellige former, enten bundet til natrium (NaMnO4) eller til kalium (KMnO4), der er lige effektive som oxidanter. Ifølge Siegrist et al. (2001) er der ikke forskel på reaktiviteten af de 2 former og det kan derfor udfra et kemisk synspunkt være underordnet hvilken af de to forbindelser, der vælges som oxidationsmiddel. Når KMnO4 reagerer med organiske eller reducerede uorganiske forbindelser under natulige forhold, dannes manganoxid (MnO2). Mange naturligt forekommende organiske forbindelser reagerer med MnO4-. Forsøg viser imidlertid at visse dele af det organisk stof ikke bliver fuldstændigt oxideret selv ved meget høje koncentrationer af permanganat eller ved langvarige forsøg, måske fordi det beskyttes af manganoxid. Permanganat kan udover frie ioner også reagere med reducerede forbindelser der er enten kompleksbundet eller adsorberet til den faste fase. Disse reaktioner foregår dog ikke ved sammen hastighed som med frie forbindelser, da bindingen af de reducerede forbindelser resulterer i en stigning i redoxreaktionens aktiveringsenergi og hvorved reaktions hastigheden nedsættes. 3.4.6 Cerium(IV)Anvendelsen af Ce(IV) salte som oxidationsmiddel har været kendt siden 1950'erne, men først i begyndelsen af 1980'erne blev Ce(SO4)2 forsøgsvis anvendt til COD målinger af spildevand (Golterman og Wisselo, 1981). Efterfølgende har Ce(SO4)2 forsøgsvis været anvendt til bestemmelse af nitratreduktionskapaciteten i sedimenter og grundvand (Kammer et al., 2000). Begge metoder er således udviklet som alternativ til de traditionelle metoder, hvor K2Cr2O7 og KMnO4 anvendes som oxidationsmiddel. Metoderne har dog aldrig fundet større anvendelse og der findes nuværende tidspunkt kun få resultater der kan dokumentere Ce(IV) egnethed som oxidationsmiddel i sedimenter og grundvand. Meget lidt vides derfor generelt om reaktioner mellem Ce(IV) og organiske og uorganiske forbindelser i jord og sediment. Opmærksomheden er i stedet vendt mod mere simple systemer, hvor Ce(IV) er anvendt til oxidationsmiddel. I kemiske sammenhænge er Ce(IV) mest kendt i forbindelse med redoxtitrering af Fe(II), hvor potentiometrisk (elektrokemisk) titrering med Ce(IV) er en standardiseret metode til bestemmelse af Fe(II) indholdet i forskellige væsker. Denne titrering involverer en elektron overførsel mellem de to redox aktive forbindelser som vist ved følgende ligning:
Reaktionen er reversibel udfra et termodynamisk synspunkt, men under normale forhold er Fe(III) ikke i stand til at omdanne Ce(III) til Ce(IV), da denne reaktion har meget høj aktiveringsenergi (Bott, 2000). Oxidationspotentialet af Ce(IV)/Ce(III) er 1,61 V (relativt til standard hydrogen elektroden) under standard betingelser. Cerium(IV) forbindelser er således en stærkere oxidant end f.eks. Au(III) og kan dermed reduceres selv af svage reducerende forbindelser, f.eks. Fe(II) salte i sur opløsning eller af H2O2 (Kolthoff og Steger, 1942). På figur 3.4 ses, et eksempel på en redoxtritrering af Ce(SO4)2 med Fe2+.
Figur 3.4. Redoxtitrering af Fe(II) med Ce(IV) (efter Kolthoff og Steger, 1942). I forbindelse med bestemmelse af jorders indhold af reducerende forbindelser er det nødvendigt at tilsætte et overskud af Cerium(IV)sulfat til jordprøverne for at sikre en høj oxidation af de reducerede forbindelser. Under normale omstændigheder skal dette overskud være mindst muligt, så den overskydende mængder af Ce(SO4)2 er så begrænset som muligt. Dette er dog ikke tilfældet for Ce(IV), idet Ce(III) dannet under oxidationen, sænker redoxpotentialet i henhold til ligningen:
hvorved overskuddet af Cerium(IV) ikke må blive for småt. Det bør derfor tilstræbes, at anvende koncentrationer af oxidationsmidlet, der er væsentligt højere end jordens indhold af reducerende forbindelser. Anvendelsen af Ce(SO4)2 til COD-bestemmelse er ifølge Golterman og Wisselo (1981), at Ce(IV) har et højere oxidationspotentiale end K2Cr2O7 og KMnO4, hvorved Ce(SO4)2 er mere termodynamisk egnet til oxidationsmiddel. Værdierne vist i tabel 3.1 antyder ligeledes at oxidationspotentialet af både Ce(IV) og K2Cr2O7 stiger med stigende syrekoncentration, hvilket bl.a. kan forklares ved den høje aktiveringsenergi der karakteriserer begge oxidationsmidler. Den høje aktiveringsenergi bevirker således at der for at sikre en total oxidation af specielt organiske forbindelser i sedimenter tilsættes en katalysator og/eller at reaktioner foregår under høj varme. I følge Golterman og Wisselo (1981) kan Ce(SO4)2 anvendes til oxidation af de fleste uorganiske forbindelser ved stuetemperatur under brug af en passende katalysator, for eksempelvis osmiumtetraoxid (OsO4) hvorimod organiske forbindelser ofte kræver både varme og en passende katalysator. Tabel 3.1. Oxidationspotentiale (V) i 1 M Ce(IV) i forskellige uorganiske syrer målt i forhold til en normal hydrogen elektrode. De viste syrer er perchlorsyre (HClO4), salpetersyre (HNO3) svovlsyre (H2SO4) og saltsyre (HCl) Til sammenligning er oxidationspotentialet af K2Cr2O7 vist i parentes hvor data foreligger (data fra Kolthoff og Belcher, 1957).
Overstiger koncentrationen af Ce(SO4)2 i opløsningen ca. > 0,1 M danner Ce(SO4)2 komplekser med svovlsyre, hvorved opløsningen bliver dyb rød. Disse komplekser er forholdsvis stabile og reagerer kun langsomt med reducerede forbindelser, og det er derfor ønskværdigt at anvende lavere koncentrationer af Ce(SO4)2. Komplekserne kan ligeledes dannes, hvis opløsningen indeholder høje koncentrationer af H2SO4. Tilstedeværelsen af katalysator eller anvendelse af opvarmning er derfor påkrævet når en stærk opløsning af Ce(SO4)2 skal reagere i opløsninger, der indeholder høje koncentrationer af H2SO4 (Golterman og Wisselo, 1981). Resultater af Sultan og Walmsley (1997), viser imidlertid, at reaktionshastighederne for reaktion imellem Ce(IV) og nogle organiske syre som eksempelvis myresyre (HCOOH) og eddikesyre (CH3COOH) nedsættes betydeligt ved stigende koncentrationer af H2SO4, som følge af kompleksdannelse mellem Ce(IV) og sulfat. Ved anvendelse af Ce(SO4)2 som oxidationsmiddel i sedimenter er det vigtigt, at tage højde for et eventuelt indhold af kalk (CaCO3) i sedimenterne, da atmosfærisk ilt (O2) kan interferere på forbruget af Ce(SO4)2. Denne interferens skyldes, at Ce(III) dannet under oxidationen af reducerende forbindelser i basiske opløsninger kan oxideres kvantitativt af opløst O2 til Ce(IV), udfra reaktionen:
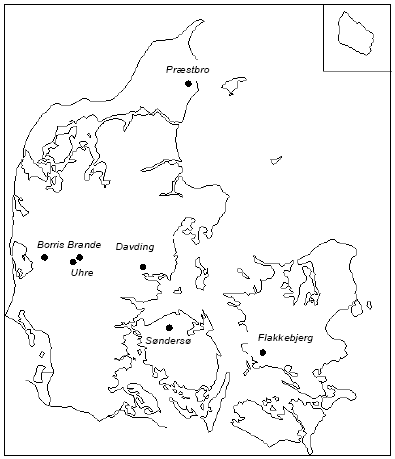
og derved regenerere oxidationsmidlet, hvormed der opstår en unøjagtig bestemmelse af jordprøvens indhold af reducerende forbindelser (Golterman og Wisselo, 1981). På denne baggrund er det nødvendigt at tilsætte syre til sedimentprøverne, så prøverne opnår en pH-værdi under 7. 3.5 Valg af mulige oxidationsmidler og oxidationsmetodeMed udgangspunkt i ovenstående gennemgang af mulige kemiske oxidationsmidler blev det besluttet at afprøve brugen af K2Cr2O7, KMnO4 og Ce(SO4)2 i udviklingen af e analysemetode. Ved brugen af K2Cr2O7 var det i høj grad muligt at bygge videre på allerede eksisterende metodebeskrivelser, mens der for KMnO4 og Ce(SO4)2 kun forelå få og ufuldstændige undersøgelsesresultater og for disse to oxidationsmidler har det været nødvendigt, at gennemføre betydelige indledende undersøgelser af mere grundlæggende karakter. 4 Lokaliteter - geologi og redoxforhold4.1 Sedimentprøver anvendt i forbindelse metodeudviklingI forbindelse med udviklingen af en egnet metode til analyse af reducerende forbindelser i sedimenter er der anvendt prøver som venligst er stillet til rådighed af Nordjyllands Amt, Ringkøbing Amt, Vejle Amt og Fyns Amt og Danmarks og Grønlands Geologiske Undersøgelse (GEUS). Desuden har de nævnte institutioner stillet geologiske oplysninger og analysedata til rådig for projektet. Placeringen af de udvalgte lokaliteter fremgår af figur 4.1. I tabel 4.1 findes en oversigt over samtlige lokaliteter med oplysninger om bl.a. DGU arkivnummer, en kort beskrivelse af geologiske forhold på lokaliteterne samt oplysninger om fordelingen af oxiderede og reducerede lag.
Figur 4.1. Sedimentprøver fra de her nævnte lokaliteter er anvendt i undersøgelsen. Tabel 4.1. Oplysninger om de udvalgte boringer med tilhørende DGU arkivnumre samt en kort beskrivelse af de geologiske forhold med fordelingen af oxiderede og reducerede lag.
5 Udvalgte oxidationsmidler til analyse af reducerende forbindelser i sedimenter
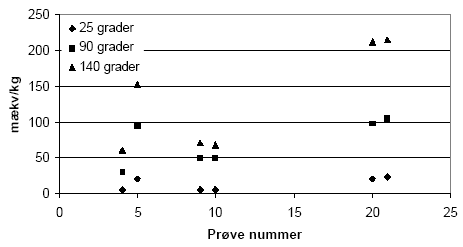
5.1 Oxidationsforsøg med kaliumdichromat (K2Cr2O7) – metode og resultater5.1.1 Optimering af forsøgsbetingelserDen i dette projekt udviklede metode til bestemmelse af reducerende forbindelser i jord- og sedimentprøver under anvendelse af oxidationsmidlet K2Cr2O7 er baseret på henholdsvis metodebeskrivelse til bestemmelse af nitratreduktionskapacitet (Pedersen, 1992) og en standardiseret metode til bestemmelse af Chemical Oxygen Demand (COD) publiceret i Standard Methods for Examination of Water and Wastewaters (APHA et. al., 1995). 5.1.1.1 Målemetode, potentiometrisk titrering eller spektrofotometrisk målingBlandt de mange forskellige analysemetoder, der findes for anvendelse af K2Cr2O7, blev der gennemført supplerende undersøgelser med dels potentiometrisk titrering af K2Cr2O7 med Fe(II) dels kolorimetrisk bestemmelse af K2Cr2O7 på spektrofotometer under brug af 1,5 diphenylcarbazid. Potentiometrisk titrering af K2Cr2O7 med Fe(II) forbindelser er den mest almindelige metode til bestemmelse af overskydende K2Cr2O7 efter reaktion. Til nærværende titreringsundersøgelse blev der anvendt en opløsning med 10 mM FeSO4 opløst i 5% H2SO4. Forsøgene blev søgt udført som en dynamisk-endepunkts-titreringer med endepunkt på 900 mV, som indikation på omslagpunktet fra oxideret til reduceret. Vanskeligheder i forbindelse med bestemmelse af koncentrationerne af K2Cr2O7 i de anvendte standardopløsninger bevirkede at denne metode forekom mindre egnet og derfor ikke søgt yderligere undersøgt. En anden meget benyttet metode til bestemmelse af K2Cr2O7 knytter sig til kolorimetrisk bestemmelse på spektrofotometer, efter reaktion mellem K2Cr2O7 og farvereagenset 1,5-diphenylcarbazid. Ved reaktionen dannes en violet farve der er kraftigt absorberende i monokromatisk lys ved 540 nm. Forsøg med koncentrationsbestemmelse af K2Cr2O7-opløsninger ved denne målemetode fundet mere velegnet i det målingen dog forudsatte at prøven havde en passende lav pH før tilsætning af 1,5 diphenylcarbazid opløst i acetone, da farvereagenset ellers ikke gav den rette farveintensitet. 5.1.1.2 ForsøgstemperaturLitteraturgennemgangen i afsnit 3.4.4 viste, at virkningen af K2Cr2O7 er meget afhængig af den anvendte temperatur i forbindelse med oxidationsprocessen. Der blev gennemført forsøg over K2Cr2O7's reaktivitet ved henholdsvis stuetemperatur (22 °C), 90 °C og 140 °C, der er den højeste temperatur der bør anvendes under hensyntagen til K2Cr2O7's dekomponering. Resultaterne af disse forsøg er vist i figur 5.1. Forsøg ved stuetemperatur med K2Cr2O7 gav kun lave indhold af reducerede forbindelser. Ved at øge reaktionstemperaturen til 90 °C, ved opvarmning i vandbad, for at overkomme K2Cr2O7's høje aktiveringsenergi måltes betydelig højere værdier for indholdet af reducerende forbindelser i forhold til resultaterne af forsøg ved stuetemperatur. Endelig blev forsøgstemperaturen hævet til 140 °C og forsøget skiftede fra batchforsøg i centrifugerør til forsøg med opslæmninger i destruktionsrør, der blev opvarmet i en varmeblok fremstillet af aluminium. Denne temperatur resulterede i højere indhold af reducerende forbindelser. Temperaturen på 140 °C blev derfor anvendt ved de efterfølgende forsøg og viste sig at være i god overensstemmelse med de anbefalinger om temperatur, der er beskrevet af bl.a. Pedersen (1992) og Eaton et al. (1995).
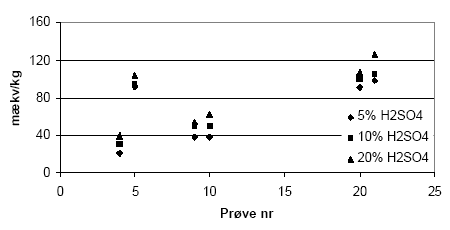
Figur 5.1. Indhold af reducerende forbindelser (beregnet som mækv/kg) i jordprøver behandlet ved forskellige temperaturer, henholdsvis stuetemperatur (ca. 25 °C), 90 °C og 140 °C. I forsøget er anvendt 10 mM K2Cr2O7 i 10 vol-% H2SO4. Prøverne, der er lufttørre (< 2mm) stammer fra Søndersø, Fyns Amt. 5.1.1.3 Koncentration af syreForuden temperaturen spiller syrekoncentrationen i opslemningen en stor betydning for reaktiviteten af K2Cr2O7 (se afsnit 3.4.4) og derfor viste forsøg med tilsætning af 10 mM K2Cr2O7 uden tilsætning af syre som ventet ikke nogen nævneværdig reaktion mellem K2Cr2O7 og sedimenterne. Effekten af forskellige syrekoncentrationer for forbruget af 0,01 M K2Cr2O7 blev undersøgt ved kogning af sedimentprøver i destruktionsrør ved den tidligere valgte temperatur på 140 °C. Resultaterne af forsøget med forskellige koncentrationer af syre for oxidationsreaktionen af reducerede forbindelser fremgår af figur 5.2. For de undersøgte sedimentprøver medførte tilsætning af stigende koncentrationer - fra 5 til 20 vol-% - H2SO4 en stigende reaktivitet og stigende forbrug af K2Cr2O7. På baggrund af denne forsøgsrække blev det besluttet at anvende en syrekoncentration på 15 vol-% H2SO4 ved de følgende forsøg.
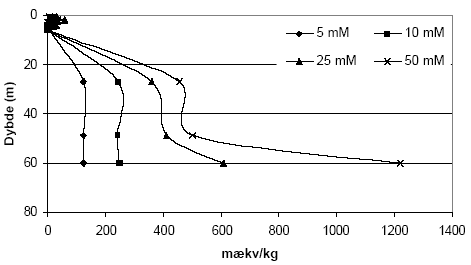
Figur 5.2. Indhold af reducerende forbindelser (beregnet som mækv/kg) i udvalgte jordprøver (lufttør < 2 mm) fra Søndersø (B1, B3 og B5), Fyns Amt, beregnet ved forbruget af 10 mM K2Cr2O7 efter tilsætning af henholdsvis 5, 10 og 20 vol-% koncentreret H2SO4. Syren er tilsat ved forsøgets begyndelse og forsøgstemperaturen er 140 °C. 5.1.1.4 Koncentration af K2Cr2O7Ligeledes med henblik på at optimere analysemetoden blev betydningen af K2Cr2O7 koncentrationen undersøgt. Der blev således gennemført undersøgelser med sedimentprøver fra Søndersø, Fyns Amt under brug af to koncentrationer, henholdsvis 10 mM og 20 mM, K2Cr2O7. Som det fremgår af figur 5.3 steg forbruget af K2Cr2O7 når koncentrationen blev fordoblet fra 10 mM til 20mM. Supplerende analyser af andre sedimentprøver (data ikke vist) synes at bekræfte at en koncentration på 20 mM K2Cr2O7 må imødekomme det forbrug der forventes for langt den overvejende del af typiske danske sedimenter. For sedimenter med meget høje indhold af organisk materiale (svarende til 4,3 % C) viste at en koncentration på 25 mM K2Cr2O7 sig utilstrækkeligt og ved analyse af denne type sedimenter vil metoden derfor kræve yderligere tilpasning.
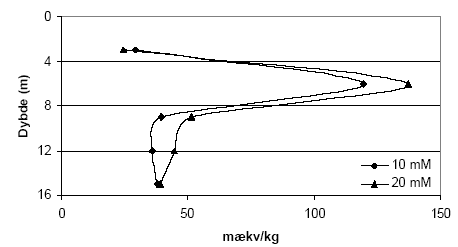
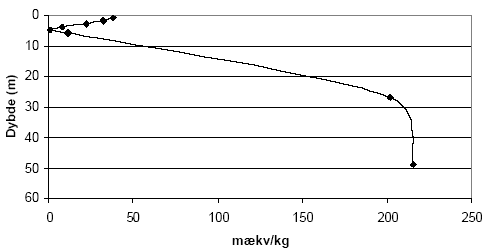
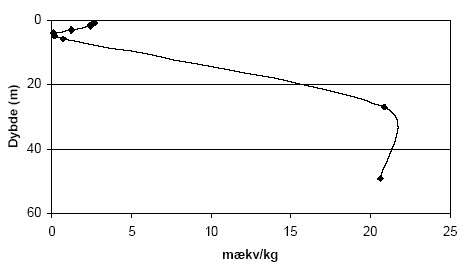
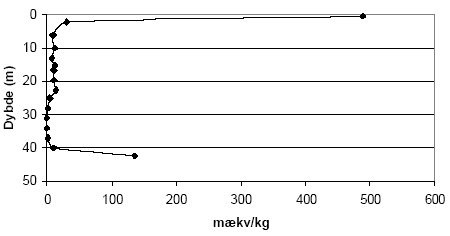
Figur 5.3. Indhold af reducerende forbindelser (beregnet som mækv/kg) i sedimentprøver (lufttøre, < 2mm) fra Søndersø (B1), Fyns Amt ved brug af henholdsvis 10 mM og 20 mM K2Cr2O7 efter opvarmning til 140 °C i 1 time. 5.1.1.5 Kalkholdige prøverAnalysemetoden blev i første omgang optimeret med henblik på analyse af kalkfrie sedimentprøver, men blev efterfølgende udviklet til ligeledes at omfatte analyse af kalkholdige prøver. Kogningen med K2Cr2O7 i destruktionsrør med tætsluttede lågene som bl.a. anbefalet af Pedersen (1992) viste sig således ikke anvendelig for kalkholdige prøver, idet kalken under kogningen med den syreholdige K2Cr2O7 afstedkommer en kraftig udvikling af kuldioxid (CO2). I forbindelse med kalkholdige prøver blev det derfor valgt i stedet at koge prøverne med løst pålagte låg hvorved den dannede CO2 kan undslippe under kogningen. Denne forsøgsopstilling medførte imidlertid tab af vand under kogningen ved 140 °C, som der blev taget hensyn til ved de efterfølgende målinger og beregninger. 5.1.2 Modificeret metode ved analyse med K2Cr2O7Resultaterne af den indledende metode optimering med K2Cr2O7 som oxidant, viser at forbruget af K2Cr2O7 er stærkt temperatur, koncentrations- og syreafhængig. Endvidere viste resultaterne, at der skal tages hensyn til sedimenternes indhold af kalk i forbindelse med analyseproceduren. Metode til analyse af kalk-frie sedimentprøver (bruser ikke ved tilsætning af fortyndet - ca. 10 % - saltsyre) I et pyrexglas (diameter, længde: 26, 200 mm) med skruelåg med teflonpakning indeholdende 3 kogekugler af glas afvejes 0,50 g jord (tørret ved 105 °C, < 2mm). Der tilsættes med dispenser 5 ml 20 mM K2Cr2O7 i 15 vol-% H2SO4 og prøven rystets forsigtigt før end låget skrues fast. Pyrexglas med indhold anbringes i en vameblok (af aluminium) og prøven varmes i 1 time ved 140 °C. Metode til analyse af kalkholdige sedimentprøver (bruser ved tilsætning af fortyndet - ca. 10 % - saltsyre) I et pyrexglas (diameter, længde: 26, 200 mm) til skruelåg med teflonpakning afvejes 0,50 g sediment (tørret ved 105 °C, < 2mm). Der tilsættes dråbevis med dispenser 5 ml 20 mM K2Cr2O7 i 15 vol-% H2SO4 og prøven rystets forsigtigt efter hver tilsætning. Herefter tilsættes 3 glaskugler og låget anbringes løst påsat på pyrexglasset. Opslemningen henstår herefter i 1 time før pyrexglas med indhold anbringes i en vameblok (af aluminium) og opvarmes i 1 time ved 140 °C. Når prøven atter er afkølet til stuetemperatur efterfyldes til oprindeligt vægt for pyrexglas med indhold for derved at kompensere for det vandtab, der måtte være sket under kogningen. Målemetode - tilberedning af kalibreringskurve Indholdet af K2Cr2O7 i prøverne efter opvarmning blev bestemt spektrofotometrisk. Til dette formål er det nødvendigt at fremstille en kalibreringskurve. Kaliberingskurven fremstilles i koncentrationsintervallet 10-6 M til 1,25 10-5 M K2Cr2O7 på baggrund af stamopløsningen på 20 mM K2Cr2O7 i 15 vol-% H2SO4. Målemetode - tilberedning af prøve Fra supernantanten overføres 1ml til en 100 ml målekolbe der fyldes med demineraliseret vand (milliQ gradient, Millipore) vand til mærket. Herfra overføres 1 ml fortyndet prøve med pipette til en 25 ml målekolbe der efterfyldes med demineraliseret vand samt 1 ml koncentreret H2SO4. Til denne prøve tilsættes 0,5 ml 1,5-diphenylcarbazid opløsning (500 mg 1,5-diphenylcarbazid i 100 ml acetone), hvorefter prøven rystes og henstår i 10 minutter for fuld farve udvikling. Prøvens absorbans måles på spectrofotometer ved 540 nm. Som blindprøve anvendes demineraliseret vand (milliQ gradient, Millipore) med svovlsyre. Yderligere informationer om målemetoden kan findes hos APHA et. al. (1995). 5.1.3 Den modificerede metode anvendt på udvalgte sedimenterForbruget af K2Cr2O7 for sedimentprøver fra Præstbro, Nordjyllands Amt, fremgår af figur 5.5. Forbrug af K2Cr2O7 er højt i prøven fra 0,45 meters dybde, hvorefter forbruget aftager markant og holder sig på dette lave niveau indtil prøven fra 42 meters dybde, hvor forbruget af K2Cr2O7 atter stiger.
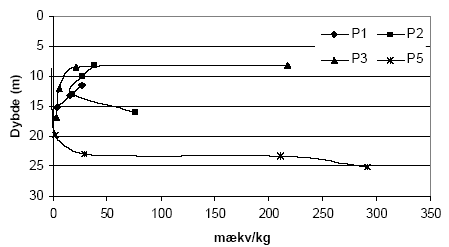
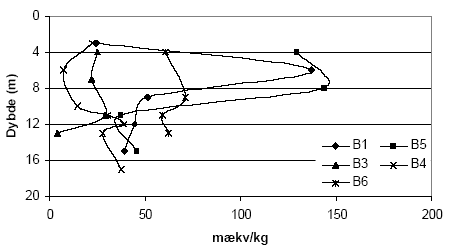
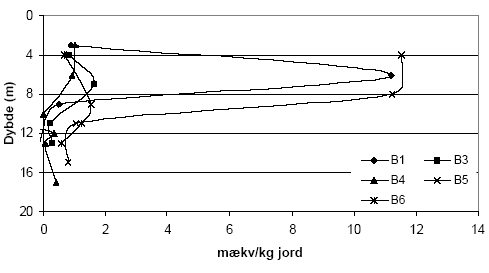
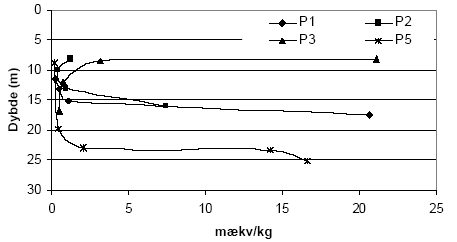
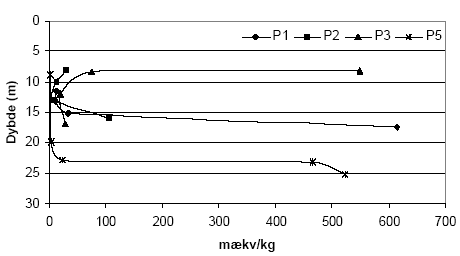
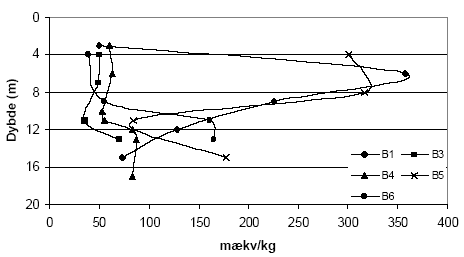
Figur 5.4. Forbrug af K2Cr2O7 (beregnet som mækv/kg) for sedimentprøver (tørret ved 105 °C, < 2 mm) fra Præstbro, Nordjyllands Amt. Koncentrationen af K2Cr2O7 er 20 mM og prøverne er opvarmet i 1 time ved 140 °C. Forbruget af K2Cr2O7 for boringerne P1, P2, P3 og P5, Ringkøbing Amt, fremgår af figur 5.5. Det fremgår af figuren, at der findes betydelige forskelle i forbruget af K2Cr2O7 for de undersøgte sedimenter. Således sker der et betydeligt forbrug af K2Cr2O7 på lokaliteterne P2, P3 og P5 sammenholdt med sedimenter fra P1, hvor forbruget af K2Cr2O7 på intet tidspunkt overstiger 50 mækv/kg. Ved P5 sker der en stigning i forbruget af K2Cr2O7 i dybderne 23 til 25 meter, med et maximalt forbrug af K2Cr2O7 i dybden 25 m på 290 mækv/kg jord. I P3 ses det et modsatrettet forløb af kurven, idet det maksimale forbrug af K2Cr2O7 findes i 8,25 m, hvorefter forbruget falder brat i resten af profilet. Det høje forbrug af K2Cr2O7 i disse prøver var sammenfaldende med høje indhold af organisk materiale. Ved P2, ses et fald i for bruget af K2Cr2O7 med til dybden 13 m, hvorefter forbruget af K2Cr2O7 atter stiger. Forbruget af K2Cr2O7 i 5 boreprofiler fra Søndersø, Fyns Amt, fremgår af figur 5.7. På figuren ses det, at der optræder betydelige forskelle mellem de enkelte lokaliteter. Således ses der et højt forbrug af K2Cr2O7 i boringerne B1 og B5 i henholdsvis 4 til 8 meters dybde, med maximalt forbrug af K2Cr2O7 på 135 til 145 mækv/kg, hvorefter forbruget aftager markant på begge lokaliteter. Forbruget af K2Cr2O7 lokaliteterne B3, B4 og B6 var under 100 mækv/kg jord, om end forbruget for B6 var højere end for både B3 og B4.
Figur 5.5. Forbrug af K2Cr2O7 (beregnet som mækv/kg) for sedimentprøver (tørret ved 105 °C, < 2mm) fra Borris (P1 og P2), Brande (P3) og Uhre (P5), Ringkøbing Amt. Koncentrationen af K2Cr2O7 er 20 mM og prøverne er opvarmet i 1 time ved 140 °C.
Figur 5.6. Forbrug af K2Cr2O7 (beregnet som mækv/kg) for sedimentprøver (tørret ved 105 °C, < 2mm) fra Davding, Vejle Amt. Koncentrationen af K2Cr2O7 er 20 mM og prøverne er opvarmet i 1 time ved 140 °C.
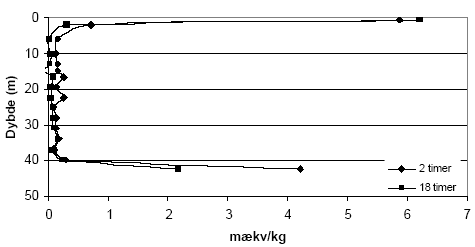
Figur 5.7. Forbrug af K2Cr2O7 (beregnet som mækv/kg) for sedimentprøver (tørret ved 105 °C, < 2mm) fra Søndersø (B1, B3-B6), Fyns Amt. Koncentrationen af K2Cr2O7 er 20 mM og prøverne er opvarmet i 1 time ved 140 °C. 5.2 Oxidationsforsøg med kaliumpermanganat (KMnO4) – metode og resultater5.2.1 Optimering af forsøgsbetingelserPermanganat findes som beskrevet i afsnit 3.4.5 ofte anvendelse som kemisk oxidationsmiddel i analytisk kemi. Permanganaten findes bundet til henholdsvis kalium og natrium og her er oxidationsforsøgene gennemført med kaliumpermanganat (KMnO4). 5.2.1.1 MålemetodeKaliumpermanganat (KMnO4) har en markant violet farve og det blev derfor undersøgt om det var muligt at bestemme de ved en kolorimetriske måling på spektrofotometer. Et scan af opløsningen viste at KMnO4 absorberer i monokromatisk lys ved bølgelængderne 525 nm og 545 nm. Da den største absorbans forekom ved 525 nm blev denne bølgelængde valgt til måling af koncentrationen af KMnO4 i de følgende undersøgelser. 5.2.1.2 Sediment:væske forholdForsøg med forskellige mængder af sediment, 1,00 g eller 2,00 g, viste ingen væsentlige forskelle i det beregnede forbrug af KMnO4 pr. kg sediment ved tilsætning af 25 ml 0,42 mM KMnO4 (data ikke vist). For at sikre den største repræsentativitet af sedimentprøven blev det besluttet at anvende den største prøvemængde, svarende til 2,00 g ved efterfølgende forsøg. 5.2.1.3 Koncentration af syreLitteraturgennemgangen i afsnit 3.4.5 viste, at KMnO4's evner som oxidationsmiddel er meget afhængig af pH i opløsningen. På den baggrund blev anvendelse af syre i forbindelse med oxidation af KMnO4 undersøgt. Der blev gennemført forsøg over KMnO4s reaktivitet ved henholdsvis 0 vol-% og 10 vol-% H2SO4. I forsøgene med 10 vol-% H2SO4 blev der observeret en tydelig brunfarvning af væsken når den koncentrerede syre blev blandet med KMnO4. Denne farvning stammer fra naturlige organiske forbindelser, bl.a. fulvosyre og huminsyre, der findes i sedimenterne og som opløses ved tilsætning af syre. Dette er i overensstemmelse med at der ikke blev konstateret en brunfarvning i forsøgene, der blev gennemført uden tilsætning af H2SO4. På denne baggrund blev det besluttet at anvende KMnO4 som oxidationsmiddel uden tilsætning af syre, selvom redoxpotentialet for reaktionen ved pH-forhold mellem 4 og12 er væsentligt lavere (0,58 V) end redoxpotentialet for KMnO4 i sur opløsning (1,68 V). 5.2.1.4 Koncentration af KMnO4Ved undersøgelser af lerholdige sedimentprøver indsamlet ved Flakkebjerg, Vestsjælland Amt, viste en koncentration på 0,42 mM KMnO4 ikke at være var tilstrækkelig høj for en fuldstændig oxidation med KMnO4 ved den anvendte prøvemængde (data ikke vist). På baggrund af dette forsøg blev det besluttet at øge koncentrationen først til 1,7 mM KMnO4 og efterfølgende til 5 mM KMnO4, da den tilførte mængde af KMnO4 ligeledes blev brugt op, ved tilsætning af 25 ml 1,7 mM KMnO4. 5.2.1.5 ReaktionstidBetydningen af reaktionstiden for forbruget af KMnO4 blev undersøgt for sandede sedimenter fra Præstbro, Nordjyllands Amt og lerholdige sedimenter fra Flakkebjerg, Vestsjællands Amt. Resultaterne over forbruget af KMnO4 efter henholdsvis 2 og 18 timer for de sandede sedimenter fremgår af figur 5.8. Forbruget af KMnO4 viser samme forløb for begge reaktionstider, men forbruget af KMnO4 efter 18 timers reaktionstid er generelt højere end for det tilsvarende forsøg med en reaktionstid på kun 2 timer. Forskellen er mest markant for sedimentprøven udtaget i 42,5 meters dybde, hvor forbruget af KMnO4 er mere end fordoblet efter en ændring i reaktionstid fra 2 til 18 timer.
Figur 5.8. Forbruget af KMnO4 (beregnet som mækv/kg) ved batchforsøg med sedimentprøver (tørret ved 105 °C, < 2 mm) fra Flakkebjerg, Vestsjællands Amt, ved reaktionstider på henholdsvis 18 og 24 timer. Den anvendte koncentration af KMnO4 var 5 mM. Forbruget af KMnO4 i hovedparten af de sandende prøverne fra Præstbro, Nordjyllands Amt, varierede ellem 0 og 1 mækv/kg og kun sedimentprøven fra 0,45 meter og 42,5 meter dybde viste et større forbrug af KMnO4, figur 5.9.
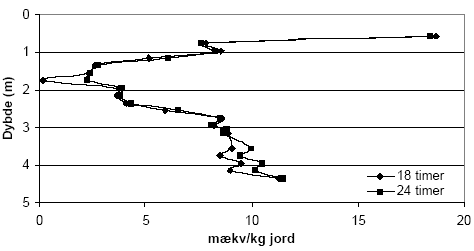
Figur 5.9. Forbrug af KMnO4 (beregnet som mækv/kg) ved batchforsøg med sedimentprøver (tøret ved 105 °C, < 2mm) fra Præstbro, Nordjyllands Amt, ved reaktionstider på henholdsvis 2 og 18 timer. Den anvendte koncentration af KMnO4 var 1,7 mM. Resultaterne fra de lerholdige sedimenter fra Flakkebjerg, Vestsjælland Amt, fremgår af figur 5.10 Grundet et generelt større forbrug af KMnO4 for prøver fra denne lokalitet blev det besluttet at undersøge reaktionstider på henholdsvis 18 og 24 timer. Det ses af figuren, at forbruget af KMnO4 var stort set ens for reaktionstider på 18 og 24 timer for sedimentprøver udtaget ned til dybde på 3,15 meter. Herefter ses der et svagt forøget forbrug af KMnO4 ved 24 timers sammenholdt med en reaktionstid på 18 timer. Det øgede forbrug af KMnO4 i sedimentprøverne under dybden 3,15 meter var sammenfaldende med gradvist mere reducerede forhold.
Figur 5.10. Forbruget af KMnO4 (beregnet som mækv/kg) ved batchforsøg med sedimentprøver (tørret ved 105 °C, < 2 mm) fra Flakkebjerg, Vestsjællands Amt, ved reaktionstider på henholdsvis 18 og 24 timer. Den anvendte koncentration af KMnO4 var 5 mM. 5.2.2 Modificeret metode til analyse ved brug af KMnO4Resultaterne af den indledende metode optimering med KMnO4 som oxidant, viste at forbruget af KMnO4 var stærkt koncentrations- og tidsafhængig. Resultaterne viste endvidere at sedimntprøver udtaget fra jordoverfladen og ned til ca. 1 meters dybde indeholder syreopløselige organiske forbindelser i et omfang som medfører misfarvning af supernantanten. Det synes derfor ikke hensigtsmæssigt at anvende KMnO4 til oxidation af overfladenære sedimentprøver med forholdsvis høje indhold af nydannet organisk stof. Metode Alle forsøg med KMnO4 blev udført i 40 ml centrifugerør af polyethylen med låg med teflon indsats. I centrifugerøret afvejes 2,00 g sediment (tørret ved 105 °C, < 2 mm) og der tilsættes med dispenser 25 ml 5 mM KMnO4 opløsning. Opslemningen henstår i 15 minutter, hvorefter låget skrues tæt til og herefter anbringes prøverne i en lysisoleret kasse. Prøverne ryster i 24 timer i en horizontal-rystende rysteapparat ved 275 rpm. Når rystetiden er tilendebragt centrifugeres prøverne ved 4500 rpm i 8 minutter og der udtages 5 ml af supernantanten, der fortyndes 10 gange forud for måling. Målingen af KMnO4 foretages på et spectrofotometer ved 525 nm. Tilberedning af kalibreringskurve Kaliberingskurven fremstilles i koncentrationsintervallet 5 10-5 M til 10-3 M KMnO4 på baggrund af stamopløsningen på 5 mM KMnO4. Standarderne behandles på samme måde som beskrevet for prøverne. 5.2.3 Den modificerede metode anvendt på udvalgte sedimenterForbruget af KMnO4 i sedimenter udtaget i 5 boringer ved Søndersø, Fyns Amt, fremgår af figur 5.11. Som det fremgår af figuren forekommer der betydelige forskelle i forbruget af KMnO4 i sedimentprøverne fra de 4 lokaliteter. Således ses et højt forbrug af KMnO4 i boringerne B1 og B5 for sedimenter udtaget i 4 til ca. 8 m dybde, med et maximalt forbrug af KMnO4 på 11 mækv/kg i dybde 6 m for B1 og et forbrug af KMnO4 på ca. 12 mækv/kg for B5. Fra ca. 6 m dybde ses der efterfølgende et nærmest parallelt forløb for forbruget af kurverne gennem resten sedimentprøverne fra de to lokaliteter. Forbruget af KMnO4 udtrykt ved formen af kurven for B3, som vist i figur 5.11, ligner det for B1 og B5, men forbruget af KMnO4 er langt mindre og overstiger ikke 2 mækv/kg sediment. Forbruget af KMnO4 er ligeledes lavt (< 2 mækv,/kg) for B4 og B6 mens der for B6 ses et maksimum i forbruget af KMnO4 ved dybden 10 m. Tilsvarende lave værdier for forbruget af KMnO4 ses i samme dybdeinterval for B3.
Figur 5.11. Forbruget af KMnO4 (beregnet som mækv/kg) ved batchforsøg med sedimentprøver (tørret ved 105 °C, < 2mm) fra Søndersø (B1 og B3-B6), Fyns Amt. Der blev anvendt 5 mM KMnO4 og reaktionstiden var 24 timer. Forbruget af KMnO4 fra boringer ved Borris, Brande og Uhre, Ringkøbing Amt, fremgår af figur 5.12. For lokaliteterne P1, P2 og P5 ses et næsten parallelt kurveforløb, mens P3 og P4 afviger fra dette mønster. I P5 ses således, et stigende forbrug af KMnO4 i dybderne 23 m til 25 m. Ligeledes steg forbruget af KMnO4 for sedimentprøver ned gennem P1, hvor der er målt et maksimalt forbrug af KMnO4 i 17,5 meters dybde. Modsat i P3, hvor forbruget af KMnO4 udviser et modsat forløb, med maksimale forbrug af KMnO4 i 8,25 meters dybde, hvorefter forbruget falder brat i resten af profilet. På lokaliteten P2 sker der et fald i forbruget af KMnO4 ned til 13 meters dybde, hvorefter forbruget af KMnO4 atter stiger. Resultaterne fra Davding i Vejle Amt er vist i figur 5.13. Forbruget af KMnO4 i de øverste 6 m af profilet er meget beskedent (< 5 mækv/kg), mens der i sedimentprøverne fra 27 og 60 meter er målt et forholdsvis højt forbrug af KMnO4 (21-22 mækv/kg). For disse prøver var det nødvendigt at ændre koncentrationen af KMnO4 fra 5 mM til 10 mM for at sikre, at der var en tilstrækkeligt mængde KMnO4 til stede i opløsningen til at reagere med prøvernes indhold af reducerende forbindelser.
Figur 5.12. Forbruget af KMnO4 (beregnet som mækv/kg) ved batchforsøg med sedimentprøver (tørret ved 105 °C, < 2mm) fra Borris (P1 og P2), Brande (P3) og Uhre (P5) i Ringkøbing amt. Der blev anvendt 5 mM KMnO4 og reaktionstiden var 24 timer.
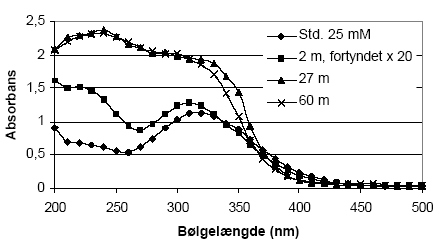
Figur 5.13. Forbruget af KMnO4 (beregnet som mækv/kg) ved batchforsøg med sedimentprøver (tørret ved 105 °C, < 2mm) fra Davding, Vejle Amt. Der blev anvendt 5 mM for prøver ned til 6 meter og 10 mM KMnO4 for sedimenterne udtaget i 27 og 60 meter. Reaktionstiden var 24 timer. 5.3 Oxidationsforsøg med Cerium(IV) – metode og resultater5.3.1 Optimering af forsøgsbetingelser5.3.1.1 Valg af cerium(IV) forbindelseDe indledende forsøg blev udført med ammonium cerium(IV)nitrat ((NH4)2Ce(NO3)6). Vanskeligheder med at finde en egnet bølgelængde til måling af denne forbindelse på spektrofotometer forhindrede dog den fortsatte anvendelse i forbindelse med metodeudviklingen. Forsøgene med (NH4)2Ce(NO3)6 gav dog nogle nyttige erfaringer som kunne anvendes ved de videre oxidationsforsøg med Ce(IV). Det har således på baggrund af reaktioner med (NH4)2Ce(NO3)6 været muligt at beskrive betydningen af Ce-opløsningers pH-værdi ved reaktionen med forskellige sedimenter. Således dannede Ce(IV) under neutrale pH-forhold et tungt opløseligt gult bundfald med karbonat ionen (CO32-) og endvidere var reaktionshastigheden meget langsom. En gentagelse af forsøgene med Ce(IV) opløst i en stærk sur opløsning (pH, 0.7) viste derimod ingen tegn på dannelse af bundfald i kalkholdige jordprøver og desuden blev reaktionshastigheden af Ce(IV) væsentligt forøget. 5.3.1.2 MålemetodeFra litteraturen er det velkendt, at Ce(SO4)2 har et absorptionsmaximun af monochromatisk lys ved 312-316 nm (Sultan og Walmsley, 1997). Et scan af en sur opløsning (5 vol-% H2SO4) af en 25 mM Ce(SO4)2-opløsning viste ligeledes et absorptionsmaximun ved 316 nm hvorefter denne bølgelængde blev valgt til de følgende målinger. Efterfølgende problemer med analyse af Ce(IV) i supernantanten fra sedimentprøver viste imidlertid, at andre forbindelser i supernantanten absorberer ved samme bølgelængde og derfor generede målingen af Ce(IV), figur 5.14. Det drejer sig bl.a. om nogle organiske forbindelser.
Figur 5.14. Bølgelængdescan fra 200 til 500 nm af en 25 mM Ce(SO4)2 opløsningen efter reaktion med 3 sedimentprøver (tørret ved 105 °C, < 2 mm) fra Davding, Vejle Amt. Prøverne er udtaget i 2, 27 og 60 meters dybde. Til sammenligning er vist et scan af en 25 mM Ce(IV) opløsning (std 25 mM). Derefter blev det besluttet at måle overskydende Ce(IV) i supernanten ved potentiometrisk titrering med ferrojernsulfat (Fe(II)SO4). Som beskrevet i afsnit 3.4.6 anvendes Ce(SO4)2 ved titrering af indholdet af ferrojern (Fe2+) i opløsninger. Det var derfor nærliggende ved de her planlagte forsøg at anvende ferrojern i form af ferrosulfat (FeSO4) til bestemmelsen ved titrering. Reaktionsforløbet er beskrevet af bl.a. Kolthoff og Steger (1942) og Golterman og Wisselo (1981). 5.3.1.3 Koncentration af syreDa Ce(SO4)2 er tungtopløseligt i vand og da mange danske sedimenter ydermere har et højt kalkindhold var det nødvendigt at tilsætte et overskud af syrer til Ce(SO4)2 - opløsningen, for at sikre at Ce(IV) forblev i opløsningen ved opblanding med sedimentprøver. Opløseligheden af Ce(SO4)2 blev dernæst undersøgt ved tilsætning af forskellige mængder H2SO4 og efterfølgende måling ved spektrometri af intensiteten af Ce(SO4)2, idet opløsningens farve blev anvendt som indikation på opløseligheden af Ce(SO4)2. Forsøgene viste tydeligt, at pH i stamopløsningen af Ce(SO4)2 skulle være pH 0,70, for at sikre en optimal farve-intensitet af Ce(IV). Yderligere blev det beregnet, på baggrund af et forventet maksimalt indhold af kalk på under 25% (beregnet som CaCO3), at syrekoncentrationen minimum skulle være 0,2 M. Ved de fortsatte forsøget blev det derfor besluttet, at opløse Ce(SO4)2 i en 5% H2SO4-opløsning for at modvirke udfældning af Ce(IV) og sikre en pH under 0.7. 5.3.1.4 Koncentration af cerium og reaktionstidForbruget af Ce(IV) i forskellige sedimentprøver blev undersøgt ved forskellige koncentrationer af Ce(SO4)2. Resultaterne fremgår af figur 5.15, hvor forbruget af Ce(IV) ses at være størst for prøverne fra henholdsvis 27, 49 og 60 meters dybde. Således opbrugte sedimentprøverne fra disse tre dybder mængden af Ce(IV) der var tilrådighed ved tilsætning af henholdsvis 5 mM og 10 mM, mens kun sedimetprøven udtaget i 60 meters dybde var i stand til at opbruge mængden af Ce(IV) ved tilsætning af henholdsvis 25 mM og 50 mM Ce(SO4)2. Figuren viser således, at sedimentprøverne fra 27 og 49 meters dybde forbrugte en stigende mængde Ce(IV) til oxidation af reducerende forbindelser når koncentrationen af Ce(SO4)2 blev forøget. Sedimentprøver udtaget indenfor de øverste 6 meter havde generelt lave forbrug af Ce(SO4)2 og fremstod upåvirket af startskoncentrationen af Ce(SO4)2.
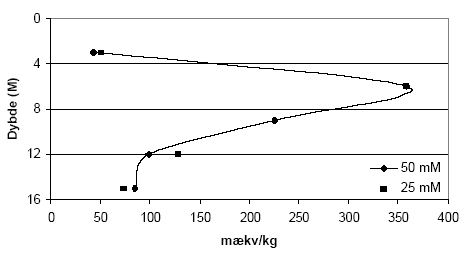
Figur 5.15. Forbrug af Ce(SO4)2 (beregnet som mækv/kg) for sedimentprøver (tørret ved 105 °C, < 2 mm) fra Davding, Vejle Amt, ved forskellige begyndelseskoncentrationer af Ce(SO4)2. Forsøgene er gennemført ved stuetemperatur og med en rystetid på 2 timer. Betydningen af rystetiden for forbruget af Ce(SO4)2 blev undersøgt for udvalgte sedimentprøver fra Vejle Amt og Fyns Amt. For sedimentprøverne 27 og 49 meter fra Vejle Amt kunne der iagttages en markant forøgelse i forbruget af Ce(IV) når rystetiden blev ændret fra 1 time til 24 timer, figur 5.16. Derimod var ingen tydelige forskelle i forbruget af Ce(IV) som følge af forøgede rystetider for sedimentprøver udtaget inden de øverste 6 meter.
Figur 5.16. Forbrug af Ce(SO4)2 (beregnet som mækv/kg) for sedimentprøver (tørret ved 105 °C, < 2 mm) fra Davding, Vejle Amt. Koncentrationen af Ce(SO4)2 er 25 mM. Forsøgene er gennemført ved stuetemperatur og med rystetider på henholdsvis 1, 2, 4 og 24 timer. Effekten af såvel forskellige Ce(SO4)2 koncentrationer som rystetider blev undersøgt for profil B1 fra Fyns Amt. Resultaterne af denne undersøgelse er vist i figur 5.17, hvor indholdet af reducerende forbindelser er bestemt ved en koncentration på 25 mM og en rystetid på 24 timer og en koncentration på 50 mM Ce(SO4)2 og en rystetid på 72 timer. Det ses på figuren, at der ikke forekommer væsentlige forskelle på indholdet af reducerende forbindelser imellem de to koncentrationer, idet begge kurver er sammenfaldende for dybderne 3, 6, 9 m, men der kun ses en begrænset afvigelse i de nederste prøver i profilet. Det må derfor forventes at alle reaktive reducerede forbindelser oxideres efter en reaktionstid på 24 timer for dette profil.
Figur 5.17. Forbrug af Ce(SO4)2 (beregnet som mækv/kg) for sedimentprøver (tørret ved 105 °C, < 2 mm) fra Søndersø (B1), Fyns Amt, ved forskellige begyndelseskoncentrationer af Ce(SO4)2. Forsøgene med 25 mM Ce(SO4)2 er gennemført ved stuetemperatur og med en rystetid på 24 timer, mens forsøgene med 50 mM er gennemført ved stuetemperatur og med en rystetid på 72 timer. På baggrund af de udførte undersøgelser med effekten af koncentrationen på indholdet af reducerende forbindelser blev det besluttet at fortsætte med en startskoncentration af Ce(SO4)2 på 25 mM idet den forventes at være tilstrækkelig ved analyse af langt de fleste naturligt forekommende sedimentprøver. Forsøgene viser imidlertid, at prøver med et ekstraordinært høj indhold af reducerende forbindelser med fordel kan undersøges med en koncentration af Ce(SO4)2 på 50 mM eller højere. Det kan dog ikke anbefales at anvende en koncentration på 50 mM i typiske danske sand- og ler prøver, idet usikkerheden på bestemmelsen af forholdsvis lave indhold af reducerende forbindelser (<50 mækv/kg) væsentligt forøget. På baggrund af forsøgende blev det desuden besluttet at gennemføre analyserne ved en rystetid på 24 timer. 5.3.2 Modiferet metode til analyse ved brug af Ce(SO4)2Resultaterne af den indledende metode optimering med cerium(IV)sulfat som oxidant, viste at forbruget af Ce(IV) afhænger af såvel koncentration, temperatur og og reaktionstid. Resultaterne viste endvidere at Ce(SO4)2 skulle opløses i en sur opløsning for at imødegå udfældninger samt for at sikre en optimal farve af opløsningen. Metode Til et 40 ml centrifugerør af polypropylen afvejes 1,00 g sediment (tørret ved 105 °C, < 2mm). Der tilsættes med dispenser 25 ml 25 mM Ce(SO4)2 opløst i en 5 vol-% H2SO4 opløsning. Prøverne henstår 30 minutter, hvorefter låget monteres og lukkes tæt til. Prøverne ryster i 24 timer på et horisontalt rysteapparat ved 275 rpm. Efter endt rystetid centrifugeres prøven ved 2000 rpm i 8 minutter. Herefter fortyndes 2 ml af supernantanten med 13 ml 5 vol-% H2SO4 opløsning. Den fortyndede prøve titreres herefter med 5 mM FeSO4 opløst i 5 vol-% H2SO4 til omslagpunkt ved dynamic-endpoint-titrering. Sammen med hver batch medtages minimum 1 standardopløsning. Denne standardopløsning er 25 ml stamopløsning (25 mM Ce(SO4)2 i 5 vol-% H2SO4 dispenseret ned i et centrifugerør. Denne standard opløsning er efterfølgende behandlet på samme måde som sedimtprøverne. Koncentrationen af stamopløsningen tjekkes med jævne mellemrum for at sikre at der ikke er sket nedbrydning af Ce(SO4)2. 5.3.3 Metoden anvendt på udvalgte sedimentprøverForbruget af Ce(SO4)2 i sedimentprøver fra Præstbro, Nordjyllands Amt er vist i figur 5.18. Figuren viser det største forbrug af Ce(SO4)2 i de øverste 0.45 m, hvorefter forbruget faldt markant ned til 40 meters dybde hvorefter der atter ses en stigning i forbruget af Ce(SO4)2. Resultaterne fra udvalgte sedimentprøver fra Borris (P1 og P2), Brande (P3) og Uhre (P5), Ringkøbing Amt, viser en variation i forbruget af Ce(IV) som vist i figur 5.19. Der ses et tilnærmelsesvis parallelt forløb af kurverne for P1 og P5, mens P2 og P3 var forskellige fra disse boreprofiler. I P5 ses således, et stigende forbrug af Ce(SO4)2 i dybderne 23 m til 25 m, med et maximalt forbrug af Ce(SO4)2 i dybden 25 m på 520 mækv/kg jord. I P1 steg forbruget af Ce(SO4)2 ligeledes gennem profilet med et maksimalt forbrug Ce(SO4)2 i 17,5 m dybde på 620 mækv/kg jord. I P3 ses det et modsatrettet forløb af kurven, idet det maksimale forbrug af Ce(SO4)2 på 560 mækv/kg jord forekom i 8,25 m, hvorefter forbruget falder markant i resten af profilet. I profilen P2, ses et fald i forbruget af Ce(SO4)2 med til dybden 13 m, hvorefter forbruget af Ce(SO4)2 igen begynder at stige.
Figur 5.18. Forbrug af Ce(SO4)2 (beregnet som mækv/kg) ved batchforsøg med sedimentprøver (tørret ved 105 °C, < 2 mm) fra Præstbro, Nordjyllands Amt, ved en koncentrationer af Ce(SO4)2 på 25 mM. Forsøgene er gennemført ved stuetemperatur og med en rystetid på 24 timer.
Figur 5.19. Forbrug af Ce(SO4)2 (beregnet som mækv/kg) for sedimentprøver (tørret ved 105 °C, < 2 mm) fra Borris (P1 og P2), Brande (P3) og Uhre (P4), Ringkøbing Amt. Koncentrationen af Ce(SO4)2 er 25 mM og forsøget er gennemført ved stuetemperatur og med en rystetid på 24 timer. Forbruget af Ce(SO4)2 i 5 udvalgte profiler i Søndersø, Fyns amt er vist på figur 5.20. Det se på figuren, at der forekommer et betydeligt forbrug af Ce(SO4)2 i dybden 6 m i profil B1 på 360 mækv/kg, hvorefter forbruget af Ce(SO4)2 mindskes gennem resten af profilet ned til en dybde på 15 m. Profilerne B3 og B4 har tilnærmelsesvis parallelle forløb gennem profilerne, dog med et højere Ce(SO4)2 forbrug i B4. Forbruget af Ce(SO4)2 i profilerne B3 og B4 overstiger på intet tidspunkt 100 mækv/kg jord. I profilet B5 ses et højt forbrug af Ce(SO4)2 i dybderne 4 m og 8 m (300-310 mækv/kg jord), hvorefter forbruget af Ce(SO4)2 falder til en værdi på 90 mækv/kg jord i dybden 11 m, for derefter igen at stige mod dybden 15 m. For profilen B6 forkommer der kun et begrænset forbrug af Ce(SO4)2 ned til en dybde på 9 m, hvorefter forbruget stiger betydeligt til et niveau på ca.170 mækv/kg jord i 11 m dybde.
Figur 5.20. Forbrug af Ce(SO4)2 (beregnet som mækv/kg) for sedimentprøver (tørret ved 105 °C, < 2 mm) fra Søndersø (B1 og B3-B6), Fyns Amt. Koncentrationen af Ce(SO4)2 er 25 mM. Forsøgene er gennemført ved stuetemperatur og med en rystetid på 24 timer. 6 Målte og beregnede indhold af reducerende forbindelser i sedimenter
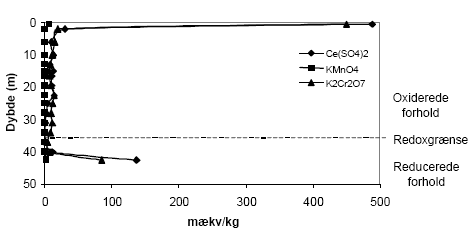
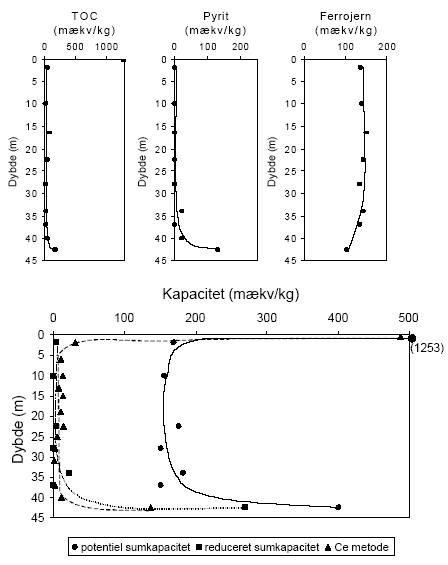
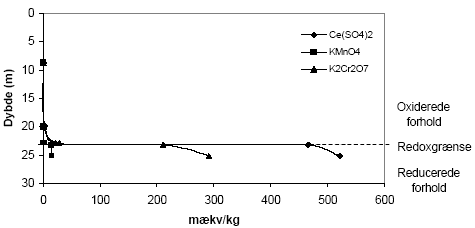
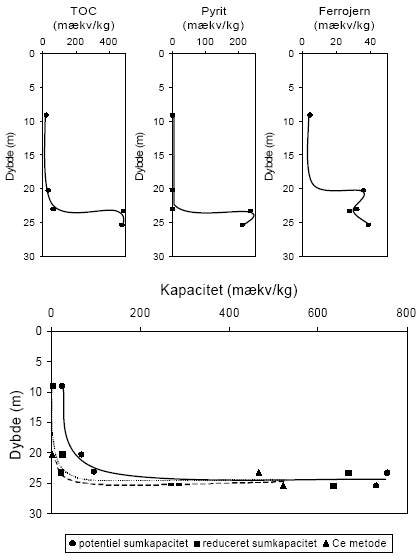
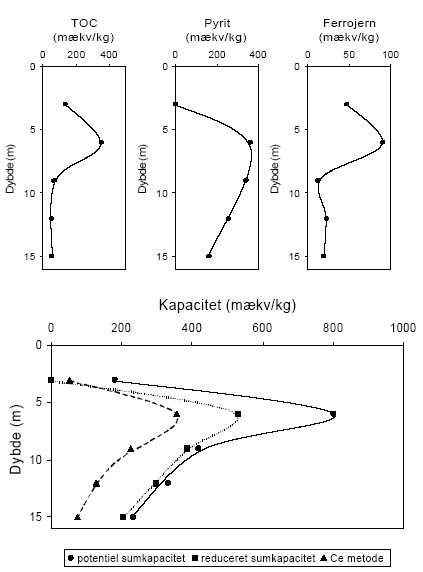
Indholdet af reducerende forbindelser i sedimenter målt efter brug af oxidationsmidlerne KMnO4, K2Cr2O7 og Ce(SO4)2 sammenholdes i det følgende for forskellige lokaliteter. Desuden foretages en sammenligning mellem det målte og det beregnede indhold af reducerende forbindelser baseret på fordelingen af organisk stof (TOC), pyrit og ferrojern. Den beregnede værdi - baseret på det totale indhold af disse 3 komponenter - benævnes i det følgende for den potentielle sumkapacitet. Til grund for beregningen antages organisk stof at bidrage med 2 mækv/mM (Barcelona og Holm, 1991), pyrit bidrager med 7 mækv/mM og ferrojern bidrager med 1 mækv/mM. Der er ydermere for hver lokalitet foretaget en vurdering af tilgængeligheden af TOC, pyrit og ferrojern og den herved beregnede værdi benævnes i det følgende for den reducerede sumkapacitet. Ved sidstnævnte beregning foretages for hver lokalitet en vurdering af den andel af TOC og ferrojern, der forventes at være aktive under netop de geologiske forhold, der er typiske for den pågældende lokalitet, idet ikke hele den potentielle pulje af reducerende forbindelser forventes at indgå i den aktive pulje (Ernstsen, 2001). 6.1 Reducerende forbindelser ved Præstbro, Nordjyllands Amt6.1.1 Måling af reducerende forbindelser ved forskellige oxidationsmidlerFor profilen ved Præstbro, der med undtagelse af et lerlag fra 41-41,5 meter, er domineret af sandede sedimenter, er forbruget af KMnO4 lavt i alle de undersøgte dybder, figur 6.1. Forbruget af de kraftigere oxidationsmidler K2Cr2O7 og Ce(SO4)2 stiger markant i det overfladenære lag til mellem 450 og 500 mækv/kg og i den dybeste prøve fra 42,5 m, hvor forbruget modsvarer et indhold af reducerende forbindelser på ca. 100 mækv/kg. For begge de nævnte dybder gav brugen af Ce(SO4)2 et lidt højere indhold af reducerende forbindelser.
Figur 6.1. Forbrug af oxidationsmidlerne Ce(SO4)2, KMnO4 og K2Cr2O7 (beregnet som mækv/kg) for sedimentprøver fra Præstbro, Nordjyllands Amt. Redoxgrænsen er beliggende i 35 meters dybde. 6.1.2 Beregning af reducerende forbindelser – potentiel og reduceret sumkapacitetDen beregnede potentielle sumkapacitet viser samme forløb som vist for Ce(SO4)2 og K2Cr2O7, med de største værdier i den øverste og nederste prøve. De beregnede værdier er generelt betydelig højere end de mængder, der blev målt ved brug af det mest effektive oxidationsmiddel, Ce(SO4)2 metoden, figur 6.2. Den beregnede potentielle sumkapacitet udgør således ca. 1300 mækv/kg i den øverste prøve hvor de med Ce(SO4)2 metoden blev målt et indhold på næsten det halve. En nærmere analyse af fordelingen af organisk stof, pyrit og ferrojern viste at det er rimeligt at antage, at den tilgængelige pulje af organisk stof (pånær ny bidrag til rodzonen) i den oxiderede del af profilen er brugt op og at den tilgængelige pulje i den reducerede del af profilen svarer til den målte værdi korrigeret for middelværdien af mængden af tilbagebleven organisk stof i den oxiderede zone. Mængden af reaktivt ferrojern vurderes at være meget ringe i hele profilen og menes ikke at bidrage til puljen af tilgængeligt reducerende forbindelser. Indholdet af pyrit, der stiger markant i prøven fra 42,5 m og antages at være fuldt tilgængeligt. Den således beregnede reducerede sumkapacitet har nu et forløb som er stort set sammenfaldende med de der er opnået med Ce(SO4)2 metoden, ligesom forskellen mellem de målte og beregnede værdier er betydelig mindre end for de potentielle sumkapaciteterne.
Figur 6.2. For oven: målte indhold af organisk stof, pyrit og ferrojern beregnet som mækv/kg. For neden: den beregnede potentielle sumkapacitet, reducerede sumkapacitet, og indhold af reducerede forbindelser målt ved Ce(SO4)2-metoden for Præstbro, Nordjyllands Amt. Redoxgrænsen er beliggende i 35 meters dybde. 6.2 Reducerende forbindelser ved Uhre, Ringkøbing Amt6.2.1 Måling af reducerende forbindelser ved forskellige oxidationsmidlerDe gelogiske forhold ved Uhre er domineret af lerholdige aflejringer, der er oxiderede ned til 23,3 meter under terræn. Herunder følger reduceret ler. Forbruget af KMnO4 er forholdsvis ringe såvel over som under redoxgrænsen, hvor det for de kraftigere oxidationsmidler Ce(SO4)2 og K2Cr2O7 ses en markant stigning i forbruget i de reducerede sedimenter under redoxgrænsen, til mellem 300 og 550 mækv/kg, figur 6.3. De største indhold opnås ved brug af Ce(SO4)2.
Figur 6.3. Forbrug af oxidationsmidlerne Ce(SO4)2, KMnO4 og K2Cr2O7 (beregnet som mækv/kg) for sedimentprøver fra Uhre (P5), Ringkøbing Amt. Redoxgrænsen er beliggende i 23,3 meters dybde. 6.2.2 Beregning af reducerende forbindelser – potentiel og reduceret sumkapacitetUdbredelsen af den oxiderede zone ved Uhre slår tydeligt igennem i fordelingen af TOC, pyrit og ferrojern, ved markant lavere indhold end i den underliggende reducerede zone. I de reducerede sedimenter er TOC dominerende (480-490 mækv/kg) sammen med pyrit (210-240 mækv/kg), mens ferrojern kun udgør en ringe del (<40 mækv/kg), figur 6.4. Den beregnede potentielle sumkapacitet er beregnet til 25 mækv/kg i den oxiderede prøve fra 9.1 meter og stiger til ca. 750 mækv/kg for sedimenterne fra den reducerede zone, figur 6.4. Ved beregning af den reducerede sumkapacitet er mængden af aktiv organisk stof reduceret med en mængde svarende til den der er tilbage i den oxiderede zone, mængden af ferrojern er reduceret under hensyn til de mængder der er tilbage i den resterende del i den oxiderede zone og pyrit er medtaget i fuldt omfang. Med denne reduktion optræder der nu en god overensstemmelse mellem det beregnede indhold og det med Ce(SO4)2 metoden målte. For de reducerede sedimenter nærmer den beregnede og den ved Ce(SO4)2-metoden målte værdi hinanden, og forskellen er omkring 100 mækv/kg for den nederste prøve udtaget i 25,4 meters dybde.
Figur 6.4. For oven: målte indhold af organisk stof, pyrit og ferrojern beregnet som mækv/kg. For neden: den beregnede potentielle sumkapacitet, reduceret sumkapacitet, og indhold af reducerede forbindelser målt ved Ce(SO4)2-metoden for Uhre (P5), Rinkøbing Amt. Redoxgrænsen er beliggende i 23,3 meters dybde. 6.3 Reducerende forbindelser ved Søndersø, Fyns Amt6.3.1 Måling af reducerende forbindelser ved forskellige oxidationsmidlerDe geologiske forhold ved Søndersø (B1) består øverst af 4.8 meter oxideret ler, hvorefter lerlaget fremstår reduceret til 8.2 meter hvorefter reduceret sand er beskrevet ned til en dybde af 17,6 meter. Det svageste af oxidationsmidlerne KMnO4 viser således kun et forbrug for den reducerede lerprøve i 6 meters dybde, figur 6.6. For de to øvrige oxidationsmidler er det maksimale forbrug ligeledes knyttet til dette lag, hvor der er målt mellem 100 og 350 mækv/kg. I prøven fra den oxiderede ler i 3 meters dybde er forbruget beregnet til mellem 20 og 50 mækv/kg, hvor den i det underliggende sandlag med prøver fra 9-15 meter variere mellem 50 og 220 mækv/kg. Ved brug af K2Cr2O7 metoden er forbruget stort set konstant for de reducerede sandprøver mens der for Ce(SO4)2-metoden ses et aftagende forbrug med tiltagende dybde.
Figur 6.6. Forbrug af oxidationsmidlerne Ce(SO4)2, KMnO4 og K2Cr2O7 (beregnet som mækv/kg) for sedimentprøver fra Søndersø (B1), Fyns Amt. De to øverste prøver stammer fra henholdsvis den oxiderede og reducerede moræneler og de efterfølgende 3 prøver fra et reduceret sandlag. 6.3.2 Beregning af reducerende forbindelser – potentiel og reduceret sumkapacitetIndholdet af TOC omregnet til mækv/kg er markant højere i der reducerede lerlag end i det oxiderede lerlag og det reducerede sand. Bidraget fra pyrit er ligeledes størst for den reducerede ler, hvorefter det næsten halveres ned gennem sandlaget. Indholdet af ferrojern i lerlaget er næsten 2 gange den for det oxiderede lerlag og 4-5 gange det der findes i det reducerede sandlag, figur 6.7. Begge de beregnede sumkapaciteter har et næsten parallelt forløb med den der er bestemt ved Ce(SO4)2-metoden, figur 6.8. Den beregnede potentielle sumkapacitet når op på omkring 800 mækv/kg for det reducerede lerlag, hvor det oxiderede lerlag og de dybere reducerede sandlag varierer mellem 200 og 400 mækv/kg. Ved beregningen af den reducerede sumkapacitet tages der hensyn til at kun ca. 2/3 af det organiske stof synes at forekomme på en tilgængelig form, at den tilgængelige pulje af ferrojern forventes at være udtømt når omkring 15% af det totale indhold er tilbage samt at hele puljen af pyrit er tilgængeligt. Disse forudsætninger bevirker at specielt mængden af sumkapacitet reduceres i lerlaget mens der for det reducerede sandlag med kun lave indhold af organisk stof og ferrojern kun forekommer mindre ændringer, og forløbet her styres i overvejende grad af fordelingen af pyrit.
Figur 6.7. For oven: målte indhold af organisk stof, pyrit og ferrojern beregnet som mækv/kg. For neden:den beregnede potentielle sumkapacitet, reducerede sumkapacitet, og indhold af reducerede forbindelser målt ved Ce(SO4)2-metoden for Søndersø (B1), Fyns Amt. De to øverste prøver stammer fra henholdsvis den oxiderede og reducerede moræneler og de efterfølgende 3 prøver fra et reduceret sandlag. 6.4 Samlet vurdering af målte og beregnede indhold af reducerende forbindelserDet generelle billede har været at forbruget af det pågældende oxidationsmidler og dermed den pulje af reducerende forbindelser der forventes at være tilgængelige stiger i den nævnte rækkefølge: KMnO4 < K2Cr2O7 < Ce(SO4)2. Dette var ligeledes forventeligt da Ce(SO4)2 med et standardelektrode potentiale (E0) på 1,44 V overstiger standardelektrode potentialet på 1,33 V for K2Cr2O7 og potentialet på 0,59 V (i neutral opløsning) for KMnO4. Det vurderes imidlertid at metoden med anvendelse af KMnO4 som oxidationsmiddel vil være uegnet for langt de fleste danske sedimenter, dersom den hermed målte pulje af reducerende forbindelser er betydelig lavere end den der beregnes ved såvel den potentielle som den aktuelle sumkapacitet. Desuden afspejler denne metode kun i mindre grad variationen i indhold af reducerende forbindelser. Brugen af K2Cr2O7 gav højere indhold af reducerede forbindelser og afspejlede også den forventede naturligt forekommende variation, men denne metode anses ikke anbefalelsesværdig på grund af dels sundhedsmæssige aspekter dels analysetekniske problemer knyttet til håndtering af kalkholdige sedimentprøver ved høj temperatur. Det vurderes derfor at Ce(SO4)2 er det af de udvalgte og afprøvede oxidationsmidler der vil være bedst egnet i forbindelse med analysen af det samlede indhold af reducerende forbindelser i typiske danske sedimenter. Med denne metode vurderes det, at er der opnået den bedste overensstemmelse mellem den målte og den teoretisk beregnede pulje af aktive reducerende forbindelser. Metoden udmærker sig dernæst ved at være forholdsvis enkelt og hurtig og vil med den anbefalede koncentration på 25 mM skønnes det at denne koncentration vil være fuldt ud tilstrækkeligt for langt de fleste danske sedimenter. For sedimenter hvor der forventes et betydeligt højere indhold af reducerende forbindelser anbefales det at øge koncentrationen af Ce(SO4)2 til 50 mM eller endnu højere. Ved brug af Ce(SO4)2-metoden er de karakteristiske værdier for den overfladenære del af rodzonen bestemt til 250-500 mækv/kg, oxideret ler 10-100 mækv/kg, reduceret ler 300-550 mækv/kg, oxideret sand <80 mækv/kg og for reduceret sand 80-220 mækv/kg. De forholdsvise høje indhold af reducerede forbindelser i rodzonen stammer antagelig fra en pulje let omsætteligt organisk stof. Målte værdier fremkommet ved brug af Ce(SO4)2 -metoden er normalt lavere end den beregnede potentielle sumkapacitet baseret på målte koncentrationer af TOC, pyrit og ferrojsamt. Ved beregningen af den reducerede sumkapacitet, hvor den forventede tilgængelighed af de reducerende forbindelser inddrages, ses et langt bedre sammenfald mellem den med metoden målte og den beregnede pulje af reducerende forbindelser. Da puljen af TOC ofte vejer tungt i den samlede sumkapacitet vil en lang række forhold knyttet til mængde og sammensætning af puljen af organisk stof i betydelig omfang kunne påvirke bidraget herfra og dermed også den beregnede aktuelle sumkapacitet. Den generelt gode sammenhæng mellem det beregnede indhold og det målte indhold bevirker at brugen af Ce(SO4)2-metoden kan være nyttig ved en kortlægning af forekomsten af reducerende forbindelser. Den forholdsvis enkle metode gør det muligt at øge detailkendskabet til fordelingen af mængden af reducerende forbindelser og supplerende analyser af udvalgte reducerende forbindelser (eksempelvis TOC, pyrit og ferrojern) vil kunne bidrage til at belyse den faktiske sammensætningen af puljen af reducerende forbindelser. 7 Metodebeskrivelse til analyse af reducerende forbindelser i sedimenter ved brug af Ce(SO4)2En kemisk oxidationsmetode med cerium(IV)sulfat (Ce(SO4)2) som oxidationsmiddel over for reducerende forbindelser i sedimenter. 7.1 AnvendelseMetoden er udviklet med henblik på at få et indblik i indholdet af reducerende forbindelser i sedimenter. I metoden bestemmes således alle de reducerende forbindelser der deltager i reaktionen med den kraftige oxidant cerium(IV)sulfat (Ce(SO4)2) der anvendes som oxidationsmiddel i metoden. Indholdet af reducerende forbindelser beregnes udfra forbruget af oxidationsmidlet. Bidraget fra indholdet af de enkelte reducerende forbindelser som eksempelvis organisk stof, pyrit og ferrojern kræver imidlertid stadig specifikke analysemetoder omfattende hver af disse forbindelser. 7.2 PrincipMetoden benyttes til at bestemme den mængde iltningsækvivalenter, der forbruges ved kemisk oxidation af reducerede forbindelser (organiske og uorganiske) i sediment. Teorien bag forsøgene er, at cerium(IV)sulfat (Ce(SO4)2) i en stærk sur opløsning vil reagere med reducerede forbindelser i sedimentet, når suspensionen rystes over tid. For at opnå en effektiv oxidation rystes sedimenterne i 24 timer med Ce(SO4)2-opløsningen. Herved reduceres Ce(IV) til Ce(III), mens de reducerede forbindelser oxideres. Den overskydende mængde Ce(IV) bestemmes ved potentiometrisk titrering med jern(II)sulfat (FeSO4) under anvendelse af en redoxelektrode som sikre en nøjagtig bestemmelse af omslagspunktet. I forsøgene er det vigtigt at der til en hver tid forekommer et overskud af H2SO4 dersom Ce(SO4)2-opløsningen ikke reagere effektivt ved pH-værdier over 0.7 og dernæst at Ce(III) kan oxideres til Ce(IV) af luftens ilt i basiske opløsninger hvilket vil influere på målingerne. Derfor indeholder alle de anvendte opløsninger i forsøget 5 vol-% H2SO4. 7.3 Materialer og apparaturLufttætte emballage til opbevaring af sedimentprøver, der efter udtagelse opbevares i frossen stand ind til analyse i laboratoriet.
7.4 Reagenser25 mM cerium(IV)sulfat opløsning i 5 vol-% svovlsyre 8,306 g Ce(SO4)2 (Acros Organics, renhed 99%) - tørret i 2 timer ved 105 °C - afvejes på analysevægt og overføres kvantitativt til en 1 liter målekolbe, der efterfyldes med ca. 800 ml demineraliseret vand (millipore gradient, Millipore). Herefter tilsættes 50 ml koncentreret H2SO4 (analysekvalitet, Merck) og efter afkøling fyldes op til mærket med demineraliseret vand og opløsningen blandes. Opløsningen overføres herefter til en 1 liter pyrex glasflaske (med teflonbelagt pakning i låget ved opbevaring) og en dispenser på 25 ml monteres. Opløsningen opbevares i pyrexflaske med låg med teflonpakning. 5 mM cerium(IV)sulfat opløsning i 5 vol-% svovlsyre til standardkurve på spectrofotometer 1,6612 g Ce(SO4)2 (Acros Organics, renhed 99%) - tørret i 2 timer ved 105 °C - afvejes på analysevægt og overføres kvantitativt til en 1 liter målekolbe, der efterfyldes med ca. 800 ml demineraliseret vand. Herefter tilsættes 50 ml koncentreret H2SO4 (analysekvalitet, Merck) og efter afkøling fyldes op til mærket med demineraliseret vand og opløsningen blandes. Opbevares i pyrexflaske med låg med teflonpakning. 5 vol-% svovlsyre (H2SO4) En målekolbe fyldes med ca. 800 ml demineraliseret vand og tilsættes 50 ml koncentreret H2SO4 (analysekvalitet, Merck). Når opløsningen er afkølet tilsættes demineraliseret vand til mærket og opløsningen blandes. 5 mM jern(II)sulfat opløsning i 5 vol-% svovlsyre 1,3901 g FeSO4, 7H2O (analysekvalitet, Merck) afvejes på analysevægt og overføres kvantitativt til en 1 liter målekolbe, der efterfyldes med ca. 800 ml demineraliseret vand. Herefter tilsættes 50 ml koncentreret H2SO4 (analysekvalitet, Merck) og efter afkøling fyldes op til mærket med demineraliseret vand og opløsningen blandes. Opløsningen overføres herefter til en 1 liter pyrex flaske (låget bør have teflonpakning ved opbevaring) der monteres på titreringsudstyret. Koncentrationen af FeSO4-opløsningen der anvendes til titreringen af prøverne undersøges ved at titrere 2 ml 25 mM Ce(SO4)2-opløsning fortyndet med 13 ml 5 vol.% H2SO4 med FeSO4-opløsning. Der titreres til omslagspunktet og koncentrationen af FeSO4 bestemmes. Koncentrationen af Ce(SO4)2–opløsningen kontrolleres jævnligt ved hjælp af spektrofotometriske målinger. Koncentrationen af Ce(SO4)2 bestemmes på baggrund af en standardkurve målt ved en bølgelængde på 316 nm. Standardkurven laves i 50 ml kolber udfra en mellemfortynding på 5 mM Ce(SO4)2 -opløsningen, fremstillet af en 25 mM Ce(SO4)2-opløsning, med koncentrationer på 0 mM, 0,05 mM, 0,075 mM, 0,1 mM, 0,125 mM og 0,2 mM. Koncentrationen af Ce(SO4)2 i prøverne kan ikke bestemmes spectrofotometrisk, da organiske forbindelser ligeledes absorbere minochromatisk lys ved denne bølgelængde. 7.5 AnalysenFra en naturfugtig sedimentprøve udtages en repræsentativ mængde, der tørres ved 105 °C indtil konstant vægt. Sedimenterne sigtes herefter gennem en 2 mm sigte og fraktionen < 2mm opbevares i lufttættende emballage indtil analysen. Afvej 1,00 g sediment til et centrifugerør og tilsættes med dispenser 25 ml 25 mM Ce(SO4)2 opløst i 5 vol-% H2SO4. Prøverne henstår herefter i 30 minutter. Fra kalkholdige sedimenter vil der ske en kraftig udvikling af kuldioxid. Herefter anbringes låget på centrifugeglasset indeholdende suspensionen på et horisontalt rysteapparat ved 275 rpm i 24 timer. En blindprøve fremstilles og behandles i øvrigt som nævnt for sedimentprøven. Når prøven har rystet i 24 timer, centrifugeres ved 2000 rpm i 8 minutter. Efter filtrering, fortyndes 2 ml af supernantanten med 13 ml 5 vol-% H2SO4.. Den fortyndede prøve fremstillet af supernantanten titreres under konstant omrøring med 5 mM FeSO4 opløst i 5 vol-% H2SO4. Der titeres til omslagspunktet med dynamic endpoint titrering. Ved omslagspunktet ændres opløsningen farve fra stærk gul til farveløs, som udryk for skiftet fra Ce(IV) til Ce(III). 7.5.1 Beregning af prøvernes indhold af reducerende forbindelserForbruget af Ce(SO4)2 under reaktion med sediment beregnes udfra følgende formel: hvor a er koncentrationen i mM af Ce(SO4)2–opløsningen der tilsættes sedimentet, b er ml FeSO4 forbrugt under titreringen af den fortyndede prøve af supernantanten, c er ml supernantant udtaget til titrering og d er koncentrationen af FeSO4. Når forbruget af Ce(SO4)2 er beregnet kan dette forbrug omregnes til iltnings ækvivalenter udfra følgende formel:
hvor d er den afvejede mængde sediment i gram.
7.6 AffaldAl affald der indeholder Ce(SO4)2 opsamles i en separat beholder. Sedimentet der har været anvendt til forsøgene opsamles sammen med andet udstyr i plastikposer og mærkes med fareklasse O, Xn. 8 ReferencerAPHA, AWWA og WEF. 1995. Standard methods for the examination of water and wastewater. 19th Edition. American Public Health Association. Washington, DC, USA. Barcelona, M.J. og Holm, T.R. 1991. Oxidation – Reduction capacities of aquifer solids. Environmental Science & Technology 25: 1565-1572. Bigham, J.M. 1996. Method of soil analysis. Part 3. Chemical methods. Soil Science Society of America, Inc. American Society of Agronomy, Inc., Madison, Wisconsin, USA. 1390 s. Bjerrum, N. 1932. Lærebog i uorganisk kemi. 3. udgave Thorsøe-Olsen & Co. København. 367 s. Bott, A.W. 2000. Electrochemical titrations. Curent separations, 18. 4. s. 129 – 132. Chambers, J., Leavitt, A., Walti, C., Schreier, C.G. og Melby, J. 2000. In-situ destruction of chlorinated solvents with KMnO4 oxidizes chromium. I Chemical oxidation and reaktive barriers. Remidation of chlorinated and recalcitrant compounds. Eds. Wickramanayake, G.B., Gavaskar, A.R. og Chen, A.S.C.The second international conference on remediation of chlorinated and recalcitrant compounds. Monterey, California, d. 22-25 maj, 2000. 470 s. Dasgupta, P.K. og Petersen, K. 1990. Kinetic approach to the measurement of chemical oxygen demand with an automated micro batch analyzer. Analytical Chemistry 62: 395- 402. Eaton, A.D., Clesceri, L.S. og Greenberg, A.E. 1995. Standard methods for examination of water and wastewater. 19. udgave. American Public Health Association, American Water Works Association og Water Environment Federation, USA. Ernstsen, V., S.V. Binnerup og Sørensen, J. 1998. Reduction of nitrate in clayey subsoils controlled by geochemical and microbial barriers. Geomicrobiology, 15: 195-207. Ernstsen, V., Henriksen, H.J. og Platen, F. von. 2001. Principper for beregning af nitratreduktion i jordlagene under rodzonen. Arbejdsrapport fra Miljøstyrelsen, nr. 24. 54 s. http://www.mst.dk/udgiv/publikationer/2001/87-7944-713-9/html/ Fresenius, W., Quentin, K.E. og Schneider, W. 1988. Water analysis. Apracticacl guide to physico-chemical, chemical and microbiological water examination and quality assurance. Springer, Heidelberg, Tyskland. 804 s. Golterman, H.L. og Wisselo, A.G. 1981. Ceriometry, a combined method for chemical oxygen demand and dissolved oxygen (with a discussion on the precision of the winkler technique). Hydrobiologia, 77: 37-42. Graversen, P., Kristiansen, H., Kelstrup, N., Petersen, K.S. 1990. Kortlægning af potentialet for nitratreduktion – Ulfborgområdet. NPo rapport nr. B16. Miljøstyrelsen. 60 s. Kammer, F., Thöming, J. og Calmano W. 2000. Redox buffer capacity concept as a tool for the assessment of long-term effects in natural attenuation / intrinsic remediation. I J. Schüring, J., Schulz, H.D., Fisher, W.R., Böttcher, J. og Duijnisveld, W.H.M. (eds.) Redox fundamentals, processes and applications. Springer, Heidelberg, Tyskland. 251 s. Kolthoff, I.M. og Belcher, R. 1957. Volumetric analysis. Volume 3. Interscience Publishers, Inc. New York, USA. 2. reviderede udgave, 3. version. 714 s. Kolthoff, I.M. og Steger, V.A. 1942. Volumetric analysis. Volume I, Theoreticcal fundamentals. 2. reviderede udgave. Interscience Publishers, Inc. New York, USA. 309 s. Murphy, A.P., Boegli, W.J., Price, M.K. og Moody, C.D. 1989. A Fenton-like reaction to neutralize formaldehyde waste solutions. Environmetal Science & Technology 23: 166-169. Pedersen, J.K. 1992. Nitratreduktion i jord og grundvand. Laboratoriet for Teknisk Hygiejne, Danmarks tekniske Højskole. 273 s. Schachtschabel, P., H.-P. Blume, G. Brümmer, K.-H. Harge, U. Schwertmann, 1989. Lehrbuch der Bodenkunde/Scheffer-Schachtscabel. 12. udgave. Stuttgart. Enke. 491 s. Siegrist, R.L., Urynowicz, M.A., West, O.R., Crimi, M.L. og Lowe, K.S. 2001. Principles and practices of in situ chemical oxidation using permanganate. Battle Press. Ohio, USA. 348 s. Sigg, L. 2000. Redox potential measurements in natural Waters: Significance, concepts and problems. I J. Schüring, Schulz, H.D., Fisher, W.R., Böttcher, J. og Duijnisveld, W.H.M (eds.): Redox fundamentals, processes and applications. Springer, Heidelberg, Tyskland. 251 s. Smith, E.B. 1995. Basic chemical thermodynamics. Oxford Science publications, Clarendon Press, Oxford. 166 s. Stones, T. 1978. Compensation for an inherent error in the determination of the chemical oxygen demand of sewage. Effluent and Water Treatment journal: 290-292. Sultan, S.M. og A.D. Walmsley 1997. Simultaneous kinetic method for the determination of vitamin C, citrate and oxalate employing the Kalman filter. Analyst, 122:1601-1604. Watts, R.J. og Smith B.R. 1991. Catalyzed hydrogen peroxide treatmentof octachlorobidenzo-pdioxin (occd) in surface soils. Chemosphere, 23, s. 949 – 955. Weast, R.C. 1985. Handbook of chemistry and physics. 66. udg. CRC press, Inc., Florida, USA.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||