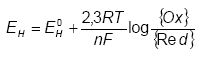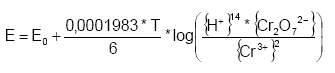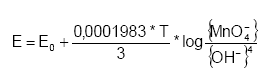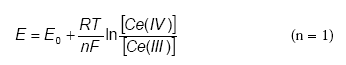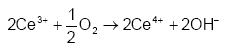|
Metode til analyse af reducerende stoffer i sedimenter 3 Kemiske oxidationsmetoder
3.1 Generel introduktion til redoxkemiI dette afsnit beskrives begreberne oxidationsmiddel, reduktionsmiddel, oxidation, reduktion, oxiderende- og reducerende forbindelser, samt oxidations- og reduktionskapacitet. Et oxidationsmiddel defineres som et stof, der er i stand til at optage elektroner, mens et reduktionsmiddel er et stof der kan afgive elektroner ved reaktion (Bjerrum, 1932). Oxidationsmidler og reduktionsmidler korresponderer til hinanden på lignende måde som syre og baser. Det vil sige at et stof, der reduceres, og har virket oxiderende kaldes et oxidationsmiddel, mens et stof, der oxideres har virket som reduktionsmiddet (eller reduktant) (Bjerrum, 1932). Forløbet kan beskrives som: Ox + ne-→ Red, hvor Ox er oxidationsmidlet, Red er reduktionsmidlet, n er antal og e- er elektroner. Denne ligning kaldes en halvcelle reaktion og skrives for nemheds skyld med den oxiderede forbindelse på venstre side i reaktionsligningen. Da der ikke forekommer frie elektroner kan den ovenstående reaktion kun forløbe, hvis en anden forbindelse stiller elektroner til rådighed for reaktionen, dvs. et reduktionsmiddel skal være tilstede. På denne baggrund kan en halvcelle reaktion ikke eksistere alene. En proces, hvor der foregår både en reduktion og en oxidation, kaldes en redoxproces. Et eksempel på en redoxproces (3) er reaktionen mellem ferrojern (Fe2+) og ilt (O2), der består af to halvreaktioner (1 + 2): Fe2+ + 3H2O →Fe(OH)3 + 3H+ + e- (1) 2H2O →O2 + 4H+ + 4e- (2) 4Fe2+ + O2 + 10H2O →4Fe(OH)3 + 8H+ (3) Processen i ligning (3) kan både benævnes oxidation og reduktion, da begge dele forekommer i ligningen (Schachtschabel et al., 1989). Om reaktionen benævnes som oxidation eller reduktion er derfor afhængig af formålet med reaktionen (Bjerrum, 1932). En forbindelses evne til at oxidere eller reducere er defineret udfra redoxpotentialet EH.
hvor EH0 er redoxpotentialet [V] (i forhold til en standard hydrogen elektrode) under standard betingelser (alle aktiviteter = 1, PH2 = 1 atm, {H+} = 1 M). F er Faradays konstant (96490 C/mol), n er antallet af ombyttede elektroner, R er gaskonstanten (8,314 J/mol*K) og T er temperaturen målt i kelvingrader (°K). Ovenstående ligning kaldes Nernsts ligning og udtrykker den mængde energi, der frigives, når et oxidationsmiddel bliver reduceret i en halvcelle reaktion (Smith, 1995). Måles redoxpotentialet til en positiv værdi er der et overskud af oxiderende forbindelser, mens negative værdier betyder at der er overskud af reducerende forbindelser, jævnfør figur 3.1. Forbindelser med høje positive redoxpotentialer er på denne baggrund gode oxidationsmidler, mens forbindelser med høje negative værdier er gode reduktionsmidler.
Figur 3.1.Typiske redoxpotentialer (mV) i naturlige systemer ved neutrale pH værdier. Redoxpotentialer i den positive retning (op til ca. +800 mV ) indikerer tilstedeværelse af stærke oxidanter, mens negative værdier indikerer tilstedeværelsen af stærke reduktanter (modificeret efter Sigg, 2000). Oxidationskapacitet defineres som det antal iltningsækvivalenter som et molekyle kan afgive når det virker oxiderende. Et ilt-atom repræsenterer 2 iltningsækvivalenter, mens et optag af et brintatom eller en elektron svarer til 1 iltningsækvivalent. Et reduktionsmiddels reduktionskapacitet er på tilsvarende vis bestemt ved antallet af iltningsækvivalenter som reduktionsmidlet optager under reduktionen (Bjerrum, 1932). Oxidations- eller reduktionskapaciteten for en given forbindelse kan variere med ydre omstændigheder, idet redoxreaktionen generelt er påvirket af pH, temperatur, og mængden af den korresponderede reducerede/oxiderede forbindelse. Disse udefra kommende påvirkningers indflydelse på oxidations- eller reduktionspotentialet kan beregnes gennem Nernsts-ligning (Smith, 1995). Tilgængeligheden af reduktanter (reducerende forbindelser) har ligeledes betydning for hvor hurtigt og effektiv den kemiske oxidationproces kan forløbe. Således vil frie reducerede forbindelser hurtigt reagere i oxidationsprocessen, mens høj ionstyrke eller kompleksdannelse betyder, at det bliver vanskeligere for oxidationsmidlet at komme i kontakt med elektrondonorer (Smith, 1995). Tilstedeværelse af reducerede forbindelser bl.a. ferrojern, der findes bygget ind i og beskyttet i lermineralstrukturen kan således have indflydelse på oxidationsprocessen, idet sandsynligheden for at kollidere med det anvendte oxidationsmiddel bliver formindsket. Sedimenter kan således være vanskelige at oxidere fuldstændigt. 3.2 Redoxkemi i sedimenterDe vigtigste redox-aktive forbindelser i forskellige sedimenter omfatter foruden O2 også nitrat, sulfat, organisk stof, forskellige reducerede former af svovl (inkl. pyrit), jern og mangan. Sammensætningen af disse forbindelser afspejles i sedimentets redoxpotentiale. I figur 3.2. er de vigtigste redoxpotentialer for ovennævnte forbindelser vist. Det ses på figuren at redoxpotentialerne er meget afhængig af pH. Samtidig ses det, at de reducerede forbindelser generelt kun har lave redoxpotentialer (0 til ca. – 300 mV), hvilket betyder der skal anvendes et stærkt oxidationsmiddel, hvis der skal ske en redoxreaktion med disse forholdsvise svage reducerende forbindelser.
Figur 3.2. Redoxpotentialer for nogle af de vigtigste redoxaktive forbindelser i sedimenter for henholdsvis pH 7 (mørke pile) og pH 8 (lyse pile). Redoxpotentialerne er beregnet for aktiviteterne {red} = 1 og {Ox} = 1; og i forbindelse med Fe(II) og Mn(II), er {Mn2+} = 1x10-6 og {Fe2+} = 1x10-6 (Sigg, 2000). 3.3 Metode til bestemmelse af reducerende forbindelser i sedimenterIndholdet af reducerende forbindelser i jord og sedimenter bestemmes i dag ofte ved enkeltanalyser af udvalgte forbindelser, som eksempelvis indholdet af ferrojern, pyrit og organisk materiale (TOC). De enkelte resultater bliver efterfølgende anvendt til at bestemme sedimenternes samlede pulje af reducerende forbindelser og den aktive pulje af de reducerende forbindelser bestemmes ved forskellen mellem indholdet i den reducerede zone og den oxiderede zone (Ernstsen et al., 2001). Der findes for indeværende kun ganske få analysemetoder, der udviklet med henblik på at undersøge den samlede pulje af reducerende forbindelser i sedimenter (Ernstsen, 2001). Den eneste metode, der så vidt vides er afprøvet på danske sedimenter er udviklet af Pedersen (1992) til bestemmelse af den totale nitratreduktionskapacitet (TRC-værdi) i jord og grundvand. Denne metode er baseret på vådkemisk oxidation af reducerende forbindelser med en blanding af kaliumdichromat (K2Cr2O7) og svovlsyre. Metoden indebærer ifølge Pedersen (1992) en total nedbrydning af organiske forbindelser, samt reducerende forbindelser, heriblandt Fe(II), pyrit og Mn(II). Resultaterne af ovennævnte metode for både sandede og lerede sedimenter viste ifølge Pedersen (1992) et sammenhæng imellem de bestemte TRC-værdier og og sedimentprøvernes geokemiske miljøer, således havde reducerede sedimentprøver højere TRC-værdier end oxiderede sedimentprøver. På baggrund af TRC-værdier og totale indhold af carbon (TOC) konkluderede Pedersen (1992) således lerholdige sedimenter hovedsageligt kan tilskrives organisk materiale udfældet på eller adsorberet til lerpartiklerne. De højere TRC-værdier der generelt findes for lerede sedimenter i forhold til sandede sedimenter tilskriver Pedersen (1992) således lerpartiklernes høje specifikke overfladeareal og øgede sorptionsegenskaber. Graversen et al. (1990) vurderede imidlertid, at metoden gav realistiske TRC-værdier for sandede sedimenter med organisk stof, men at metoden for øvrigte sedimenter gav urealistiske høje værdier. I begrænset omfang har kaliumpermanganat (Siegrist et al., 2002, Chambers et al., 2000) og forskellige cerium(IV)-holdige forbindelser (Kammer et al., 2000) været anvendt til bestemmelse af sedimenters indhold af reducerende forbindelser. Ligeledes har persulfat været anvendt ved analyse af sedimenters indhold af reducerende forbindelser - og dermed mulige reduktionskapacitet over for nitrat (Pedersen, 1992). 3.4 Analysemetoder til bestemmelse af reducerende forbindelser i sedimentprøver3.4.1 Oxygen (O2)Ilt (O2) er fra et termodynamisk synspunkt en stærk oxidant i en vandig opløsning: O2 + 4H+ +4e- →2H2O (1,229 V) Reaktionshastigheden af reaktionerne mellem O2 og redoxaktive metaller som Fe(II), og Mn(II) er derimod langsom, da reaktionshastigheden hovedsagelig afhænger af O2's partialtryk i den vandige opløsning, hvilket som udgangspunkt er lavt under normale omstændigheder. Ilt er på denne baggrund ikke anvendelig som oxidationsmiddel, når en hurtig metode ønskes og dette oxidationsmiddel blev derfor ikke yderligere i dette projekt. 3.4.2 Hydrogenperoxid (H2O2)Anvendelse af hydrogenperoxid (H2O2) sker bl.a. ved in situ oxidation af især forurenede grunde med forbindelser efter oliespild, hvor bl.a. napthalen, phenantren, pyren og phenoler er almindeligt forekommende. Hydrogenperoxid har ligeledes forsøgsvis været anvendt ved in situ remediering af chlorerede forbindelser som trichlorethylen og perchlorethylenb (Siegrist et al. 2001; Watts og Smith, 1991; Murphy et al.,1989; Barcelona og Holm, 1991). Undersøgelsen er udført med henblik på at undersøge dels sedimenters forbrug af H2O2 ved in situ oxidation af forurenende organiske forbindelser og dels kinetikken af H2O2 reduktionen. Resultaterne viste at nettoreduktionen af H2O2 er på samme niveau eller svagt overstiger den målte reduktionskapacitet bestemt ved oxidation med kaliumdichromat (K2Cr2O7) i en stærk sur opløsning. Oxidationen af organiske forbindelser under forbrug af H2O2 forløber let i sedimenter ved tilstedeværelsen af jernholdige forbindelser. Hydrogenperoxid danner ved tilstedeværelsen af jern det såkaldte Fenton reagens, der frigiver frie hydroxyl radikaler, som er stærke men ikke specifikt virkende oxidanter, som hurtigt nedbryder mange forskellige organiske forbindelser. Generelt deltager Fe2+/Fe3+ eller andre redoxaktive metaller som henholdsvis elektrondonorer og elektronacceptorer i mange af reaktionerne. Det vil derfor være vanskeligt at anvende H2O2 ved bestemmelse af sedimenters indhold af reducerende forbindelser, da disse redoxaktive metaller vil kunne udgøre en del af de reducerende forbindelser. Herved vil brugen af den metode være begrænset til analyse af organisk stof, som eneste komponent i beregningerne, dersom andre redoxaktive metaller med stor sandsynlighed både vil kunne oxideres og reduceres. Hydrogenperoxid vil under tilsætning af ferrojern oxidere bedst under meget sure forhold (pH 2 – 4). Reagenset bliver ineffektiv under neutrale til basiske forhold og/eller under forhold hvor H2O2 er i stand til at autonedbryde (til H2O og O2) ved kontakt med mineraloverflader, bl.a. karbonater (Siegrist et al., 2001). Brugen af oxidationsmidlet H2O2 under samtidig tilsætning af ferrojern blev ikke undersøgt yderligere dersom ferrojern indgår i puljen af potentielle reducerende forbindelser i sedimenter. 3.4.3 Ozon (O3)Ozon (O3) har ligesom H2O2 været forsøgt anvendt ved in situ remediering og dermed iltning af flere forskellige organiske forureninger. Her knyttes brugen af O3 gerne til teknikker med gennemblæsning af luft i forbindelse med grundvandsforureninger af f.eks. chlorerede forbindelser (Siegrist et al. 2001). Ozon er almindeligt anvendt ved rensning af drikkevand. Da O3 er svagt opløseligt i vand kan denne oxidere forurenende forbindelser direkte eller gennem dannelse af hydroxyl radikaler. Nedbrydningen af O3 er stærkt påvirket af pH-forhold, koncentrationen af O3 og tilstedeværelsen af forbindelser der destruere frie radikaler, primært bikarbonat. I dybereliggende jordlag kan uorganiske forbindelser som OH- og Fe2+ starte eller fremskynde nedbrydningen af O3. For at sikre en effektiv dannelse af radikaler bliver H2O2 ofte tilsat ved anvendelse af O3 (Siegrist et al. 2001). Dersom O3 er meget reaktivt og ustabilt, skal produktionen af O3 helst foregå tæt på brugsstedet hvilket gør det uhensigtsmæssig at anvende i laboratoriet. Dertil kommer, at O3 er giftigt i selv små mængder (Siegrist et al. 2001). På denne baggrund blev det besluttet at brugen af O3 ikke skulle undersøges yderligere som aktiv komponent i den ønskede metode. 3.4.4 Kaliumdichromat (K2Cr2O7)Kaliumdichromat (K2Cr2O7) har tidligere været et alment benyttet oxidatinsmiddel men anvendes nu i langt mindre omfang dersom K2Cr2O7 er giftig og kræftfremkaldende, selv i små doser. Meget forskning vedrørende kemisk oxidation har dog været udført med K2Cr2O7, da denne forbindelse i kemiske sammenhænge er et bedre oxidationsmiddel end for eksempel kaliumpermanganat (KMnO4). Oxidation med dichromat (Cr2O72-) i sur opløsning kan beskrives udfra følgende halvreaktion: Cr2O7- + 14H+ + 6e- ? 2Cr3+ + 7H2O Oxidationspotentialet E (V) kan ved ligevægt beregnes udfra Nernsts ligning:
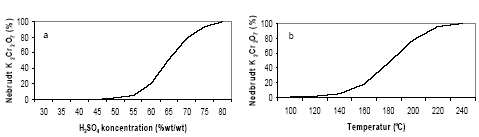
Hvor E0 er standardelektrodepotentialet (V) og T er temperaturen i kelvingrader (°K) (Kolhoff og Steger, 1942). Det fremgår af Nernstes ligning, at ligevægten afhænger af temperatur, pH, samt aktiviteterne af dichromat ionen (Cr2O72-) og chrom-ionen (Cr3+). Temperaturens indflydelse på reaktionen med Cr2O72- er stor og der vil ske en ufuldstændig oxidation af de reducerede forbindelser i prøven ved lave temperaturer, mens der ved høje temperature ske en fuldstændig oxidation. Når K2Cr2O7 udsættes for stærk opvarmning vil der ske en dekomponering af molekylet efter følgende: 4K2Cr2O7 ? 4K2CrO4 + 2Cr2O3 + 3O2 Udfra reaktionsligningen ses det, en opvarmning af K2Cr2O7 i sur opløsning kan producere O2, der forlader opløsningen uden at medvirke til oxidationen af reducerende forbindelser (Pedersen, 1992) og risikoen for dekomponering stiger med stigende temperatur. Således fandt Pedersen (1992), at dekomponering af K2Cr2O7 fandt sted i begrænset omfang ved temperaturer under 140 °C, hvorefter dekomponeringen af K2Cr2O7 stiger eksponentielt til ca. 95% ved 220 C. Ligeledes Stones (1978) har beskrevet denne sammenhæng, figur 3.3. Det vil derfor ikke være hensigtsmæssigt at opvarme K2Cr2O7 til temperaturer højere end 140 °C.
Figur 3.3. Dekomponering af K2Cr2O7 i forbindelse med syrekoncentration og temperatur. a) K2Cr2O7 nedbrydning som funktion af H2SO4 koncentrationen. b) Nedbrydning af K2Cr2O7 i 78,3% H2SO4 som funktion af temperatur (Stones, 1978). Foruden temperaturen har koncentrationen af syre ligeledes stor betydning for dekomponering af K2Cr2O7. Som det fremgår af figur 3.3 stiger dekomponeringen med stigende syrekoncentration. Endvidere er dekomponeringen, som allerede nævnt, også afhængig af Cr2O72- koncentrationen. Således vil koncentrationen af Cr2O72- falde, efterhånden som de reducerede forbindelser oxideres. Hermed bliver dekomponeringen af K2Cr2O7 også afhængig af prøvens indhold af reducerede forbindelser. Da resultatet af analysen således afhænger af forbruget at oxidationsmiddel vil det være hensigtsmæssigt at anvende en temperatur og en syrekoncentration, der begrænser dekomponeringen af Cr2O72- samtidig med at oxidationsforholdene tilgodeses. Under hensyntagen til de nævnte forhold omkring dekomponering findes der i litteraturen flere metoder til oxidation af organisk materiale med K2Cr2O7 som afviger fra hinanden ved forskellige reaktionstider og –temperaturer samt forskelle i syre- og Cr2O72- koncentrationer (Eaton et al., 1995; Pedersen, 1992, Barcelona og Holm, 1992). Således variere de typiske koncentrationer af Cr2O72- mellem 0,05 og 0,2 M og for (svovl)syre mellem på 5– 10 M. Kaliumdichromats evne til at oxidere forskellige organiske forbindelser afhænger meget af de organiske forbindelsers opbygning, idet ikke alle organiske molekyler vil opnå samme grad af oxidation under ens forsøgsbetingelser. Således vil reducerede sukker forbindelser, som eksempelvis glykose, kunne oxideres af K2Cr2O7 uden opvarmning, mens visse heterocycliske forbindelser, som eksempelvis pyridin, kun i ringe grad vil bliver oxideret efter kogning i 2 timer ved 140 °C (Dasgupta og Petersen, 1990). Endvidere er oxidationshastigheden af reducerede forbindelser af første orden med hensyn til både Cr2O72- og den reducerede forbindelse. Dette betyder, at medmindre Cr2O72- er tilstede i et meget stor overskud under en reaktion, vil den mængde reducerede forbindelser, der vil blive oxideret over et givent tidsinterval afhænge af begyndelseskoncentrationen af K2Cr2O7 (Dasgupta og Petersen, 1990). Ved oxidation af sedimenter vil der derfor kunne optræde betydelige forskelle i graden af oxidation bestemt ved især sammensætningen af det organiske materiale og samt begyndelseskoncentrationen af Cr2O72-. 3.4.5 Kaliumpermanganat (KMnO4)Kaliumpermanganat (KMnO4) er et yndet oxidationsmiddel i industrien, idet permanganat ikke bryder carbon skelettet i mange organiske forbindelser under oxidationsprocessen. Når KMnO4 kan anvendes til denne form for oxidation skyldes det at KMnO4 ikke danner frie radikaler. I stedet sker oxidationsprocessen med KMnO4 ved direkte elektron overførsel til reducerede forbindelser. Dette betyder at der skal skabes en direkte kontakt mellem MnO4- og den reducerede forbindelse før reaktionen kan finde sted. Udover de mange anvendelsesformer i industrien, bliver KMnO4 også brugt til in situ remediering af forurende grunde med organiske forbindelser som olie og chlorerede forbindelser (Siegrist et al. 2001). Kaliumpermanganat anvendes desuden også ved måling eksempelvis spildevands kemiske oxygen behov (Chemical Oxgen Demand eller COD) (Eaton et al., 1995, Bigham, 1996, Fresenius et al., 1988). Kaliumpermanganat har potentiale til at oxidere både organiske og uorganiske forbindelser, såsom ferrojern (Fe2+) og reduceret managan (Mn2+) samt naturligt forekommende organisk materiale. Oxidationsprocessen med KMnO4 variere med pH-forholdene og kan beskrives udfra følgende halvcelle reaktioner: MnO4- + 8H + 5e- ? Mn2+ + 4H2O (pH < 3,5) MnO4- + 2H2O + 3e- ? MnO2(s) + 4OH- (pH 3,5 – 12) MnO4- + e- ? MnO42- (pH > 12) Under naturlige forhold kan oxidationspotentialet for KMnO4 beregnes udfra Nernsts ligning:
Hvor E0 er standardelektrodepotentialet (V) og T er temperaturen i kelvingrader (°K) (Kolhoff og Steger, 1942). Det fremgår af Nernsts ligning, at ligevægten afhænger af temperaturen, samt aktiviteterne af MnO4- og den dannede OH-. MnO4--ionen har under naturlige forhold et redoxpotentiale på 0,58 V og et redoxpotentiale på 1,68 V under sure betingelser (Weast,1985), hvilket betyder at KMnO4 er et bedre oxidationsmiddel under sure betingelser end under neutrale forhold Reaktionen mellem MnO4- og reaktanten er meget temperatur afhængige. Forsøg med trichlorethylen viser, at reaktionshastigheden for KMnO4 tredobles når temperaturen øges fra 10 °C til 20 °C (Siegrist et al. 2001). Temperaturafhængigheden skyldes en høj aktiveringsenergi ved redoxreaktioner med KMnO4. Udover temperaturen har især koncentrationen af MnO4- stor betydningen for oxidationen af de reducerede forbindelser. Således viser forsøg at en øget koncentration af MnO4- øger oxidationen af reducerede organiske forbindelser. Årsagen til et øget forbrug af KMnO4 ved 3000 mg/L er ifølge Siegrist et al. (2001) en øget chance for kollision mellem MnO4- og de reducerede forbindelser. Permanganats nedbrydning er relativt upåvirket af pH indenfor intervallet 4 til 8. Ved værdier over 8 har produktionen af OH- en stigende betydning for redoxpotentialet, idet jorden over denne pH-værdi selv indeholder så mange OH- ioner, at reaktionen mellem KMnO4 og reducerede forbindelser begynder at aftage. Derimod vil der ofte i forbindelsen med redoxprocesser dannes brint-ioner (H+), som i ikke bufferede systemer med lav pH, kan medføre en yderligere sænkning af pH, til værdier på 2 til 3 (Siegrist et al., 2001). Dette drastiske fald i pH kan påvirke redoxreaktionerne for en række forbindelser, der findes naturligt i jorden. Permanganat findes i to forskellige former, enten bundet til natrium (NaMnO4) eller til kalium (KMnO4), der er lige effektive som oxidanter. Ifølge Siegrist et al. (2001) er der ikke forskel på reaktiviteten af de 2 former og det kan derfor udfra et kemisk synspunkt være underordnet hvilken af de to forbindelser, der vælges som oxidationsmiddel. Når KMnO4 reagerer med organiske eller reducerede uorganiske forbindelser under natulige forhold, dannes manganoxid (MnO2). Mange naturligt forekommende organiske forbindelser reagerer med MnO4-. Forsøg viser imidlertid at visse dele af det organisk stof ikke bliver fuldstændigt oxideret selv ved meget høje koncentrationer af permanganat eller ved langvarige forsøg, måske fordi det beskyttes af manganoxid. Permanganat kan udover frie ioner også reagere med reducerede forbindelser der er enten kompleksbundet eller adsorberet til den faste fase. Disse reaktioner foregår dog ikke ved sammen hastighed som med frie forbindelser, da bindingen af de reducerede forbindelser resulterer i en stigning i redoxreaktionens aktiveringsenergi og hvorved reaktions hastigheden nedsættes. 3.4.6 Cerium(IV)Anvendelsen af Ce(IV) salte som oxidationsmiddel har været kendt siden 1950'erne, men først i begyndelsen af 1980'erne blev Ce(SO4)2 forsøgsvis anvendt til COD målinger af spildevand (Golterman og Wisselo, 1981). Efterfølgende har Ce(SO4)2 forsøgsvis været anvendt til bestemmelse af nitratreduktionskapaciteten i sedimenter og grundvand (Kammer et al., 2000). Begge metoder er således udviklet som alternativ til de traditionelle metoder, hvor K2Cr2O7 og KMnO4 anvendes som oxidationsmiddel. Metoderne har dog aldrig fundet større anvendelse og der findes nuværende tidspunkt kun få resultater der kan dokumentere Ce(IV) egnethed som oxidationsmiddel i sedimenter og grundvand. Meget lidt vides derfor generelt om reaktioner mellem Ce(IV) og organiske og uorganiske forbindelser i jord og sediment. Opmærksomheden er i stedet vendt mod mere simple systemer, hvor Ce(IV) er anvendt til oxidationsmiddel. I kemiske sammenhænge er Ce(IV) mest kendt i forbindelse med redoxtitrering af Fe(II), hvor potentiometrisk (elektrokemisk) titrering med Ce(IV) er en standardiseret metode til bestemmelse af Fe(II) indholdet i forskellige væsker. Denne titrering involverer en elektron overførsel mellem de to redox aktive forbindelser som vist ved følgende ligning:
Reaktionen er reversibel udfra et termodynamisk synspunkt, men under normale forhold er Fe(III) ikke i stand til at omdanne Ce(III) til Ce(IV), da denne reaktion har meget høj aktiveringsenergi (Bott, 2000). Oxidationspotentialet af Ce(IV)/Ce(III) er 1,61 V (relativt til standard hydrogen elektroden) under standard betingelser. Cerium(IV) forbindelser er således en stærkere oxidant end f.eks. Au(III) og kan dermed reduceres selv af svage reducerende forbindelser, f.eks. Fe(II) salte i sur opløsning eller af H2O2 (Kolthoff og Steger, 1942). På figur 3.4 ses, et eksempel på en redoxtritrering af Ce(SO4)2 med Fe2+.
Figur 3.4. Redoxtitrering af Fe(II) med Ce(IV) (efter Kolthoff og Steger, 1942). I forbindelse med bestemmelse af jorders indhold af reducerende forbindelser er det nødvendigt at tilsætte et overskud af Cerium(IV)sulfat til jordprøverne for at sikre en høj oxidation af de reducerede forbindelser. Under normale omstændigheder skal dette overskud være mindst muligt, så den overskydende mængder af Ce(SO4)2 er så begrænset som muligt. Dette er dog ikke tilfældet for Ce(IV), idet Ce(III) dannet under oxidationen, sænker redoxpotentialet i henhold til ligningen:
hvorved overskuddet af Cerium(IV) ikke må blive for småt. Det bør derfor tilstræbes, at anvende koncentrationer af oxidationsmidlet, der er væsentligt højere end jordens indhold af reducerende forbindelser. Anvendelsen af Ce(SO4)2 til COD-bestemmelse er ifølge Golterman og Wisselo (1981), at Ce(IV) har et højere oxidationspotentiale end K2Cr2O7 og KMnO4, hvorved Ce(SO4)2 er mere termodynamisk egnet til oxidationsmiddel. Værdierne vist i tabel 3.1 antyder ligeledes at oxidationspotentialet af både Ce(IV) og K2Cr2O7 stiger med stigende syrekoncentration, hvilket bl.a. kan forklares ved den høje aktiveringsenergi der karakteriserer begge oxidationsmidler. Den høje aktiveringsenergi bevirker således at der for at sikre en total oxidation af specielt organiske forbindelser i sedimenter tilsættes en katalysator og/eller at reaktioner foregår under høj varme. I følge Golterman og Wisselo (1981) kan Ce(SO4)2 anvendes til oxidation af de fleste uorganiske forbindelser ved stuetemperatur under brug af en passende katalysator, for eksempelvis osmiumtetraoxid (OsO4) hvorimod organiske forbindelser ofte kræver både varme og en passende katalysator. Tabel 3.1. Oxidationspotentiale (V) i 1 M Ce(IV) i forskellige uorganiske syrer målt i forhold til en normal hydrogen elektrode. De viste syrer er perchlorsyre (HClO4), salpetersyre (HNO3) svovlsyre (H2SO4) og saltsyre (HCl) Til sammenligning er oxidationspotentialet af K2Cr2O7 vist i parentes hvor data foreligger (data fra Kolthoff og Belcher, 1957).
Overstiger koncentrationen af Ce(SO4)2 i opløsningen ca. > 0,1 M danner Ce(SO4)2 komplekser med svovlsyre, hvorved opløsningen bliver dyb rød. Disse komplekser er forholdsvis stabile og reagerer kun langsomt med reducerede forbindelser, og det er derfor ønskværdigt at anvende lavere koncentrationer af Ce(SO4)2. Komplekserne kan ligeledes dannes, hvis opløsningen indeholder høje koncentrationer af H2SO4. Tilstedeværelsen af katalysator eller anvendelse af opvarmning er derfor påkrævet når en stærk opløsning af Ce(SO4)2 skal reagere i opløsninger, der indeholder høje koncentrationer af H2SO4 (Golterman og Wisselo, 1981). Resultater af Sultan og Walmsley (1997), viser imidlertid, at reaktionshastighederne for reaktion imellem Ce(IV) og nogle organiske syre som eksempelvis myresyre (HCOOH) og eddikesyre (CH3COOH) nedsættes betydeligt ved stigende koncentrationer af H2SO4, som følge af kompleksdannelse mellem Ce(IV) og sulfat. Ved anvendelse af Ce(SO4)2 som oxidationsmiddel i sedimenter er det vigtigt, at tage højde for et eventuelt indhold af kalk (CaCO3) i sedimenterne, da atmosfærisk ilt (O2) kan interferere på forbruget af Ce(SO4)2. Denne interferens skyldes, at Ce(III) dannet under oxidationen af reducerende forbindelser i basiske opløsninger kan oxideres kvantitativt af opløst O2 til Ce(IV), udfra reaktionen:
og derved regenerere oxidationsmidlet, hvormed der opstår en unøjagtig bestemmelse af jordprøvens indhold af reducerende forbindelser (Golterman og Wisselo, 1981). På denne baggrund er det nødvendigt at tilsætte syre til sedimentprøverne, så prøverne opnår en pH-værdi under 7. 3.5 Valg af mulige oxidationsmidler og oxidationsmetodeMed udgangspunkt i ovenstående gennemgang af mulige kemiske oxidationsmidler blev det besluttet at afprøve brugen af K2Cr2O7, KMnO4 og Ce(SO4)2 i udviklingen af e analysemetode. Ved brugen af K2Cr2O7 var det i høj grad muligt at bygge videre på allerede eksisterende metodebeskrivelser, mens der for KMnO4 og Ce(SO4)2 kun forelå få og ufuldstændige undersøgelsesresultater og for disse to oxidationsmidler har det været nødvendigt, at gennemføre betydelige indledende undersøgelser af mere grundlæggende karakter.
|
||||||||||||||||||||||||||||||