Serietitel nr. XX, 2000
Udvikling af analysemetode til bestemmelse af Polycykliske Aromatiske Hydrocarboner (PAH'er) i jord
Indholdsfortegnelse
1. Indledning
| 1.1 | Miljøstyrelsens oplæg |
| 1.2 | Overordnede rammer for projektet |
Miljøstyrelsen har under Teknologiprogrammet for jord- og grundvandsforurening udbudt
følgende projekt:
Udvikling af analysemetode til bestemmelse af Polycykliske Aromatiske Hydrocarboner (PAH’er) i jord.
VKI afgav i oktober 1998 tilbud på dette projekt. Miljøstyrelsen har i brev dateret den 20. november 1998 betroet VKI udførelsen af opgaven.
Denne rapport er udarbejdet på grundlag af projektoplægget fra Miljøstyrelsen, arbejdsgruppens indstillinger til styregruppen samt VKI's erfaring med metodeudviklingsprojekter, herunder også for Miljøstyrelsen, og vort brede kendskab til analyser af jordprøver.
1.1 Miljøstyrelsens oplæg
I Miljøstyrelsens oprindelige udbudsmateriale var følgende krav til udviklingsopgaven fastsat:
Der skulle udvikles en standardmetode til analyse af PAH i jord, som vil være i stand til at opfylde kravene til jordkvalitetskriteriet for 7 udvalgte PAH-forbindelser. De 7 PAH-forbindelser fluoranthen, benz(b+k+j)fluoranthen, benz(a)pyren, dibenz(a,h)anthracen og indeno(1,2,3-cd)pyren skulle kunne bestemmes således, at jordkvalitetkriteriet på 1,5 mg/kg TS for summen af de 7 samt de individuelle jordkvalitetskriterier på 0,1 mg/kg TS for benz(a)pyren og dibenz(a,h)anthracen kunne overholdes.
Der skulle udvikles en robust ekstraktionsmetode for PAH i jord, hvor flere metoder skulle undersøges med henblik på at minimere ekstraktionstiden og forbedre de arbejdsmiljømæssige forhold i forbindelse med ekstraktions-solventet specielt med henblik på at erstatte brugen af toluen og dichlormethan.
Der skulle fastlægges en målemetode for PAH i de opnåede ekstrakter.
Endelig skulle der fremstilles minimum ét referencemateriale, som kunne danne basis for kontrollen af ekstraktionsmetodens effektivitet.
1.2 Overordnede rammer for projektet
For at tage hensyn til de procedurer og apparaturforhold, der eksisterer på de danske miljølaboratorier samtidig med arbejdet for at udvikle og optimere analysemetoden, foreslog VKI i projektoplægget at tilknytte en arbejdsgruppe bestående af erfarne analysekemikere til projektet. I forbindelse med Miljøstyrelsens accept af denne arbejdsgruppe blev der udarbejdet nogle retningslinier for gruppen, som fastlægger dens formål og kompetence.
Kommissorium for den rådgivende arbejdsgruppe
Arbejdsgruppen bør bestå af 4 udvalgte fagligt kvalificerede personer fra relevante og repræsentative danske miljølaboratorier samt 2 personer fra VKI, som varetager funktionerne som henholdvis formand (projektlederen) og sekretær.
Arbejdsgruppen har ingen reel kompetence til at træffe beslutninger for PAH projektet, men skal alene rådgive projektets styregruppe, som træffer de endelige beslutninger på grundlag af arbejdsgruppens indstillinger samt den kontrakt, som er indgået mellem VKI og Miljøstyrelsen.
Arbejdsgruppens primære arbejdsområder
| 1) | Rådgive styregruppen med hensyn til GC-MS-parametre (dvs. GC-kolonnevalg, MS-indstillinger, opnåelig analysetid og injektionsteknik, samt hvilke PAH-forbindelser der fortrinsvis bør moniteres foruden de af Miljøstyrelsen allerede fastsatte). |
| 2) | Rådgive med hensyn til hvilke interne standarder hhv. surrogatstandarder der bør bruges i metoden. |
Arbejdsgruppens sekundære arbejdsområder
| 3) | Vurdere, hvorvidt der bør bruges oprensningstrin i metoden og i givet fald hvilke (på baggrund af de udførte eksperimenter). |
| 4) | Vurdere de valgte ekstraktionsmetoders anvendelighed og relevans. |
| 5) | Vurdere forsøgsplanlægningen i forbindelse med optimeringsforsøg for ekstraktions- og GC-MS-metoderne. |
Det er forudsat, at gruppen skal afholde 4 møder i løbet af projektperioden fra 20/11-98
til 20/5-99, men antallet af møder kan dog beskæres eller forøges afhængigt af
arbejdsgruppens skønnede behov. Formandens opgave er at lede arbejdsgruppens møder og
tilrettelægge gruppens arbejde. Sekretæren forbereder dagsordenen og udarbejder korte
beslutningsreferater fra møderne, som tilsendes medlemmerne af arbejdsgruppen samt
styregruppen. De fire øvrige gruppemedlemmer skal sikre en faglig kvalitetssikring af den
kommende analysemetode samt varetage de øvrige danske miljølaboratoriers interesser.
Arbejdsgruppens medlemmer
Arbejdsgruppens medlemmer er:
Nis Hansen, Miljø-Kemi, Albertslund
Jens Rasmussen, Miljø- og Levnedsmiddelkontrollen, Helsingør
Pia Sjelborg, Danmarks Jordbrugsforskning, Flakkebjerg, Slagelse
Tore Vulpius, MS Consult, Skovlunde
Hans Peter Dybdahl, VKI, Hørsholm
Susan Bennetzen, VKI, Hørsholm
Søren Bøwadt, VKI, Hørsholm
Denne gruppe har afholdt tre møder i projektperioden og har i den tid formuleret nogle anbefalinger til projektets styregruppe, som i store træk er blevet fulgt, og som afspejles i de følgende afsnit. Heraf er de vigtigste anbefalinger at benytte GC-MS til bestemmelsen af PAH, hvilke surrogatstandarder der bør bruges, og hvilke PAH-forbindelser metoden bør medbestemme.
2. Fremskaffelse og fremstiling af referencematerialer
Indsamling
Fra 2 jordrensefirmaer har VKI modtaget et udvalg af 5 naturlige jordprøver forurenet med PAH og andre hydrocarboner. Ud af disse fem prøver er så udvalgt to prøver af en sandet jordtype fra Jylland, med et relativt lavt indhold af PAH (men stadig højere end jordkvalitetskriteriet) og hydrocarboner (olie), som er blandet sammen til en prøve for at få en tilstrækkelig mængde. Yderligere er der udtaget en prøve af en leret jordtype fra Sjælland med et højt indhold af PAH og hydrocarboner (olie). I alt modtog VKI ca. 200 L af hver type til videre forarbejdning.
Formaling og homogenisering af jordprøver
De to prøver blev derefter behandlet hver for sig på følgende måde:
| Sten og grenstumper større end ca. 25 mm blev fjernet ved håndsortering og bortskaffet. Derefter resterede ca. 75 % af de oprindelige prøver. | |
| Den resterende våde jord blev tørret i varmeskab på bakker ved max. 105 şC | |
| Den tørrede jord blev derefter knust til < 8 mm i et valseværk, blandet og derefter formalet i en ATOX 3.5 mølle, således at > 99 % af jorden har en partikelstørrelse mindre end 90 µm. Ved hver formaling blev 10 – 30 kg af det første materiale bortkastet for at sikre mod forurening og sammenblanding fra tidligere prøver samt for at tillade indstilling af møllen til den ønskede partikelstørrelse. Under formalingen er der samtidig sket en vis opblanding af prøverne. | |
| Efter formalingen er hver prøve blandet yderligere og neddelt i 8 delprøver. |
Sterilisering
De formalede og homogeniserede prøver blev derefter steriliseret med gamma-bestråling for at nedbringe kimtallet i prøverne. Før bestrålingen var kimtallet i prøverne omkring 2-3 x 105 pr gram, mens det efter behandlingen lå under detektionsgrænsen på 100 kim pr. 1,6 g jord.
Påfyldning
Jorden blev derefter fyldt på 250 mL metalbeholdere med 100 g ± 1 g i hver beholder. Påfyldningen foregik i rentrum for at undgå forurening og krydskontaminering af prøverne. Herunder har man taget specielle hensyn for at undgå afblanding (lagdeling) af prøverne.
Den sandede jordtype med lavt forureningsniveau er mærket med følgende label:
Reference Material |
Den lerede jordtype med højt forureningsniveau er mærket med følgende label:
Reference Material |
Til fastlæggelse af en certificeret værdi skal der gennemføres en ekstern certificering af materialet med deltagelse af kvalificerede laboratorier. Databehandlingen kan foretages efter de metoder, VKI har udviklet i forbindelse med produktion af andre referencematerialer.
En certificering er ikke omfattet af denne udviklingsopgave.
3. Fremgangsmåde
3.1 Udvikling og afprøvning af GC-MS-metode til bestemmelse af PAH i jord
Formålet med udvælgelsen og validering af målemetoden for PAH i jord har været at opnå en analysemetode, som er generelt anvendelig for så mange PAH-forbindelser som muligt, og som har de lavest mulige detektionsgrænser og den bedst mulige reproducerbarhed.
Til vurdering af ekstraktionsmetoderne skulle målemetoderne være fastlagt på forhånd, og der måtte ikke ske ændringer i målemetoden undervejs. Derfor var arbejdsgruppens første og primære opgave at fastlægge de generelle retningslinier for en målemetode. På baggrund af den brede anvendelse af GC-MS i danske miljølaboratorier og kravene til minimering af solventforbrug, tid og detektionsgrænser besluttede arbejdsgruppen at anbefale brugen af GC-MS til målemetode for analysen.
Der blev opstillet følgende krav til målemetoden:
| Detektionsmetoden skal gøre det muligt at opnå en detektionsgrænse på en 1/10 del af jordkvalitetskriteriet på 0,1 mg/kg TS for enkeltforbindelser | |
| Metoden skal som minimum kunne bestemme de 7 PAH’er omfattet af jordkvalitetskriteriet, men bør desuden kunne analysere for de 16 EPA PAH’er, som bruges over næsten hele verden | |
| Reproducerbarheden for metoden skal være bedre end ± 10% | |
| Analysetiden bør være maksimalt en time pr. prøve, men det tilstræbes at opnå en analysetid på ca. 30 min. |
VKI skulle beskrive kravene til en eller flere analysemetoder på en sådan måde, at analyserne kan gennemføres på andre laboratorier med en tilsvarende analysekvalitet, herunder specificere de generelle forhold, der har betydning for resultaterne, og de krav til instrumentets/metodens ydeevne, som laboratorierne skal opfylde i forbindelse med analyserne. Denne metode er vedlagt som Bilag 1 til denne rapport.
3.1.1 Udvikling af GC-MS metode
Følgende instrumenter har været benyttet under udviklingen af denne analysemetode:
Et gaskromatografisk system med temperaturstyring, kapillarkolonne og splitless injection (pulsed splitless injector samt trykstyring af bæregassen anbefales for bedre reproducerbarhed). On-column injection ved hjælp af deaktiveret forkolonne kan også benyttes, blot det sikres, at ekstrakterne har tilstrækkelig renhed, og de kromatografiske betingelser er tilpasset denne injektionsteknik.
Et massespektrometer med mulighed for SIM (single ion monitoring) og tilkoblet datasystem, der tillader dataopsamling og lagring af alle data, der optages i det kromatografiske forløb. Massespektrometre, der fungerer efter ion-trap-princippet, kan også benyttes, såfremt tilstrækkelig følsomhed og specificitet dokumenteres. Datasystemet skal kunne søge datafilerne for ioner med specifikke masser og skal kunne udskrive ionresponset i forhold til tiden eller scan-nummer. Datasystemet skal kunne integrere såvel som re-integrere signalet for ethvert ekstraheret ion.
Anvendelsesområde og analyseforbindelser
Den udviklede metode kan benyttes til at bestemme indholdet af PAH i jord. PAH omfattet af denne metode er de 7 forbindelser som kontrolleres i forbindelse med kravene til jordkvalitetskriteriet fastlagt i Vejledningen fra Miljøstyrelsen: Prøvetagning og analyse af jord, nr. 13, 1998.
| US-EPA PAH | Jordkvalitets-PAH |
| Naphthalen | |
| Acenaphthylen | |
| Acenaphthen | |
| Fluoren | |
| Phenanthren | |
| Anthracen | |
| Fluoranthen | Fluoranthen |
| Pyren | |
| Benz(a)anthracen | |
| Chrysen/triphenylen | |
| Benzo(b+j+k)fluoranthen | Benzo(b+j+k)fluoranthen |
| Benz(a)pyren | Benz(a)pyren |
| Indeno(1,2,3-cd)pyren | Indeno(1,2,3-cd)pyren |
| Dibenzo(a,h)anthracen | Dibenzo(a,h)anthracen |
| Benzo(ghi)perylen |
Metoden kan endvidere benyttes til de 16 ovenstående EPA Priority Pollutants. Reelt er
det 18 PAH, da benzo(j)fluoranthen ikke hører til EPA PAH-forbindelserne, men kun meget
vanskeligt kan adskilles fra henholdvis benzo(b)fluoranthen og benzo(k)-fluoranthen.
Desuden kan chrysen under normale omstændigheder ikke adskilles fra triphenylen – se
afsnit 4.
I figur 3.1.1a er vist et GC-MS-kromatogram med PAH-standarder, hvoraf de nævnte 16 (18)
EPA PAH’er er markerede med navn.
Interferenser
Da mange PAH-forbindelser har identisk molekylevægt og struktur samt fysisk-kemiske egenskaber, der ligner hinanden meget, er det vanskeligt fuldstændig at undgå interferens i bestemmelsen af PAH (Bjørseth et al. (1985). Det er derfor af stor vigtighed, at laboratoriet ved GC-MS-bestemmelsen benytter optimale kromatografiske betingelser for at opnå størst mulig separation mellem muligt interfererende forbindelser (Hyver et al., 1989). Da forskellige kromatografiske kolonner har forskellig separation af individuelle PAH-forbindelser, og da separationen desuden er stærkt afhængig af det anvendte temperaturprogram, er det op til det enkelte laboratorium at sikre mindst mulig interferens i bestemmelserne. I de tilfælde, hvor interferens ikke kan undgås, bør der angives en sum af de individuelle forbindelser samt klart angives, hvilke forbindelser det drejer sig om. De bedst kendte interferenser på normale GC-kolonner er chrysen og triphenylen samt benzo(b)fluoranthen, benzo(j)fluoranthen og benzo(k)fluoranthen. Disse forbindelser angives normalt som en sum af benzo(b+j+k)fluorenthen og chrysen/triphenylen (se figur 3.1.1a).
Figur 3.1.1a Se her
GC-MS-kromatogram af PAH-standarder med markering af de 16 (18) EPA PAH-forbindelser.
Kalibreringsstandarder
Autentiske PAH-standarder for de 16 (18) EPA PAH eller de 7 jordkvalitets-PAH (se figur 3.1.1a) sammenblandes og fortyndes til fem eller seks kalibreringsopløsninger i intervallet 0,05 til 10 mg/L. Det er vigtigt at benytte det samme solvent, som ekstraktionen ender op i, for at undgå varierende diskriminering i GC-injektionen (Hyver et al. 1989).
Surrogatstandarder, fortrinsvis i acetone eller et andet letflygtigt og polært solvent, sættes til prøver inkl. kontrol- og blindprøver inden ekstraktion, svarende til en koncentration i prøven på 0,5 - 1 mg/kg (5-10 µg pr. surrogat PAH til 10 g prøve). Tilsætningen foretages minimum 10 min. inden ekstraktionen for at lade opløsningsmidlet fordampe og surrogatstandarderne absorberes i prøven.
Surrogatstandardblandingen skal indeholde følgende forbindelser:
Stof |
Normal surrogat |
Alternativ |
| Naphthalen | Naphthalen-d8 | Biphenyl-d10 |
| Acenaphthylen Acenaphthen |
Biphenyl-d10 | Phenanthren-d10 |
| Fluoren Phenanthren Anthracen |
Phenanthren-d10 | Biphenyl-d10 |
| Fluoranthen Pyren |
Fluoranthen-d10 | Pyren-d10 |
| Benz(a)anthracen Chrysen/triphenylen Benz(b+j+k)fluoranthen Benz(a)pyren |
Benz(a)pyren-d12 | Dibenzo(ah) anthracen-d14 |
| Indeno(1,2,3-cd)pyren Dibenzo(ah)anthracen Benzo(ghi)perylen |
Dibenzo(ah) anthracen-d14 |
Benz(a)pyren-d12 |
I figur 3.1.1b er vist et GC-MS-kromatogram af de i tabellen anførte surrogatstandarder.
samt sprøjtestandarden.
Sprøjtestandard
Tilsætningen af sprøjtestandard er frivillig i denne metode, men anbefales for at kunne skelne mellem problemer ved ekstraktion og GC-MS-bestemmelse (Means, 1998).
Anthracen-d10 opløst i acetone eller cyclohexan afhængig af ekstraktionssolvent, sættes til ekstrakter og standarder til en slutkoncentration i ekstraktet på 3 mg/L, f.eks. 50 µL af en opløsning på ca. 60 mg/L til 1,00 mL ekstrakt.
GC-MS parametre
Ekstraktet analyseres ved gaskromatografi med massespektrometrisk detektion ved anvendelse af single ion monitoring (GC-MS-SIM) (VKI, 1996).
For hver komponent moniteres 2 ioner. Primær-ionen anvendes ved beregning, sekundær-ionen anvendes til verifikation af identiteten.
Figur 3.1.1b Se her!
GC-MS-kromatogram af deutererede PAH-surrogatstandarder
og sprøjtestandard.
De benyttede gaskromatografiske betingelser:
| Gaskromatograf: | Temperaturprogrammerbar og trykstyret |
| Injektor: | Pulsed splitless 1µL |
| Injektionspuls: | 35 psi/2 min., ved 280° C |
| Kolonne: | 5% phenyl methyl silicone eller tilsvarende, 30 m, 0,25 mm ID, 0,25 µm film |
| Bæregas: | He, 1,6 ml/min., svarende til en bæregashastighed på 46 cm/s, som holdes konstant ved hjælp af elektronisk trykregulering |
| Kolonneindgangstryk: | 12,8 psi ved 40şC (trykket afpasses efter starttemperaturen) |
| Detektortemperatur: | 280° C |
| Temperaturprogram: | 40-50° C (acetone) eller 65-75 ° C (cyclohexan), 2 min. 10° C/min., til 285° C30° C/min., til 310° C, 5 min. |
| Total analysetid: | ca. 32 min. |
| Detektion: | Karakteristiske masser (m/z), SIM mode (se det medfølgende metodeudkast i Bilag 1) |
Bemærk: Det er vigtigt at benytte en starttemperatur, der svarer til det solvent, som
benyttes til injektionen, for at opnå de bedst mulige kromatografiske betingelser. Hvis
der ikke benyttes elektronisk trykstyring af bæregassen, er det af afgørende betydning
for separationen af de enkelte PAH-forbindelser at sikre sig, at der holdes et starttryk
ved lav temperatur, som resulterer i en bæregashastighed på over 20 cm/s ved
sluttemperaturen (Hyver et al., 1989).
I figur 3.1.1c og d er vist et GC-MS-kromatogram af henholdsvis referencematerialet på højt og lavt niveau, hvor de beskrevne GC-MS-parametre har været benyttet.
Kalibrering
Der analyseres kalibreringsopløsninger for alle komponenter inkl. surrogatstandarder. Der anvendt 6 niveauer, da standardkurven ofte ikke er lineær ved GC-MS. Kalibreringen er foretaget ved en non-lineær kalibrering ved brug af en kvadratisk kalibreringsfunktion hørende til softwaret til instrumentet.
Efter kalibreringen kontrolleres denne ved genberegning af de brugte standarder. Afvigelsen fra den teoretiske koncentration bør efter genberegning ikke overstige 10% generelt og må ikke overstige 25% i hele det beregnede interval.
Laboratoriet har sikret sig, at koncentrationerne i de analyserede ekstrakter ligger inden for det valgte kalibreringsinterval. Ellers blev ekstrakterne fortyndet og analyseret på ny.
3.1.2 Validering af VKI-standarder
De standarder, som VKI har benyttet ved alle GC-MS-bestemmelser, er blevet valideret over for et certificeret referencemateriale fra NIST i USA. Som det ses i nedenstående Tabel 3.1.4a, er der god overensstemmelse mellem NIST's SRM 2260 og VKI's standarder, således at man vha. VKI's standarder gennemsnitligt finder 103% af den certificerede værdi. På denne baggrund skønnes det påvist, at de på VKI brugte standarder lever op til de kvalitetskrav, der kræves for at kunne dokumentere og validere en reference-metode.
Figur 3.1.1c Se her!
GC-MS-kromatogram af ekstrakt fra referencematerialet på højt niveau.
Figur 3.1.1d Se her!
GC-MS-kromatogram af ekstrakt fra referencematerialet på lavt niveau.
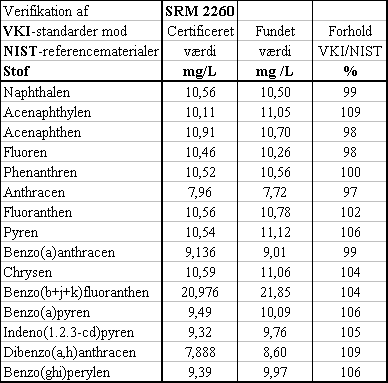
Tabel 3.1.2
Sammenligning mellem VKI standarder og SRM 2260 fra NIST
Linearitet
I forbindelse med alle analyser er kalibrering af GC-MS foretaget vha. ovenstående VKI-standarder. Kalibreringen er foretaget efter en kvadratisk non-lineær kalibreringsprocedure i intervallet 0,05 – 10 mg/L for at undgå kalibreringsfejl i det højeste og laveste niveau, hvor GC-MS for PAH ofte viser tegn på non-linearitet. I hvert tilfælde har alle de analyserede forbindelser haft en regressionskoefficient på over 0,99. Foruden de normale kalibreringsstandarder er også surrogatstandardernes linearitet undersøgt efter de samme retningslinier og med samme resultat (r > 0,99). Det vil sige at metodens kalibreringsforhold er i orden, hvilket også vises af den førnævnte validering af VKI's standarder.
Den beskrevne GC-MS-procedure har været benyttet under hele projektperioden til omkring 300-400 analyser af standarder, kontrol og blindprøver samt prøveekstrakter uden andre forholdsregler end rutinemæssig service af det benyttede instrument med brug af den samme GC-kolonne uden synlig forringelse af performance og separationsevne.
3.2 Udvikling af ekstraktionsprocedure for PAH i jord
For ekstraktion af faste prøver for PAH’er er der beskrevet mange forskellige ekstraktionsprocedurer med anvendelse af forskellige solventer/solventblandinger og med anvendelse af forskellige teknikker (Schantz et al., 1997; Heemken et al., 1997; Fisher et al., 1997; Dupeyron et al., 1999; Letellier et al.,1999).
Formålet med undersøgelsen af allerede kendte og udvikling af nye ekstraktionsmetoder til PAH i jord har været at opnå en så generelt effektiv og pålidelig ekstraktionsprocedure som muligt ved brug af den mindst mulige tid og arbejdskraft.
De miljø- og arbejdsmiljømæssige forhold ved de afprøvede metoder har indgået i overvejelserne om, hvilke metoder der i sidste ende kan anbefales. Herunder er inddraget opløsningsmidlernes egenskaber, mængder, toksicitet og mulige eksponeringstrin.
VKI har i forbindelse med dette projekt afprøvet en række forskellige procedurer, herunder de mere traditionelle med Soxhlet og rystebordsmetoder, samt mere moderne metoder med anvendelse af automatiserede instrumenter og begrænset solventforbrug. VKI har afprøvet Superkritisk Ekstraktion (SFE) med anvendelse af CO2 som solvent, Accelerated Solvent Extraction (ASE), mikrobølgeovnsekstraktion (MAE) samt en moderne variant af Soxhlet’en, nemlig Soxtec. VKI har under udførelsen af projektet draget nytte af samarbejdet med nogle af verdens førende forskere inden for brugen af disse metoder og har dermed erhvervet "State of the Art"-viden, som er kommet projektet til nytte.
Til de traditionelle ekstraktioner ved brug af rystebordsmetoder har de normale solventer været afprøvet som referencegrundlag samtidig med alternative solventblandinger (eksempelvis pentan:acetone og cyclohexan:isopropanol) med henblik på at skaffe dokumentation for disses anvendelighed. Endvidere er forholdene omkring tilsætning af vand eller vandig pyrophosphatopløsning i forbindelse med rystemetoderne inddraget. I forbindelse med valg af ekstraktionsprocedurer har det været inddraget, hvorvidt proceduren kan anvendes som rutinemetode på laboratorier, der gennemfører rutineanalyser for PAH’er.
De følgende generelle krav har været retningsvisende for udvælgelsen af de metoder, der benyttes i det endelige metodeforslag:
| Genfinding af total og individuelle PAH’er skal være højere end 85% i forhold til den mest effektive metode | |
| Reproducerbarheden ved metoden bør være bedre end ± 10% | |
| Ekstraktionstiden skal være mindre end 4 timer | |
| Der skal anvendes de mindst mulige solventmængder | |
| Der vælges de arbejdsmiljømæssigt mest hensigtsmæssige solventer | |
| Metoderne må ikke anvende chlorerede solventer | |
| Surrogat-genfindingen bør være så høj som mulig for at øge metodens robusthed | |
| Det tilstræbes at ekstraktionsmetoderne automatiseres så meget som muligt med henblik på at mindske de arbejdsmiljømæssige påvirkninger |
3.2.1 Rystemetoder
Ekstraktionsforsøg
Rystebord benyttet: IKA HS 501 Digital, 250 ryst/min.
De følgende metoder blev udvalgt på baggrund af deres generelle udbredelse i analytiske laboratorier til brug for flere forskellige forbindelser samt for at dække så mange forskellige solventblandinger som muligt. Da de fleste af metoderne er forskellige, beskrives disse hver for sig (bortset fra acetone:pentan og acetone:hexan, hvor metoderne er identiske) med angivelse af antallet af udførte ekstraktionsforsøg.
For alle ekstraktionsforsøg gælder følgende procedure for tilsættelse af surrogatstandarder og intern sprøjtestandard:
Surrogatstandarder: Alle prøver tilsættes deuteriummærkede surrogatstandarder før ekstraktionen, svarende til en slutkoncentration i ekstraktet på 1 ppm (100m L af en opløsning på ca. 100 ppm).
Intern sprøjtestandard: Der udtages 1 mL ekstrakt, som tilsættes 50 m L anthracen d10, 60 ppm i acetone, svarende til 3 ppm i slutkoncentration.
Metode 1
Rystebordsmetode med brug af cyclohexan:isopropanol (2:1) som ekstraktionsmiddel
| Prøvemængde | Ekstraktionsmiddel | Ekstraktionstid | Analyser udført |
| 10g + 7 mL H2O | Cyclohexan:iso- Propanol (2:1), 8 mL x 3 |
30 min., gentages x 3, vaskes med H2O | 2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind |
Fremgangsmåde: Der afvejes ca. 10 g prøve i en 50 mL flaske med skruelåg.
Prøven tilsættes 7 mL H2O og 8 mL cyclohexan/2-propanol (2:1). Prøven
behandles 5 min. på ultralydsbad + 30 min. på rystebord (250 ryst/min.) Derefter
centrifugeres prøven i 10 min. (3000 RPM). Den ovenstående væske hældes i en
skilletragt, vandet føres tilbage til jordprøven, og ekstraktionen gentages 2 gange med
8 mL cyclohexan/2-propanol (2:1). De samlede ekstrakter vaskes med 2 x 15 mL analyserent H2O.
Ekstraktet filtreres herefter gennem faseseparationsfilter med glødet Na2SO4
og inddampes forsigtigt under påblæsning af nitrogen til ca. 5 mL.
Prøven overføres til 10 mL målekolbe, og der fyldes op til mærket med cyclohexan.
Metode 2
Rystebordsmetode med brug af dichlormethan som ekstraktionsmiddel
| Prøvemængde | Ekstraktionsmiddel | Ekstraktionstid | Analyser udført |
| 10 g + 40 mL H2O, pH<2 |
dichlormethan, 150 mL |
2 timer | 2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind |
Fremgangsmåde: Der afvejes ca. 10 g prøve i en 500 mL red cap-flaske. Der
tilsættes 40 mL analyserent H2O, og der justeres til pH<2 med koncentreret
H2SO4. Der tilsættes 150 mL dichlormethan, og prøven behandles 5
min. på ultralydsbad + 2 timer på rystebord (250 ryst/min.). Derefter centrifugeres
prøven i 5 min. (1500 RPM). Prøven overføres til skilletragt, og dichlormethan-fasen
filtreres gennem faseseperationsfilter med glødet Na2SO4 til en
rundbundet kolbe. Prøven inddampes til ca. 5 mL på rotationsfordamper ved forsigtig
indblæsning under nitrogen. Prøven overføres til 10 mL målekolbe, og der fyldes efter
til mærket med dichlormethan.
Metode 3
Rystebordsmetode med brug af toluen og natriumpyrophosphat (0,05 M) som ekstraktionsmiddel
| Prøvemængde | Ekstraktionsmiddel | Ekstraktionstid | Analyser udført |
| 50 + 20 mL 0,05 M natriumpyro-phosphat | Toluen 20 mL |
1 time | 2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind |
Fremgangsmåde: Der afvejes ca. 50 g prøve i en 80 mL centrifugeglas, og der
tilsættes 20 mL 0,05 M natriumpyrophosphat og 20 mL toluen. Prøven ekstraheres 1 time
på rystebord (250 ryst/min.). Prøven centrifugeres og ekstraktet hældes direkte på
samplerglas.
Metode 4
Rystebordsmetode med brug af acetone:pentan (1:1) eller acetone:hexan (1:1) som ekstraktionsmiddel
| Prøvemængde | Ekstraktionsmiddel | Ekstraktionstid | Analyser udført |
| 10 g + 40 mL H2O, pH 10-12 | acetone:pentan (1:1) eller acetone: hexan (1:1), 150 mL |
2 timer | 2 jord som triplikat x 2, 2 kontrolprøver, 1 blind, 2 reagensblind |
Fremgangsmåde: Der afvejes ca. 10 g prøve i en 500 mL red cap-flaske, og der
tilsættes surrogatstandard. Efter mindst 10 min. henstand tilsættes 40 mL H2O,
og der justeres til pH 10-12 med 4M NaOH. Der tilsættes 150 mL
ekstraktionsmiddel, og prøven behandles 5 min. på ultralydsbad + 2 timer på rystebord
(250 ryst/min.). Derefter centrifugeres prøven i 5 min. (1500 RPM). Væskefasen
overføres til skilletragt, og den organiske fase filtreres gennem faseseperationsfilter
med 10 – 15 g glødet Na2SO4 til en rundbundet kolbe.
Ekstraktet inddampes til ca. 5 mL på rotationsfordamper ved forsigtig indblæsning under
nitrogen. Ekstraktet overføres til 10 mL målekolbe, og der fyldes efter til mærket med
acetone. Heraf udtages 1 mL ekstrakt, som overføres til en GC-vial, og der tilsættes en
sprøjtestandard før analyse på GC-MS.
Resultater
Figur 3.2.1a se her
Sammenligning af rysteekstraktionsmetoder for referencejord på højt niveau.
Figur 3.2.1b se her
Sammenligning af rysteekstraktionsmetoder for referencejord på lavt niveau.
I de to ovenstående figurer ses det tydeligt, at ekstraktionseffektiviteten med acetone:hexan og acetone:pentan for begge de to referencematerialer er signifikant bedre end de andre solventblandinger. Da samtidig metoden med cyclohexan:isopropanol er betydelig mere arbejdskrævende, og metoderne med DCM og toluen arbejdsmiljømæssigt er uønskede, betyder det, at kun metoderne med acetone:hexan og acetone:pentan vil være acceptable. Da også hexan som solvent har vist sig at have en højere toksicitet end andre alifatiske hydrocarboner, giver det sig selv, at metoden, som benytter acetone:pentan, vil være at foretrække. Dette er nok en lille overraskelse, da man tidligere altid har benyttet metoder, som brugte DCM og toluen som ekstraktionssolvent.
Det er dog værd at bemærke, at metoden med toluen og pyrophosphat kun gør brug af 20 mL toluen til ekstraktion af 50 g jord, og at metoden samtidig er den nemmeste rent arbejdsmæssigt. Dette gør, at denne metode muligvis kunne foretrækkes i forbindelse med feltarbejde, hvor der er krav om brug af større jordprøver og mindre solventmængder, samtidig med at kravet om kvantitativ genfinding er mere lempeligt.
På baggrund af ekstraktionsforsøgene for de traditionelle rystemetoder er metoden, som gør brug af acetone:pentan, blevet udvalgt på grund af sin høje ekstraktionseffektivitet, moderate arbejdsmiljøbelastning og den simple håndtering.
3.2.2 Soxtec-metoder
Instrument benyttet: FOSS Soxtec Avanti Manuel System 230V.
De følgende metoder blev udvalgt med baggrund i US EPA's metode 3541, som er en ny metode, der er optimeret efter de seneste års videnskabelige litteratur, samt for at dække så mange forskellige solventblandinger som muligt. Da alle metoderne er identiske bortset fra solventblandingen, beskrives disse samlet i nedenstående tabel med angivelse af antallet af udførte ekstraktionsforsøg.
For alle ekstraktionsforsøg gælder følgende procedure for tilsættelse af surrogatstandarder og intern sprøjtestandard:
Surrogatstandarder: Alle prøver tilsættes deuteriummærkede surrogatstandarder før ekstraktionen, svarende til en slutkoncentration i ekstraktet på 1 ppm (100 m L af en opløsning på ca. 100 ppm).
Intern sprøjtestandard: Der udtages 1 mL ekstrakt, som tilsættes 50 m L anthracen d10, 60 ppm i acetone, svarende til 3 ppm i slutkoncentration.
Ekstraktionsforsøg
| Betingelser: | |
| Ekstraktion: | "Boiling" (ekstraktionshylster neddyppet i solvent) 1 time |
| Ekstraktion: | "Rinsing" (solvent drypper ned i ekstraktionshylsteret) 1 time |
| Ekstraktionshylster: | 33 x 80 mm |
| Inddampning: | Til omkring 10 mL (ca. 5 min. afhængig af det brugte solvent) |
| Ekstraktionsmiddelmængde: | 50 mL |
| Prøvemængde | Ekstraktions- temperatur |
Ekstraktionsmiddel | Analyser udført |
| 10 g | 140° C | acetone:hexan (1:1), 50 mL |
2 jord som triplikat, 2 kontrolprøver, 2 blind, 1 reagensblind |
| 10 g | 165° C | cyclohexan, 50 mL |
2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind |
| 10 g | 165° C | cyclohexan:iso- propanol (1:1), 50 mL | 2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind |
| 10 g | 160° C | acetone:cyclohexan (2:1), 50 mL | 2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind |
| 10 g | 140° C | dichlormethan, 50 mL |
2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind |
| 10 g | 140° C | acetone:pentan (1:1), 50 mL |
2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind |
Foruden de ovenfor beskrevne metoder er Soxtec, med brug af acetone:hexan (1:1), benyttet
til ekstraktion af 1 certificeret referencemateriale og er desuden benyttet til alle
stabilitets- og homogenitetstest for de to referencematerialer, der er produceret i
forbindelse med metode-udviklingen for PAH i jord. De i den forbindelse udførte
ekstraktioner er beskrevet i den efterfølgende tabel.
| Prøvemængde | Ekstraktions- temperatur |
Ekstraktions- middel |
Analyser udført |
| 10 g | 140° C | acetone:hexan (1:1), 50 mL | 1 reference HS 4B-Canada x 6 |
| 10 g | 140° C | acetone:hexan (1:1), 50 mL | Stabilitetstest. Dag 0: 3 x Loamy soil HL, 3 x sandy soil LL |
| 10 g | 140° C | acetone:hexan (1:1), 50 mL | Stabilitetstest. Dag 15A: Loamy soil HL 3 dåser, sv. til 3 temp. 20, 0, 37, 37, 20, 0. Dag 15 B: Sandy soil LL: Som dag 15A Dag 15C: Soil HL: 1 dåse 1 best. 2 dåser 1 best. 1 dåse 2 best. Soil LL: Som dag 15C soil HL |
| 10 g | 140° C | acetone:hexan (1:1), 50 mL | Stabilitetstest. Dag 45A: Soil HL:4 dåser, sv til 3 temp. 1
dåse 20, 1 dåse 37, 1 dåse 0, 2 dåser 37, 1 dåse 20, 1 dåse 0. Dag 45B: Soil LL: Som dag 45A Dag 45C: Soil HL: 1 dåse 1 best. 2 dåse. 1 best. 1 dåse 2 best. Soil LL: Som dag 45C soil HL |
| 10 g | 140° C | acetone:hexan (1:1), 50 mL | Segmenteringstest: Dag 80A: 1 dåse ved 37° C. Soil HL: Top-1, Bund-1, Top-2, Midt-1, Bund-2, Midt-2. Dag 80B: Soil LL: Som dag 80A soil HL. |
| 10 g | 140° C | acetone:hexan (1:1), 50 mL | Stabilitetstest. Dag 90A: Soil HL: 4 dåser, sv. til 3 temp.
1 dåse 0, 1 dåse 20, 1 dåse 0, 1 dåse 37, 1 dåse 37, 2 dåser 20. Dag 90B: Soil LL: Som dag 90A |
Fremgangsmåde: Der afvejes ca. 10 g prøve i ekstraktionshylster, som blandes med ca. 10
g vandfrit Na2SO4, og der tilsættes surrogatstandard.
Derefter tilsættes 50 mL ekstraktionsmiddel, og ekstraktionshylsteret anbringes i
Soxtec-apparatet. Prøverne koges 1 time ved 140 - 165°C, hvorefter der skylles 1 time
(reflux), og opløsningsmidlet inddampes i ca. 5 min. til omkring 10 mL.
Efter ekstraktion inddampes prøven forsigtigt under påblæsning af nitrogen til ca. 5 mL. Ekstraktet overføres kvantitativt til 10 mL målekolbe, og der fyldes op til mærket med acetone, hexan eller cyclohexan afhængig af den ekstraktionsmetode, der er brugt. Heraf udtages 1 mL ekstrakt, som overføres til en GC-vial, og der tilsættes evt. en sprøjtestandard før analyse på GC-MS.
Figur 3.2.2a se her
Sammenligning af Soxtec-ekstraktionsmetoder for referencejord på højt niveau.
Figur 3.2.2b se her
Sammenligning af Soxtec ekstraktionsmetoder for referencejord på lavt niveau
På baggrund af resultaterne vist i de ovenstående to figurer kunne det se ud til, at ekstraktionen med cyclohexan er den mest effektive, og at de 5 andre metoder er nogenlunde ligeværdige. Billedet snyder dog lidt, fordi cyclohexan for det første har en stor standardafvigelse og for det andet har en dårlig genfinding af surrogatstandarden, hvilket oftest betyder, at resultatet overkompenseres og derved bliver for stort. Det vil derfor ikke være cyclohexan, der bør foretrækkes, men i stedet enten acetone:hexan, acetone:cyclohexan eller cyclohexan:isopropanol.
Selvom DCM og acetone:pentan giver næsten lige så gode resultater som de tre andre, er effektiviteten dog knap så god, og DCM har desuden en relativt stor standardafvigelse, mens acetone:pentan diskriminerer lidt mod de tunge PAH-forbindelser (se Bilag 2 over resultaterne af de enkelte PAH-forbindelser).
For de tre andre ekstraktionsmetoder, som alle viser næsten identisk ekstraktionseffektivitet, er acetone:cyclohexan lige en anelse bedre og har marginalt bedre standardafvigelse, hvorfor denne foretrækkes. Denne metode har desuden den bedste genfinding af surrogatstandarder og er derfor mere robust i tilfælde, hvor PAH-forbindelserne er bundet stærkere til jorden end i de to fremstillede referencematerialer.
På baggrund af ekstraktionsforsøgene med Soxtec er metoden, som gør brug af acetone:cyclohexan, blevet udvalgt på grund af sin høje ekstraktionseffektivitet, moderate arbejdsmiljøbelastning, og fordi den ser ud til at kunne være mere robust ved ekstraktion af jord, der har stor bindingsenergi.
3.2.3 Accelerated Solvent Extraction-metoder (ASE)
Instrument benyttet: Dionex ASEtm 200 Accelerated Solvent Extractor.
De følgende metoder er blevet udvalgt med baggrund i US EPA's metode 3545a, som er en ny metode, der er optimeret efter de seneste års videnskabelige litteratur (Schantz et al., 1997; Heemken et al., 1997; Fisher et al., 1997). I modsætning til de andre metoder var det ikke for ASE muligt at dække så mange forskellige solventblandinger som for de foregående metoder. Dette skyldes dels, at vi kun havde instrumentet til rådighed i relativ kort tid, dels, at disse ekstraktionsforsøg var blandt de første, der blev udført, hvor effektiviteten af acetone:pentan blandingen endnu ikke var blevet opdaget. Da alle metoderne er identiske bortset fra solventblandingen, beskrives disse samlet i nedenstående tabel med angivelse af antallet af udførte ekstraktionsforsøg.
For alle ekstraktionsforsøg gælder følgende procedure for tilsættelse af surrogatstandarder og intern sprøjtestandard:
Surrogatstandarder: Alle prøver tilsættes deuteriummærkede surrogatstandarder før ekstraktionen, svarende til en slutkoncentration i ekstraktet på 1 ppm (100m L af en opløsning på ca. 100 ppm).
Intern sprøjtestandard: Der udtages 1 mL ekstrakt, som tilsættes 50 m L anthracen d10, 60 ppm i acetone, svarende til 3 ppm i slutkoncentration.
| Betingelser: | |
| Ovntemp: | 100° C. |
| Tryk: | 2000 psi |
| Statisk tid: | 5 min. |
| Ekstraktions-volumen: | 60% af cellevolumen |
| Skyllevolumen: | 60% af cellevolumen |
| Nitrogen-purge: | 60 sek. ved 150 psi |
| Prøvemængde | Cellestr. | Ekstraktions- middel |
Analyser udført |
| 10 g | 11 mL | acetone: dichlormethan |
2 jord som triplikat, 2 kontrolprøver, 2 blind, 1 reagensblind (hydromatrix) |
| 10 g | 11 mL | acetone: hexan |
2 jord som triplikat, 2 kontrolprøver, 2 blind, 1 reagensblind (hydromatrix) |
| 10 g + 1 mL H2O 30 g |
11 mL 33 mL |
acetone: hexan
|
2 jord som triplikat (10 g + 1 mL H2O), 2 jord som triplikat (30 g), 3 kontrolprøver (10 g ), 1 reagensblind (certif. sand) |
| 10 g + 1 mL H2O 30 g |
11 mL 33 mL |
acetone: cyclohexan |
2 jord som duplikat (10 g + 1 mL H2O), 2 jord som triplikat (30 g), 3 kontrolprøver (10 g ), 1 reagensblind (certif. sand) |
Fremgangsmåde: Læg et filter i bunden af cellen, den fyldes den helt op med jord, og der
tilsættes surrogatstandard. Stram cellen godt og sæt den på instrumentets karrusel.
Isæt opsamlingsglas på apparatet, svarende til celle størrelsen. Ekstraktionen foregår
automatisk efter de ovenfor stående parametre. Efter ekstraktion overføres prøven,
efter inddampning med nitrogen, til henholdvis 10 mL målekolbe (10 g prøve), eller 25 mL
målekolbe (30 g prøve). Der fyldes efter til mærket med ekstraktionsmiddel. Hvis
prøven indeholder H2O, tørres prøven med glødet Na2SO4
inden inddampning.
Figur 3.2.3a se her
Sammenligning af ASE-ekstraktionsmetoder for referencejord på højt niveau
*) Resultatet er et gennemsnit af 2 prøver
Figur 3.2.3b se her
Sammenligning af ASE-ekstraktionsmetoder for referencejord på lavt niveau
I forbindelse med ekstraktionsforsøgene med Accelerated Solvent Extraction var udgangspunktet, som med alle de andre moderne metoder, at finde i den nye U.S. EPA-metode, som gør brug af enten acetone:hexan eller acetone:DCM. Men da ingen af de to solventblandinger er acceptable i arbejdsmiljømæssig henseende i Danmark (specielt ikke DCM), blev det besluttet at benytte en blanding af acetone:cyclohexan som alternativ på prøvemængder på 30 gram i stedet for de 10 gram, der ellers var benyttet tidligere. Dette skyldes, dels at der hidtil ikke har været rejst problemer i arbejdsmiljøhenseende, og dels at cyclohexan tidligere ofte har været anvendt til ekstraktion af PAH i jord. Som det ses af de to ovenstående figurer, resulterede det i ekstraktionseffektiviteter, som var lige så gode som for de to andre solventblandinger fra U.S. EPA-metoden.
Som et sidste forsøg blev der tilsat 1 mL vand til 10 g prøve inden ekstraktion med både acetone:hexan og acetone:cyclohexan for at eftervise, om vandet ville påvirke ekstraktionseffektiviteten for ASE, som det har været nævnt fra flere sider (Heemken et al., 1997; Fisher et al., 1997). Umiddelbart virkede det, som om tilsætningen af vand gav en øget genfinding, men ved nærmere eftersyn skyldes det højere resultat en lavere surrogatgenfinding. Forsøget blev derefter gentaget uden brug af surrogatstandarder for at vurdere den ukorrigerede genfinding.
Disse forsøg gav en gennemsnitlig genfinding på ca. 60 –70% eller samme niveau som genfindingen af surrogater i de første forsøg. Det vil sige: Hvis man korrigerer med surrogatgenfindingen, får man resultater, som var meget tæt på resultaterne for de tørrede prøver. Dette er vist i den sidste kolonne i begge figurerne, hvor surrogatgenfindingen er estimeret på baggrund af genfindingen i de første forsøg. Dette betyder, at ekstraktionseffektiviteten for prøver tilsat vand i forbindelse med ekstraktionen generelt er lavere end for tørre prøver. Selvom vand, som er bundet naturligt i jord, ikke virker på samme måde som vand tilsat umiddelbart før ekstraktionen, betyder dette, at man skal være forsigtig med at ekstrahere prøver, der har et naturligt indhold af vand på mere end 10%, medmindre prøverne blandes med et effektivt tørringsmiddel for at opsuge det overskydende vand.
På baggrund af ekstraktionsforsøgene med Accelerated Solvent Extraction er metoden, som gør brug af acetone:cyclohexan, blevet udvalgt på grund af sin høje ekstraktionseffektivitet og moderate arbejdsmiljøbelastning, som samtidig giver mulighed for at benytte større prøvemængder uden tab af ekstraktionseffektivitet.
3.2.4 Mikrobølgeekstraktionsmetoder
Instrument benyttet: CEM MARS 5 Microwave Accelerated Reaction System.
De følgende metoder blev udvalgt med baggrund i US EPAs metode 3546, som er en ny metode, der er optimeret efter de seneste års videnskabelige litteratur, samt for at dække så mange forskellige solventblandinger som muligt (Dupeyron et al., 1999; Letellier et al., 1999). Da alle metoderne er identiske bortset fra solventblandingen, beskrives disse samlet i nedenstående tabel med angivelse af antallet af udførte ekstraktionsforsøg.
For alle ekstraktionsforsøg gælder følgende procedure for tilsættelse af surrogatstandarder og intern sprøjtestandard:
Surrogatstandarder: Alle prøver tilsættes deuteriummærkede surrogatstandarder før ekstraktionen, svarende til en slutkoncentration i ekstraktet på 1 ppm (100 m L af en opløsning på ca. 100 ppm).
Intern sprøjtestandard: Der udtages 1 mL ekstrakt, som tilsættes 50 m L anthracen d10, 60 ppm i acetone, svarende til 3 ppm i slutkoncentration.
| Betingelser: | |
| Mikrobølgeenergi: | 1200 Watt (100%), (14 ekstraktionsbeholdere) |
| Ekstraktionstemperatur: | 110° C |
| Opvarmningstid til operationstemperatur: | 10 min., temperatur holdes: 10 min. |
| Ekstraktionscelletype: | XP-1500, 100 mL |
| Prøvemængde | Ekstraktionstid | Ekstraktionsmiddel | Analyser udført |
| 10 g + 1 eller 2 mL H2O |
10 min. | acetone:hexan (1:1), 25 mL |
2 jord som duplikat (1 mL H2O), 2 jord som
duplikat (2 mL H2O), 2 kontrol (1 mL H2O), 2 kontrol (2 mL H2O) 1 reagensblind (25 mL solvent + 1 mL H2O) |
| 10 g + 2 mL H2O |
10 min. | acetone:hexan (1:1), 25 mL |
2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind (25 mL solvent) |
| 10 g + 2 mL H2O |
10 min. | dichlormethan, 25 mL |
2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind (25 mL solvent) |
| 10 g + 2 mL H2O |
10 min. | cyclohexan, 25 mL |
2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind (25 mL solvent) |
| 10 g + 2 mL H2O |
10 min. | cyclohexan:isopropanol (1:1), 25 mL | 2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind (25 mL solvent) |
| 10 g + 2 mL H2O |
10 min. | acetone:pentan (1:1), 25 mL |
2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind (25 mL solvent) |
| 10 g + 2 mL H2O 30 g + 6 mL H2O |
10 min.
10 min. |
acetone:hexan (1:1), 25 mL acetone:hexan (1:1), |
1 reference HS 5-Canada x 6 2 jord som triplikat |
| 10 g + 2 mL H2O 30 g + 6 mL H2O |
20 min.
20 min. |
acetone:hexan (1:1), 25 mL acetone:hexan (1:1), |
1 reference HS 5-Canada x 6 2 jord som triplikat |
Fremgangsmåde: Prøven afvejes direkte i ekstraktionbeholderen og tilsættes
surrogatstandard. Efter henstand i mindst 10 min. tilsættes først 2 mL vand og dernæst
ekstraktionsmiddel. Efter ekstraktion afventes nedkøling af ekstraktionscellerne i ca. 30
min., således at trykket igen er gået af ekstraktionscellerne, og prøven tørres med
ca. 10 g vandfrit Na2SO4. Herefter overføres ekstraktet
kvantitativt, efter inddampning med nitrogen, til henholdvis 10 mL målekolbe (10 g
prøve) eller 25 mL målekolbe (30 g prøve). Der fyldes efter til mærket med
ekstraktionsmiddel. Heraf udtages 1 mL ekstrakt som overføres til en GC-vial, og der
tilsættes en sprøjtestandard før analyse på GC-MS.
Resultater
Figur 3.2.4a se her
Sammenligning af mikrobølgeekstraktionsmetoder for referencejord på højt niveau.
Figur 3.2.4b se her
Sammenligning af mikrobølgeekstraktionsmetoder for referencejord på lavt niveau.
På baggrund af resultaterne vist i de ovenstående to figurer kunne det se ud til, at ekstraktionerne med acetone:hexan, DCM, cyclohexan og acetone: pentan er nogenlunde ligeværdige. Billedet snyder dog lidt, fordi cyclohexan for det første har en dårlig genfinding af surrogatstandarden og dernæst en stor standardafvigelse for surrogaten, hvilket betyder, at metoden må betragtes som værende ustabil. Det vil derfor ikke være cyclohexan, der bør foretrækkes, men i stedet enten acetone:hexan eller acetone:pentan. Ekstraktionen med DCM ser ud til at give gode og reproducerbare resultater, men da solventet er helt uacceptabelt, kan metoden kun bruges som reference for de andre metoder. Da resultaterne for acetone:hexan og acetone:pentan er praktisk taget identiske og deruden har gode og stabile surrogatgenfindinger, er det klart, at sidstnævnte foretrækkes på grund af mindre problemer for arbejdsmiljøet.
På baggrund af ekstraktionsforsøgene ved hjælp af mikrobølgeovn (MAE) er metoden, som gør brug af acetone:pentan, blevet udvalgt på grund af sin høje ekstraktionseffektivitet, moderate arbejdsmiljøbelastning og en god metoderobusthed.
3.2.5 Superkritiske ekstraktionsmetoder
Instrument benyttet: Hewlett Packard HP 7680T.
Oprindelig var det meningen at SFE-forsøgene skulle tage udgangspunkt i US EPAs metode 3560, som er en metode, der benytter instrumentet HP 7680T, som har været benyttet af en del laboratorier over hele verden. Metoden bygger på tre ekstraktioner med CO2 under forskellige betingelser og brug af små mængder af vand og organisk solvent. Desværre er der sket det, at Hewlett Packard har taget instrumentet af markedet i foråret 1999. Strategien måtte derfor ændres, da der kun eksisterer 4-5 af disse instrumenter i Danmark (heraf det ene på VKI), og instrumentet kan derfor ikke danne basis for en standardmetode til brug her i landet.
Metoden blev derfor ændret, så den kunne udføres på HP 7680T, som VKI har til rådighed, men også umiddelbart kunne overføres til andre instrumenter, som stadig sælges. Superkritisk Ekstraktion (SFE)af PAH har ofte det problem, at de lette PAH’er relativt let ekstraheres med ren CO2 som ekstraktionsmiddel, mens de tunge kun vanskeligt ekstraheres, medmindre der benyttes meget høje temparaturer (Bøwadt et al., 1995; Benner, 1998). Hvis man derimod benytter CO2 iblandet en modifier, ekstraheres de tunge PAH’er meget bedre, men derved mistes let de flygtige PAH’er (detaljeret teknisk forklaring som afhænger af instrumentet, der benyttes).
Derfor benyttedes under denne metodeafprøvning to metoder: en metode, som benytter ren CO2, og en metode, som benytter CO2 iblandet en lille mængde methanol som ekstraktionsmiddel. Som for alle ekstraktionsforsøg gælder følgende procedure for tilsættelse af surrogatstandarder og intern sprøjtestandard:
Surrogatstandarder: Alle prøver tilsættes deuteriummærkede surrogatstandarder før ekstraktionen, svarende til en slutkoncentration i ekstraktet på 1 ppm (20m L af en opløsning på ca. 100 ppm).
Intern sprøjtestandard: Der udtages 1 mL ekstrakt, som tilsættes 50 m L anthracen d10, 60 ppm i acetone, svarende til 3 ppm i slutkoncentration.
Instrument HP 7680T Supercritical Fluid Extractor.
| Betingelser: | |
| Ekstraktionstryk: | 355 bar |
| Ekstraktions-temperatur: | 150şC |
| Ekstraktionstid: | 5 min. statisk og 30 min. dynamisk ekstraktion med et flow på 1mL/min. (som flydende CO2) |
Opsamling på fastfasefælde af ODS-materiale med en fældetemperatur på 20şC (ved brug
af ren CO2) og 65şC (ved brug af modifier).
Fastfasefælden elueres med 2 x 1,4 mL acetone til to GC-vials.
| Prøvemængde | Ekstraktionsmiddel | Ekstraktionstid | Analyser udført |
| 2 g 2 g |
Ren CO2, 1mL/min. CO2 + 5% methanol |
5 min. statisk og 30 min. dynamisk for begge metoder |
2 jord x 7, 2 kontrolprøver, 1 blind, 2 reagensblind |
Fremgangsmåde: En prøve på ca. 2 g blandes med 8-10 g granuleret Na2SO4 og fyldes forsigtigt på en 7 mL ekstraktionscelle forsynet med en glasfiberfilterskive (med en porediameter på ca. 1 µm) i begge ender. Ekstraktionscellen forsegles og sættes i instrumentets karrusel. Efter ekstraktion ender prøven op i to 1,8 mL GC-vials. Heraf er den ene vial hældt op i et spidsglas og tilsættes acetone op til 2 mL. Hvis man ønsker at undlade at kontrollere for restindhold af PAH i den anden vial, vil det kunne betale sig at hælde de to vials sammen i et spidsglas og inddampe til 2 mL.
Figur 3.2.5a se her
Sammenligning af superkritiske ekstraktionsmetoder for referencejord på højt niveau.
Figur 3.2.5b se her
Sammenligning af superkritiske ekstraktionsmetoder for referencejord på lavt niveau.
På baggrund af resultaterne vist i de to ovenstående figurer kan det være svært entydigt at vælge den bedste af de to SFE-metoder, da resultaterne er temmelig ens. Generelt giver metoden, som gør brug af ren CO2, dog en anelse bedre genfinding og har desuden har en større robusthed. De to metoder har dog modsatrettet trend mht. ekstraktionseffektivitet for de enkelte PAH-forbindelser. Således giver ren CO2 en høj genfinding af de lette PAH’er og lav genfinding af de tunge PAH’er, mens det modsatte er tilfældet, når der tilsættes methanol som modifier.
Dette har en relativ teknisk forklaring, som har relation til det instrument, der er brugt til ekstraktionsforsøgene. Kort sagt skyldes det, at ren CO2 ikke har tilstrækkelig ekstraktionseffektivitet til at frigøre de tunge PAH’er fra jorden, mens additionen af methanol som modifier øger ekstraktionseffektiviteten for de tunge PAH’er på bekostning af dårlig opsamling af de lette PAH’er (Bøwadt et al., 1995; Benner, 1998; Bøwadt et al., 1997). Dette ville muligvis kunne undgås ved brug af andre instrumenttyper.
I efteråret 1999 vil der blive lavet en nordisk interlaboratorieafprøvning med deltagelse af en del forskellige instrumenttyper, hvoraf nogle burde være i stand til at give bedre resultater end det i denne undersøgelse brugte. En evt. introduktion af SFE i den resulterende metode til ekstraktion af PAH fra jord vil derfor være nødt til at afvente resultaterne fra denne metodeafprøvning.
På baggrund af ekstraktionsforsøgene for Superkritisk Ekstraktion (SFE) er metoden, som gør brug af ren CO2, blevet udvalgt til sammenligning med de andre udvalgte metoder, men den er ikke blevet udvalgt til at indgå i den endelige metode, da resultaterne ikke er tilstrækkelig gode. En evt. introduktion af SFE i den endelige metode for bestemmelse af PAH i jord vil være nødt til at afvente en interlaboratorieafprøvning i Norden i efteråret 1999.
3.2.6 Kontrolanalyser ved hjælp af Soxhlet
Den følgende metode blev udvalgt med baggrund i US EPA's metode 3540c, som er en metode, der benyttes af utallige laboratorier over hele verden. Den bruger normalt enten dichlormethan eller acetone:hexan (1:1) som ekstraktionsmiddel, men på baggrund af erfaringer fra NIST's laboratorium i Gaithersburg i USA er kun den sidste solventblanding benyttet i disse forsøg. Dr. Michele Schantz og Dr. Steve Wise fra NIST, som tæller blandt de førende eksperter i analyse og certificeringer af PAH, har erfaring for, at acetone:hexan har mindst lige så stor ekstraktionseffektivitet som dichlormethan for praktisk taget alle miljømatricer. Som for alle ekstraktionsforsøg gælder følgende procedure for tilsættelse af surrogatstandarder og intern sprøjtestandard:
Surrogatstandarder: Alle prøver tilsættes deuteriummærkede surrogatstandarder før ekstraktionen svarende til en slutkoncentration i ekstraktet på 1 ppm (100m L af en opløsning på ca. 100 ppm).
Intern sprøjtestandard: Der udtages 1 mL ekstrakt, som tilsættes 50 m L anthracen d10, 60 ppm i acetone, svarende til 3 ppm i slutkoncentration.
| Prøvemængde | Ekstraktionstid | Ekstraktionsmiddel | Analyser udført |
| 10 g | 16 timer | acetone:hexan (1:1), 300 mL |
2 jord som triplikat, 2 kontrolprøver, 1 blind, 2 reagensblind |
Fremgangsmåde: Der afvejes ca. 10 g prøve i et ekstraktionshylster, der
tilsættes ca. 10 g Na2SO4. Efter ekstraktion inddampes prøven til
ca. 5 mL på rotationsfordamper ved forsigtig indblæsning under nitrogen. Prøven
overføres til 10 mL målekolbe, og der fyldes efter til mærket med acetone:hexan (1:1).
Resultater
Figur 3.2.6 se her
Ekstraktionsudbytter ved Soxhlet-ekstraktion for begge referencejordtyper
3.2.7 Sammenligning af ekstraktionsmetoder
I det følgende afsnit laves en sammenligning af de udvalgte metoder inden for hver ekstraktionstype for at vurdere dem i forhold til hinanden både med hensyn til ekstraktionseffektivitet for summen af PAH’er samt for individuelle PAH-forbindelser. Desuden gøres der et forsøg på at vurdere alle aspekter af de valgte metoder, såsom arbejdstid, solventforbrug og automatiseringsmuligheder.
Sammenligning af ekstraktionsmetoder for summen af PAH-forbindelser
Ved sammenligning af de 6 metoder vist i de to følgende figurer ses det helt tydeligt, at rystemetoden, Soxtec, ASE og MAE giver resultater, som er praktisk taget identiske for summen af de 16 (18) EPA PAH’er og de 7 MST PAH’er for begge referencejordtyper. Således er der, for summen af EPA og MST PAH’er en relativ standardafvigelse (% RSD) på den opnåede middelværdi for de fire metoder på mellem 1,7 og 4,5%, for begge de to referencematerialer (se Tabel 3.2.7c).
Dette er en hidtil uset høj metodesammenlignelighed for fire uafhængige metoder, som givetvis vil skabe international opmærksomhed i organisk-analytiske kredse. Soxhlet og specielt SFE skiller sig ud med lidt ringere resultater. For Soxhlet er dette dog ikke signifikant i forhold til et 95% konfidensinterval, mens det for SFE er tilfældet for begge jordtyper. Eftersom Soxhlet bruges som reference for kvantitativ ekstraktion i praktisk talt alle laboratorier over hele verden, er det meget positivt, at både rystemetoden, Soxtec, ASE og MAE giver mindst lige så gode eller bedre resultater. Derved opnås en til vished grænsende sandsynlighed for, at de fire nævnte metoder er optimale mht. ekstraktionseffektiviteten for PAH i jord, når der betragtes en sum af 16 (18) eller 7 forbindelser.
Figur 3.2.7a se her
Sammenligning af ekstraktionsmetoder for referencejord på højt niveau; sum af 16 og
7 PAH.
Figur 3.2.7b se her
Sammenligning af ekstraktionsmetoder for referencejord på lavt niveau; sum af 16 og 7
PAH
Sammenligning af ekstraktionsmetoder for individuelle PAH-forbindelser
Ved sammenligning af de 6 metoder vist i de tre følgende tabeller (3.2.7b og d) ses det helt tydeligt, at rystemetoden, Soxtec, ASE og MAE generelt har meget lille spredning mellem metoderne for de individuelle 16 (18) EPA og 7 MST PAH’er for begge referencejordtyper. Kun for sammenligningen af naphtalen ser spredningen af resultaterne ud til at være uacceptabelt stor, idet specielt MAE giver et noget højere resultat for begge referencejordtyper. Dette er dog ikke statistisk signifikant på grund af den store spredning for resultaterne på ASE (skyldes, at der kun er to resultater) som gør, at konfidensintervallerne for de fire metoder overlapper. Den store standardafvigelse mellem metoderne for naphtalen kan dog afhjælpes ved at benytte den alternative surrogatstandard biphenyl d10 i stedet for naphtalen d8 for ASE- og MAE-metoderne. Det er dog ikke gjort i dette projekt for at få den størst mulige metodesammenlignelighed.
For de individuelle EPA og MST PAH’er der en relativ standardafvigelse (% RSD) for de fire metoder på mellem 2 og 40% med en middel-RSD på 10% for begge de to referencematerialer. Også dette er en hidtil uset høj metodesammenlignelighed for fire uafhængige metoder, som givetvis vil skabe international opmærksomhed i organisk-analytiske kredse. Specielt skaber dette opmærksomhed, når det bemærkes at kvantificeringen har et meget stort dynamisk område med en forskel på en faktor ~50 mellem højeste og mindste koncentration i begge referencematerialer. Desuden bemærkes den relative standardafvigelse (% RDS) på ~10% helt nede ved koncentrationer omkring jordkvalitetskriteriet på 0,1 ppm (undtagen for naphthalen og acenaphtylen).
Soxhlet og specielt SFE skiller sig ud med lidt ringere resultater. For Soxhlet er dette dog ikke signifikant i forhold til et 95% konfidensinterval, mens det for SFE er tilfældet for begge jordtyper, specielt for de tunge PAH’er. At de tunge PAH’er giver problemer for SFE, skyldes givetvis problemer med surrogatforbindelserne for disse forbindelser, et forhold, som muligvis hænger sammen med brugen af fastfaseopsamling og elueringen af denne. Det vil sige, at en bedre metodeoptimering for ekstraktion af PAH ved hjælp af SFE sandsynligvis ville kunne løse problemerne for denne metode også, men dette ligger uden for formålet med denne undersøgelse.
Eftersom Soxhlet bruges som reference for kvantitativ ekstraktion i praktisk talt alle laboratorier over hele verden er det meget positivt, at både rystemetoden, Soxtec, ASE og MAE giver mindst lige så gode eller bedre resultater. Derved opnås en til vished grænsende sandsynlighed for, at de fire nævnte metoder er optimale mht. ekstraktionseffektiviteten for PAH i de to referencejordtyper, når man betragter de individuelle 16 (18) eller 7 forbindelser.
Genfinding af kontrolprøver
Tabel 3.2.7a se her!
Gennemsnitlig genfinding fra kontrolprøver for de forskellige metoder
Tabel 3.2.7b se her
Sammenligning af udvalgte ekstraktionsmetoder for referencejord på højt niveau;
individuelle PAH-forbindelser
Tabel 3.2.7c se her!
Sammenligning af udvalgte ekstraktionsmetoder for referencejord på lavt niveau;
individuelle PAH-forbindelser
Tabel 3.2.7d se her!
Sammenligning af de 4 udvalgte ekstraktionsmetoder, rystemetoden, Soxtec-ekstraktion,
Accelerated Solvent Extraction og mikrobølgeekstraktion for referencejord på højt og
lavt niveau; individuelle PAH-forbindelser.
Tabel 3.2.7e se her!
Oversigt over ekstraktionsmetoder
3.2.8 Konklusion for ekstraktionsprocedurerne
På baggrund af de viste resultater for de enkelte ekstraktionsmetoder og sammenligningen mellem dem, er det tydeligt, at fire metoder skiller sig ud ved at have høj ekstraktionseffektivitet, være reproducerbare, have stor sammenlignelighed og give fordele såvel arbejdstidsmæssigt som arbejdsmiljømæssigt. De fire metoder er: Rystemetoden ved brug af pentan:acetone (1:1), Soxtec-ekstraktion ved brug af cyclohexan:acetone (1:1), Accelerated Solvent Extraction (ASE) ved brug af cyclohexan:acetone (1:1) samt mikrobølgeekstraktion (MAE) ved brug af pentan:acetone (1:1).
I forhold til en betragtning om, at den foreslåede metode bør kunne praktiseres i de fleste normalt udstyrede laboratorier i Danmark, er det klart, at den udviklede rystemetode må være hovedmetoden, mens de tre andre metoder kun kan være alternativer (omend ligeværdige), da de kræver instrumentinvesteringer i størrelsesordenen fra 150.000 – 500.000 kr. På den anden side vil det betyde en udviklingsmæssig begrænsning for danske miljølaboratorier, hvis man ikke tillader brugen af de 3 alternative metoder, da disse som vist i Tabel 3.2.7e har fordele rent arbejdstids- og arbejdsmiljømæssigt. Dette kunne betyde store konkurrencemæssige ulemper for danske miljølaboratorier på europæisk plan.
Det anbefales derfor, at metodeforslaget "Bestemmelse af PAH i jord" gennemføres med rystemetoden som hovedmetode og med de tre moderne ekstraktionsmetoder som ligeværdige alternativer. Det anbefales desuden, at metoden medbestemmer både de 7 PAH’er, der benyttes til bestemmelse af Miljøstyrelsens jordkvalitetskriterier (MST PAH’er) og de 16 (18) PAH-forbindelser, der traditionelt bestemmes i henhold til US EPAs metoder.
Hver af de fire metoder har fordele og ulemper, og en empirisk oversigt over disse er givet i Tabel 3.2.7e. Foruden de lidt grove betragtninger i tabellen skal her lige uddybes nogle få ting. Fra tabellen ses det tydeligt, at ASE og MAE nok er de metoder, der i løbet af en arbejdsdag vil kunne resultere i det størst mulige antal færdige ekstrakter. I praksis gælder det for alle metoderne, at det størst mulige antal ekstrakter ligger mellem det beregnet ud fra selve ekstraktionstiden og den tid, det tager for en total prøveforberedelse på en enkelt prøve.
Desuden bør man huske, at sekventielle ekstraktioner i automatiserede systemer oftest har mulighed for at foregå uden opsyn og derfor ikke kræver så megen arbejdskraft. Det bør også betænkes, at selvom nogle af de viste metoder vil kunne producere et meget stort antal færdige ekstrakter pr. arbejdsdag, så vil det i praksis kun være muligt at udføre ~ 40 GC-MS-bestemmelser pr. dag ved hjælp af den i metodeudkastet foreslåede GC-MS-metode (medmindre flere instrumenter benyttes). Det vil derfor være op til det enkelte laboratorium, deres normale opgaver, fremtidige visioner og personalemæssige prioriteter, hvilken metode der benyttes til de rutinemæssige bestemmelser af PAH i jord.
3.3 Stabilitet og homogenitet af referencematerialer
Indledning
En del af projektet har været at fremstille og teste 2 referencematerialer til analyse af PAH i jord.
Homogenitets- og stabilitetstest
Prøver
To prøver er undersøgt for homogenitet og stabilitet i projektperioden: HL SOIL og LL SOIL. Der er gennemført en kombineret stabilitets- og homogenitetstest over 90 dage. Hver prøve er testet som følger:
Vurdering af datamaterialet
Datamaterialet indikerer, at der er enkelte resultater, som afviger fra det øvrige datamateriale. Disse resultater kan påvirke den statistiske vurdering, hvorfor der er taget højde for dette ved datatolkningen nedenfor.
Homogenitet
Homogenitet, svarende til forskel mellem dåser, er undersøgt ved en ensidig F-test på 95%-niveau. Varians mellem dåser er testet over for varians inden for dåser bestemt ved replikatbestemmelser på enkeltdåser. Undersøgelse omfatter tests på 18 dåser i 7 analyseserier, med 22 replikatbestemmelser på 9 dåser, svarende til i alt 11 frihedsgrader for varians mellem dåser og 13 frihedsgrader inden for dåser.
Homogenitet svarende til forskel inden for dåser er undersøgt ved at analysere en dåse, med dobbeltprøver udtaget i toplaget, i midterlaget og bundlaget af prøven, efter opbevaring af prøven uden omrystning. Der er udført F-test på variansen mellem lag over for variansen på replikater af analyser udført ved hvert lag.
Stabilitet
Stabilitet er undersøgt over 90 dage (0, 15, 45, 90 dage) ved prøver opbevaret i køleskab (prøve id. "0"), ved stuetemperatur (prøve id. "25"), og 37 grader (prøve id. "37"). Der er gennemført F-test for forskel i temperatur med bestemmelse af variansen mellem resultater ved de tre temperaturer, poolet over dag 15, 45 og 90, svarende til 2 frihedsgrader. Variansen er testet over for variansen på replikater, bestemt som ved homogenitetstesten. Varians for stabilitet er påvirket af varians for forskel mellem dåser, hvorfor det ikke kan afgøres i denne undersøgelse, om variation i prøvekoncentrationer mellem dåser opbevaret ved forskellig temperatur skyldes inhomogenitet eller ustabilitet. Data er afbildet grafisk med resultater som funktion af tiden for hver temperatur. Herudover er der lavet grafisk afbildning af RT, der er koncentrationsbestemmelse for prøver opbevaret ved henholdsvis 37 og 25 grader i forhold til koncentrationsbestemmelser for prøver ved 5 grader. Herudover er det dertilhørende konfidensinterval (2*UT) indtegnet, hvor
RT = Koncentration af prøve opbevaret ved temperaturen T/Koncentration af prøver opbevaret på køl ("0")
UT=(variansT + varians0)1/2 * RT/100
| VariansT: | Varians for bestemmelse af koncentrationer i prøver opbevaret ved T grader |
| Varians0: | Varians for bestemmelse af koncentration i prøver opbevaret ved referencetemperaturen "0" (køl) |
Denne procedure svarer til BCR's procedure for vurdering af referencematerialers stabilitet.
Konklusioner og rekommandationer
HL SOIL
Tests for homogenitet og stabilitet samt den grafiske optegning af resultaterne indikerer, at prøven HL SOIL er homogen og stabil for næsten alle parametre. Tests for lagdeling indikerer ligeledes, at der ikke er forskel mellem prøver udtaget forskellige steder i en dåse. Der vurderes derfor, at prøven er egnet til en certificering.
De relative standardafvigelser for replikater ligger på omkring 7 pct. ved denne undersøgelse. Det bør sikres ved en evt. opfølgende metodeundersøgelse og certificering på laboratorier, om denne standardafvigelse er tilstrækkelig lav til at teste materialet. Såfremt laboratorierne har en relativ lav standardafvigelse (<3 gange større), bør tests for homogenitet og evt. stabilitet vurderes nærmere og evt. suppleres med yderligere tests.
LL SOIL
De statistiske tests indikerer, at prøven LL SOIL kan være inhomogen og/eller ustabil. Der er en tendens til, at koncentrationen af de enkelte komponenter og summen af PAH (de 16 og de 5) i prøver opbevaret ved høj temperatur falder en anelse i koncentration over perioden (0-20 pct.). Dette er kun en tendens; der er ikke signifikant forskel mellem varians for stabilitet og for forskel mellem dåser. Testene viser herudover, at der ikke er forskel på prøver udtaget forskellige steder i dåsen.
Standardafvigelserne for replikater ved testene er omkring 2-3 pct. Disse standardafvigelser vurderes umiddelbart at være ekstremt lave i forhold til laboratoriernes standardafvigelser ved rutineanalyser af analoge prøver. Det vurderes derfor, at materialet kan være egnet til en certificering, såfremt videre homogenitetstests viser, at standardafvigelsen for eventuel inhomogenitet og/eller ustabilitet er mere end tre gange mindre end laboratoriernes standardafvigelser. Disse undersøgelser kan f.eks. inkorporeres i laboratorieanalyser ved en metodeafprøvning og/eller en certificering.
Tabel 3.3a se her!
Oversigt over stabilitets- og homogenitetstest af referencematerialet på højt niveau (HL
Soil)
Tabel 3.3b se her!
Oversigt over stabilitets- og homogenitetstest af referencematerialet på lavt niveau (LL
Soil).
3.4 Oprensningsteknikker
Ved en omfattende undersøgelse af de udførte analyser for de forskellige ekstraktionsteknikker er det blevet klarlagt, at det ikke er nødvendigt at indføre et oprensningstrin i metoden for at opnå en god reproducerbarhed. Det blev derfor besluttet ikke at undersøge oprensninger i dette projekt. Principielt giver det bedre muligheder for at opnå gode betingelser for målemetoden. Men en mere kompleks analyseprocedure øger muligheden for fejl undervejs, foruden at analyseusikkerheden øges ved flere analysetrin. Såfremt det alligevel måtte ønskes at bruge en oprensningsmetode, findes et stort antal beskrevet i litteraturen.
Et eksempel på en simpel procedure er den følgende (Guerin, 1999): En Pasteur-pipette stoppes med en smule silyleret glasuld og pakkes med ca. 50 mm neutral aluminiumoxid (aktiveret ved 400şC i 4 timer) efterfulgt af 10 – 20 mm tør Na2SO4 som topdække. Pipetten bankes ganske let mod et fast underlag for at sikre en god pakning og renses før brug med ca. 10 mL hexan. Pipetten elueres med 10 mL hexan.
Behovet for en oprensning vil afhænge af både prøvens karakter, ekstraktionsproceduren og målemetoden, hvorfor det ikke kan fastslås på forhånd, hvorvidt det er nødvendigt.
3.5 Validering af den samlede metode til bestemmelse af PAH i jord
Foruden valideringen af de standarder, som VKI har benyttet til GC-MS-bestemmelserne over for en certificeret standardopløsning, og de kontrolforsøg (tilsætningsforsøg med 1 ppm standarder), som er udført kontinuerligt i forbindelse med udvælgelsen af ekstraktionsmetoden, er der yderligere udført forsøg til bestemmelse af detektionsgrænser, og hele metoden er verificeret over for to certificerede referencematerialer.
3.5.1 Bestemmelse af detektionsgrænser og genfinding på lavt niveau
Der benyttes normalt to forskellige metoder til bestemmelse af detektionsgrænser: bestemmelse ved hjælp af blindværdier og bestemmelse ved hjælp af tilsætningsforsøg på højst 5 gange den forventede detektionsgrænse. I dette projekt er begge metoder blevet brugt. Det skyldes dels, at der for hver analyseserie medbestemmes blindværdier, hvorfor disse naturligt kan danne baggrund for en detektionsgrænsebestemmelse, og dels, at det var ønskeligt at få bestemt en genfindingsprocent for prøver med et niveau omkring 5 gange detektionsgrænsen.
Det blev derfor besluttet indledningsvis at bruge blindværdierne fra ekstraktionsforsøgene til at lave en foreløbig detektionsgrænsebestemmelse og så senere at lave den mere arbejdskrævende, men også mere nøjagtige, bestemmelse ved hjælp af tilsætningsforsøg for hovedmetoden. Normalt benytter man mindst 6 blindværdier til bestemmelse af detektionsgrænsen, men i dette tilfælde, hvor der er foretaget et meget stort antal forskellige ekstraktionsforsøg, har vi været nødt til at begrænse antallet af GC-MS-kørsler, og dermed er antallet af medbestemte blindværdier i de forskellige ekstraktionsserier kun på 4-5 for hver metode. Det betyder selvfølgelig, at detektionsgrænsebestemmelsen er behæftet med større usikkerhed (denne baseres på spredningen af de målte værdier og er derfor stærkt afhængig af antallet af målinger), og at den efterfølgende skal verificeres i de laboratorier, der benytter sig af metoderne.
Tabel 3.5.1a se her!
Bestemmelse af detektionsgrænser på baggrund af blindværdier
Tabel 3.5.1b se her!
Bestemmelse af detektionsgrænser og genfindinger for rystemetoden på baggrund af
tilsætning på lavt niveau (5 x detektionsgrænsen)
Som det ses i Tabel 3.5.1a, ligger hovedparten af detektionsgrænserne på mellem 0,01 og 0,02, hvilket på baggrund af nøjagtigheden af bestemmelsen må siges at være tilfredsstillende, da det klart indikerer, at detektionsgrænserne for metoden generelt er omkring 0,01 mg/kg TS eller 1/10 af kravet til jordkvalitetskriteriet. Som tidligere nævnt kræves det dog stadig, at de laboratorier, som benytter metoden verificerer den endelige detektionsgrænse.
At den i Tabel 3.5.1a bestemte detektionsgrænse er usikker, ses tydeligt af bestemmelsen af detektionsgrænsen for rystemetoden beskrevet i Tabel 3.5.1b, da denne tabel verificerer detektionsgrænsen til at være mindre end eller lig med 0,01 mg/kg TS for alle de 16 EPA PAH’er. Dermed er forudsætningen for opretholdelse af et jordkvalitetskriterium på 0,1 mg/kg TS opfyldt.
Af Tabel 3.5.1b ses også den gennemsnitlige genfinding af den seksdobbelte ekstraktion på lavt niveau. En genfinding på i gennemsnit 84% for de 7 MST PAH’er og 88% for de 16 (17) EPA PAH’er er absolut respektabelt, når man betragter det lave tilsætningsniveau samt det faktum, at der er benyttet en naturlig jord til ekstraktionerne. Metoden opfylder derfor de af Miljøstyrelsen fastsatte krav til en referencemetode for bestemmelse af PAH i jord.
3.5.2 Verifikation af den samlede metode ved brug af certificerede referencematerialer
Som en endelig verifikation af den samlede udviklede metode til bestemmelse af PAH i jord er der gennemført en seksdobbelt analyse af to forskellige referencematerialer fra National Research Council i Canada. Begge materialerne er sedimenter, som stammer fra havne i forskellige dele af Canada, og disse materialer siges normalt at være nogle af de vanskeligst ekstraherbare materialer, der kan købes.
Ved det ene referencematreriale HS-4B er der brugt Soxtec-ekstraktion, fordi denne teknik fra starten var forventet at ville give værdier tættest på referencemetoden Soxhlet. De fremkomne ekstrakter er efterfølgende analyseret på GC-MS med to forskellige kalibreringer. En kalibrering på baggrund af VKI's egne standarder og en kalibrering lavet ud fra en certificeret referencestandard fra NIST (SRM 2260).
Som det ses af Tabel 3.5.2a ligger de fundne middelværdier i de fleste tilfælde lidt lavere end certificeringsintervallet for materialet. Men når man bestemmer 95%-konfidensintervallet for de af VKI opnåede resultater, ligger så godt som alle værdier inden for det certificerede interval. Det er desuden værd at bemærke, at der er god overensstemmelse mellem de 95%-konfidensintervaller, der er opnået for VKI- og NIST-standarderne.
Tabel 3.5.2a se her!
Verifikation af Soxtec-metoden ved hjælp af HS-4B-materialet fra NRC.
For referencematerialet HS-5 er der udført seksdobbelte ekstraktioner for både mikrobølgemetoden og rystemetoden for at verificere, at der ligesom for de to af VKI udviklede referencematerialer ikke er signifikant forskel på ekstraktionseffektiviteten mellem metoderne. Resultaterne for HS-5 er vist i Tabel 3.5.2b. Også for dette referencemateriale er der nogle værdier som ligger uden for 95%-konfidensintervallet, men generelt er der et godt overlap med de certificerede værdier. Kun for to PAH-forbindelser (benzo(a)pyren og benzo(g,h,i)perylen) ligger de fundne værdier uden for de certificerede intervaller for begge referencematerialer.
Det er dog ikke usædvanligt, at der er et par forbindelser, for hvilke det kan være meget vanskeligt at opnå de samme resultater som i certifikatet. Det gør sig specielt gældende, når materialerne stammer fra en institution, som udfører certificeringen "in-house" (hvilket er tilfældet for begge de brugte materialer) og ikke benytter flere uafhængige laboratorier til certificeringen, som det praktiseres af bl.a. BCR og NIST. De observerede afvigelser er derfor heller ikke bekymrende, så længe værdierne generelt ligger godt, hvilket er tilfældet her.
På baggrund af de opnåede resultater for de to referencematerialer må det konkluderes, at metoden lever op til de krav, som man må forvente af en moderne analysemetode til rutinebrug i miljølaboratorier, samt lader sig verificere både i forhold til de brugte PAH-standarder og de anvendte referencematerialer.
Tabel 3.5.2b se her!
Verifikation af ruste- og mikrobølgemetoden ved hjælp af HS 5-materialet fra NRC.
4. Konklusion og anbefalinger
I forbindelse med udviklingen af en samlet analysemetode til bestemmelse af PAH i jord er de følgende procedurer og "delafsnit" gennemgået og verificeret:
Der er
| udviklet en GC-MS-metode baseret på single ion monitoring og brug af surrogatstandarder med en detektionsgrænse på 0,01 mg/L. |
Der er
| udviklet og verificeret en hovedekstraktionsmetode og 3 ligeværdige alternativer med usædvanlig stor reproducerbarhed mellem metoderne. |
Der er
| fremstillet 2 kandidatreferencematerialer for PAH i jord med et indhold i intervallet på henholdsvis 0,2-13 mg/kg TS (højt niveau, leret jord) og 0,03-1,6 mg/kg TS (lavt niveau, sandet jord). For materialet på højt niveau er homogenitet og stabilitet blevet verificeret i løbet af projektperioden, mens materialet på lavt niveau kræver yderligere stabilitetstest, før en endelig verificering for stabilitet og homogenitet kan finde sted. |
| Den samlede metode er valideret over for 2 certificerede referencematerialer fra National Research Council i Canada med tilfredsstillende resultater. |
| Standarderne brugt af VKI til alle undersøgelser er valideret mod en certificeret standardopløsning fra NIST (SRM 2260). |
| Detektionsgrænsen for den samlede analysemetode er verificeret for rystemetoden til at være 0,01 mg/kg TS, hvilket er krævet for at kunne opretholde et jordkvalitetskriterium på 0,1 mg/kg TS for individuelle PAH-forbindelser. |
| Den samlede metode bestående af prøveforbehandling, ekstraktionsmetode (1 hovedekstraktionsmetode og 3 ligeværdige alternativer), analysemetode ved hjælp af GC-MS samt kvalitetskontrolprocedurer er beskrevet i et metodeudkast dateret den 6.10.1999. |
| Metodeudkastet er skrevet i en form, som benyttes i forbindelse med internationale standarder, og kræver kun mindre modifikationer samt oversættelse til engelsk for at kunne foreslås som kandidat til en fremtidig ISO-standard. |
Det anbefales, at metoden gælder for 16 (18) EPA PAH-forbindelser, heraf de 7 PAH’er fra Miljøstyrelsens jordkvalitetskriterium i alle jordtyper.
Det anbefales, at metodens reproducerbarhed og nøjagtighed verificeres ved en interlaboratoriemetodeafprøvning ved brug af de udviklede referencematerialer. Denne metodeafprøving vil sandsynligvis kunne benyttes til en verificering af de to materialers homogenitet samt til en certificering af begge materialer, såfremt der opnås tilfredsstillende resultater.
5. Litteratur
Bjørseth, A., Ramdahl, T. (editor) (1985.). Handbook of Polycyclic Aromatic Hydrocarbons, Volume 2. Marcel Dekker inc., New York,.
Hyver, K.J., Sandra, P. (editor) (1989) High Resolution Gas Chromatography, 3. Ed. Hewlett-Packard Co., Delaware.
Means, J.C. (1998). Compound-Specific Gas Chromatographic/Mass Spectrometric Analysis of Alkylated and Parent Polycyclic Aromatic Hydrocarbons in Waters, Sediments, and Aquatic Organisms. Journal of AOAC International, 81 s. 657- 672.
Bestemmelse af PAH i jord og vand ved GC-MS, VKI-metode 0-32. 3. udgave, 20/10-96.
Schantz, M.M., Nichols, J.J., Wise, S.A. (1997) Evaluation of Pressurized Fluid Extraction for the Extraction of Environmental Matrix Reference Materials. Analytical Chemistry, 69, 1997, s. 4210-4219.
Heemken, O.P., Theobald, N.,. Wenclaviak, B.W (1997). Comparison of ASE and SFE with Soxhlet, Sonication, and Methanolic Saponification Extractions for the Determination of Organic Micropollutants in Marine Particulate Matter. Analytical Chemistry, 69, 1997, s. 2171-2180.
Fisher, J.A., Scarlett, M.J., Stott; A.D. (1997). Accelerated Solvent Extraction: An Evaluation for Screening of Soils for Selected U.S. EPA Semivolatile Organic Priority Pollutants. Environmental Science and Technology, 31, 1997, s. 1120-1127.
Dupeyron, S., Dudermel, P-M., Couturier, D., Guarini, P., Delattre, J-M. (1999). Extraction of Polycyclic Aromatic Hydrocarbons from Soil: A Comparison Between Focused Microwave Assisted Extraction, Supercritical Fluid Extraction, Subcritical Solvent Extraction and Soxhlet Techniques. International Journal of Environmental Analytical Chemistry, 73, 1999, s. 191-210.
Letellier, M., Budzinski; H. (1999). Influence of Sediment Grain Size on the Efficiency of Focused Microwave Extraction of Polycyclic Aromatic Hydrocarbons. The Analyst, 124, 1999, s. 5-14.
Bøwadt, S., Hawthorne; S.B. (1995). Supercritical Fluid Extraction in Environmental Analysis. Journal of Chromatography A, 703, 1995, s. 549-571.
Benner, B.A. (1998). Summarizing the Effectiveness of Supercritical Fluid Extraction of Polycyclic Aromatic Hydrocarbons from Natural Matrix Environmental Samples. Analytical Chemistry, 70,1998, s. 4594-4601.
Bøwadt, S., Mazeas, L., Miller, D.T., Hawthorne S.B. (1997). Field-Portable Determination of Polychlorinated and Polynuclear Aromatic Hydrocarbons in Soil Using Supercritical Fluid Extraction. Journal of Chromatography A, 785, 1997, s. 205-217.
Guerin, T.F. (1999). The Extraction of Aged Polycyclic Aromatic Hydrocarbon (PAH) Residues From a Clay Soil Using Sonification and a Soxhlet Procedure: A Comparative Study. Journal of Environmental Monitoring, 1, 1999, s. 63-67.
Bilag 1: Bestemmelse af PAH i jord
Indhold
1 Bestemmelse af PAH i jord
Bestemmelse af polycyklisk aromatisk hydrocarbon (PAH) i jord ved hjælp af gaskromatografi – massespektrometri (GC-MS) efter ekstraktion med Rystemetode (pentan:acetone), Soxtec, Accelerated Solvent Extraction (ASE) eller Mikrobølgeekstraktion (MAE).
2 Referencer
Vejledning fra Miljøstyrelsen: Prøvetagning og analyse af jord, nr. 13, 1998.
Udvikling af analysemetode til bestemmelse af Polycykliske Aromatiske Hydrocarboner (PAH'er) i jord. Rapport fra VKI til Miljøstyrelsen, februar 2000.
| EPA 3540c: | 1996 Soxhlet Extraction |
| EPA 3541: | 1994 Automated Soxhlet Extraction |
| EPA 3545a: | 1998 Pressurized Fluid Extraction |
| EPA 3546: | Microwave Assisted Solvent Extraction (proposed) |
3 Orientering og anvendelsesområde
Denne metode kan benyttes til at bestemme indholdet af PAH i jord. PAH omfattet af denne metode er de 7 forbindelser, som kontrolleres i forbindelse med jordkvalitetskravene fastlagt i Vejledningen fra Miljøstyrelsen: Prøvetagning og analyse af jord, nr. 13, 1998.
| US-EPA PAH | Jordkvalitets-PAH |
| Naphthalen | |
| Acenaphthylen | |
| Acenaphthen | |
| Flouren | |
| Phenanthren | |
| Anthracen | |
| Flouranthen | Flouranthen |
| Pyren | |
| Benz(a)anthracen | |
| Chrysen/triphenylen | |
| Benzo(b+j+k)fluoranthen | Benzo(b+j+k)flouranthen |
| Benz(a)pyren | Benz(a)pyren |
| Indeno(1,2,3-cd)pyren | Indeno(1,2,3-cd)pyren |
| Dibenzo(a,h)anthracen | Dibenzo(a,h)anthracen |
| Benzo(ghi)perylen |
Metoden kan endvidere benyttes til de 16 ovenstående EPA Priority Pollutants. Reelt er
det 18 PAH, da benzo(j)fluoranthen ikke hører til EPA PAH-forbindelserne, men kun meget
vanskeligt kan adskilles fra henholdvis benzo(b)fluorenthen og benzo(k)-fluorenthen.
Desuden kan chrysen under normale omstændigheder ikke adskilles fra triphenylen – se
afsnit 4.
Oversigt over ekstraktionsmetoderne
| Parameter | Rystemetode |
Soxtec |
ASE |
MAE |
| Ekstraktionstid | 2 timer |
2 timer |
15 min. |
20 min. |
| Min. prøveforberedelsestid for enkeltprøve | 3-3,5 timer |
2,5-3 timer |
45 min. |
1-1,5 timer |
| Instrumenttype | Batch |
Batch |
Sekventiel |
Batch |
| Prøvemængde | 10 g a |
10 g a |
10 – 30 g |
10 g |
| Antal prøver pr. kørsel b | 16-20 |
6 |
1 |
14 |
| Automatisering b | Manuel |
Manuel |
Automatisk |
Manuel |
| Antal prøver pr. instrumentopsætning b | 16-20 |
6 |
24 |
14 |
| Ekstraktionssolvent c | pen:ace (1:1) |
cyc:ace (1:1) |
cyc:ace (1:1) |
pen:ace (1:1) |
| Solventmængde pr. prøve | 150 mL |
50 mL |
10-12 mL |
25 mL |
| Tørring med Na2SO4 | Ja |
Nej |
Nej |
Ja |
| Centrifugering | Ja |
Nej |
Nej |
Ja |
| Opkoncentrering efter ekstraktion | Ja |
Ja |
Ja |
Ja |
| Teknikertid | Høj |
Medium |
Medium |
Medium |
| Analysetid | Høj |
Medium |
Lav |
Lav |
| Forbrug af tilbehør | Lav |
Høj |
Lav |
Lav |
a
Det er sandsynligt, at der vil kunne benyttes større prøvemængder, men det er ikke verificeret.b Afhængig af det brugte instrument, da flere versioner forefindes.
c De brugte forkortelser er: pen = pentan, cyc = cyclohexan og ace = acetone.
4 Interferenser
Da mange PAH-forbindelser har identisk molekylevægt og struktur samt fysisk-kemiske egenskaber, der ligner hinanden meget, er det vanskeligt fuldstændig at undgå interferens i bestemmelsen af PAH (Bjørseth et al., 1985). Det er derfor af stor vigtighed, at laboratoriet ved GC-MS-bestemmelsen benytter optimale kromatografiske betingelser for at opnå størst mulig separation mellem muligt interfererende forbindelser (Hyver et al., 1989).
Da forskellige kromatografiske kolonner har forskellig separation af individuelle PAH-forbindelser, og da separationen desuden er stærkt afhængig af det brugte temperaturprogram, er det op til det enkelte laboratorium at sikre mindst mulig interferens i bestemmelserne. I de tilfælde, hvor interferens ikke kan undgås, bør der angives en sum af de individuelle forbindelser samt klart angives, hvilke forbindelser det drejer sig om. De bedst kendte interferenser på normale GC-kolonner er chrysen og triphenylen samt benzo(b)fluoranthen, benzo(j)fluoranthen og benzo(k)fluoranthen (Bjørseth et al., 1985). Disse forbindelser angives normalt som en sum af benzo(b+j+k)fluorenthen og chrysen/triphenylen.
5 Princip
En jordprøve ekstraheres ved hjælp af en af fire alternative ekstraktionsmetoder: Traditionel rystemetode ved brug af en blanding af pentan:acetone, Soxtec, Accelerated Solvent Extraction (ASE) eller Mikrobølgeekstraktion (MAE). Den traditionelle rystemetode betragtes som hovedmetoden, da den ikke forudsætter specielt udstyr, mens de tre moderne metoder kan benyttes som ligeværdige alternativer.
En homogeniseret jordprøve opslemmes i vand, og der tilsættes en blanding af pentan:acetone (1:1), hvorefter prøven kort gives ultralyd og ekstraheres på rystebord. Ekstraktet centrifugeres, dekanteres og tørres efterfulgt af inddampning til passende volumen.
Ekstraktet analyseres ved GC-MS SIM (single ion monitoring). Indholdet beregnes ud fra responset af den specificerede primære ion, og resultatet korrigeres med genfindelsen af de tilsatte surrogatstandarder. Identiteten kontrolleres ved det relative respons af den specificerede sekundære ion.
6 Reagenser og standarder
| 6.1 | Generelt |
| 6.2 | Kemikalier |
| 6.3 | Stamopløsninger |
| 6.4 | Kalibreringsstandarder |
| 6.5 | Surrogatstandarder |
| 6.6 | Sprøjtestandarder |
| 6.7 | Kontrolanalyser |
| 6.7.1 | Blindprøver |
| 6.7.2 | Kontolanalyser |
6.1 Generelt
Alle brugte reagenser skal være af analytisk renhedsgrad og velegnede til formålet. Renheden af reagenser og opløsninger kontrolleres ved analyse af reagens- og procedure blind i hver analyseserie.
6.2 Kemikalier
| 6.2.1 | Natriumsulfat, Na2SO4, eller magnesiumsulfat, Mg2SO4: analytisk renhedsgrad, vandfrit, aktiveret ved opvarmning til 450°C i 16 timer |
| 6.2.2 | Acetone, pentan, cyclohexan - skal være af HPLC eller pesticid renhedsgrad |
| 6.2.3 | Acetone:pentan, i forholdet 1:1 ved sammenblanding af lige store volumen |
| 6.2.4 | Acetone:cyclohexan, i forholdet 1:1 ved sammenblanding af lige store volumen |
| 6.2.5 | Vand, Lichrosolv eller tilsvarende renhedsgrad |
| 6.2.6 | Natriumhydroxid, NaOH: 4 M vandig opløsning |
| 6.2.7 | Autentiske PAH-standarder af dokumenteret renhed eller certificerede opløsninger af blandinger af PAH-forbindelser |
6.3 Stamopløsninger
Fremstil stamopløsninger ved afvejning af omkring 0,0100 g rent stof (6.2.7). Opløs stoffet i acetone eller cyclohexan (6.2.2) og fortynd til 10 mL i en målekolbe (1000 mg/L). Hvis renheden af stoffet er 96% eller større, kan den afvejede mængde bruges uden korrektion til beregning af koncentrationen i stamopløsningen. Kommercielt tilgængelige stamopløsninger kan bruges, hvis de er certificerede af leverandøren eller en uafhængig kilde.
Overfør stamopløsningerne til glasflasker med PTFE-forede skruelåg eller til glasampuller, der lukkes. Opbevares i mørke ved –18°C, eller som leverandøren anbefaler.
Stamopløsninger er normalt holdbare 1 år (afhængig af laboratoriernes kvalitetskrav). Hvis kontrolanalyser i henhold til laboratoriernes interne kvalitetskontrol indikerer et problem, skal stamopløsningerne erstattes tidligere.
6.4 Kalibreringsstandarder
Autentiske PAH-standarder for de 16 (18) EPA PAH eller de 7 jordkvalitet PAH (se afsnit 3 og 6.2.7) sammenblandes og fortyndes til fem eller seks kalibreringsopløsninger i intervallet 0,05 til 10 mg/L. Det er vigtigt at benytte det samme solvent, som ekstraktionen ender op i, for at undgå diskriminering i GC-injektionen.
6.5 Surrogatstandarder
Surrogatstandarder, fortrinsvis i acetone eller et andet letflygtigt og polært solvent, sættes til prøver inkl. kontrol- og blindprøver inden ekstraktion, svarende til en koncentration i prøven på 0,5 - 1 mg/kg (5-10 µg pr. surrogat PAH til 10 g prøve). Tilsætningen foretages minimum 10 min. inden ekstraktionen for at lade opløsningsmidlet fordampe og surrogatstandarderne absorberes i prøven.
Surrogatstandarden skal indeholde følgende forbindelser:
| Stof | Normal surrogat | Alternativ surrogat |
| Naphthalen | Naphthalen-d8 | Biphenyl-d10 |
| Acenaphthylen Acenaphthen |
Biphenyl-d10 | Phenanthren-d10 |
| Fluoren Phenanthren Anthracen |
Phenanthren-d10 | Biphenyl-d10 |
| Fluoranthen Pyren |
Fluoranthen-d10 | Pyren-d10 |
| Benz(a)anthracen Chrycen/triphenylen Benz(b+j+k)fluoranthen Benz(a)pyren |
Benz(a)pyren-d12 | Dibenzo(ah)anthracen-d14 |
| Indeno(1,2,3-cd)pyren Dibenzo(ah)anthracen Benzo(ghi)perylen |
Dibenzo(ah)anthracen-d14 | Benz(a)pyren-d12 |
6.6 Sprøjtestandarder
Tilsætningen af sprøjtestandard er frivillig i denne metode, men anbefales for at kunne skelne mellem problemer ved ekstraktion og GC-MS-bestemmelse (Means, 1998).
Anthracen-d10 opløst i acetone eller cyclohexan afhængig af ekstraktionssolvent sættes til ekstrakter og standarder til en slutkoncentration i ekstraktet på 3 mg/L, f. eks. 50 µL af en opløsning på ca. 60 mg/L til 1,00 mL ekstrakt.
6.7 Kontrolanalyser
6.7.1 Blindprøver
Der skal analyseres mindst to blindprøver i forbindelse med hver analyseserie. Blindprøverne skal indeholde samtlige elementer af analysen startende med punkt 8.4
6.7.2 Kontrolprøver
I hver analyseserie skal medanalyseres to kontrolprøver bestående af det af Miljøstyrelsen krævede referencemateriale til beregning af nøjagtighed samt sW og sB. Desuden laves et kontrolforsøg ved tilsætning af PAH til en ren jordprøve på et niveau mindre end 10 gange detektionsgrænsen for at bestemme genfindingen. Endvidere skal der udføres en dobbeltbestemmelse på en af prøverne til beregning af CVW på naturlige prøver i henhold til Miljøstyrelsens krav i NOVA 2003 moniteringsprogrammet.
7 Apparatur
| 7.1 | GC-MS, gaskromatografi med massespektrometrisk detektion |
| 7.2 | Rysteapparat |
| 7.3 | Soxtec-ekstraktor |
| 7.4 | Accelerated Solvent Extractor |
| 7.5 | Mikrobølgeekstraktor |
| 7.6 | Ekstraktionsglas |
7.1 GC-MS, gaskromatografi med massespektrometrisk detektion
Et gaskromatografisk system med temperaturstyring, kapillarkolonne og splitless injektion (pulsed splitless injektor samt trykstyring af bæregassen anbefales for bedre reproducerbarhed). On-column injektion ved hjælp af en deaktiveret forkolonne kan også benyttes, blot det sikres, at ekstrakterne har tilstrækkelig renhed, og de kromatografiske betingelser er tilpasset denne injektionsteknik.
Et massespektrometer med mulighed for SIM (single ion monitoring) og tilkoblet datasystem, der tillader dataopsamling og lagring af alle data, der optages i det kromatografiske forløb. Massespektrometre, der fungerer efter ion-trap-princippet, kan også benyttes, såfremt tilstrækkelig følsomhed og specificitet kan dokumenteres. Datasystemet skal kunne søge datafilerne for ioner med specifikke masser og skal kunne udskrive ionresponset i forhold til tiden eller scan-nummer. Datasystemet skal kunne integrere såvel som re-integrere signalet for ethvert ekstraheret ion.
Kapillarkolonne med 5% phenyl methyl silicone eller tilsvarende apolær fase samt høj temperaturstabilitet og lav blødning anbefales for at sikre tilstrækkelig stort signal/støj forhold. Kolonnekrav er minimum 25 – 30 m længde og maksimum 0,25 mm indre diameter med en fasetykkelse på 0,1 – 0,25 µm.
7.2 Rysteapparat
Til ekstraktion af prøver i ekstraktionsglas. Ekstraktionsglassene skal kunne fastspændes i liggende position, og der skal kunne ekstraheres i langsgående retning. Minimum 250 ryst pr. minut.
7.3 Soxtec-ekstraktor
Sammenkoblet, automatiseret eller manuel, Soxhlet-instrument med lavt solventforbrug. I det indledende ekstraktionstrin er prøven nedsænket i det kogende solvent for at sikre effektiv kontakt mellem prøven og solventet, hvilket sikrer hurtig ekstraktion af de organiske forbindelser. Herefter løftes prøvehylstret op af solventet og skylles ved reflux som ved Soxhlet. I det tredie trin begyndes inddampning af solventet automatisk for at reducere volumen. Der er to principielle forskelle fra traditionel Soxhlet: Prøven er nedsænket i det kogende solvent og begyndende inddampning, som sker i forbindelse med ekstraktionen. Herved nedsættes analysetiden til ca. 2 timer, og solventforbruget sænkes.
7.4 Accelerated Solvent Extractor
Accelerated Solvent Extractor med 11 mL ekstraktionsceller af rustfrit stål eller andet materiale, der kan modstå de krævede tryk på ca. 2000 psi. Ekstraktionsceller af andre størrelser kan også benyttes, blot det sikres, at cellen fyldes helt enten med prøvemateriale eller med et inert materiale, som begrænser dødvolumen. Andre tilsvarende systemer, som kan demonstrere den nødvendige ydeevne for ekstraktion af PAH i jord, vil principelt også kunne anvendes (Schantz et al., 1997; Heemken et al., 1997; (Fisher et al., 1997).
7.5 Mikrobølgeekstraktor
En mikrobølgeovn med lukkede ekstraktionsceller, der har kemiske, resistente overflader, og som er gennemtrængelige for mikrobølger, så solventet kan absorbere mikrobølge-energien. Mikrobølgeovnen skal være forsynet med en temperaturstyring, så temperaturen i ekstraktionscellerne kan kontrolleres. Ekstraktionen foregår under forhøjet temperatur og tryk. Andre tilsvarende systemer, som kan demonstrere den nødvendige ydeevne for ekstraktion af PAH i jord, vil også kunne anvendes. Der har således været demonstreret gode ekstraktionsevner for instrumenter, der benytter såkaldt fokuserede mikrobølger i åbne systemer med brug af en refluxkondenser (Dupeyron et al., 1999; Letellier et al., 1999). Dette instrument vil også kunne bruges, såfremt ekstraktionseffektiviteten demonstreres.
7.6 Ekstraktionsglas
Det i metoden anvendte glasudstyr skal være renset efter laboratoriets interne regler fulgt af glødning ved 450şC i minimum 16 timer. Til rystemetoden skal anvendes 500 mL red cap-flasker (eller tilsvarende glasflasker med teflonforet låg).
8 Fremgangsmåde
8.1 Prøveudtagning
Prøveudtagning foretages i henhold til Vejledning fra Miljøstyrelsen: Prøvetagning og analyse af jord nr. 13, 1998.
8.2 Opbevaring af prøver
Jordprøver opbevares i en lufttæt beholder i køleskab ved 4° C i mørke, indtil igangsættelse af analysen. Ved længere tids opbevaring af prøver (over en uge) er det ofte en fordel at fryse disse ned ved – 20° C for at undgå mikrobiel aktivitet.
8.3 Forbehandling
Jordprøver findeles og lufttørres i stinkskab i ca. 18 – 24 timer. Herefter homogeniseres prøverne grundigt og sigtes gennem en 2 mm si for at fjerne træstykker, sten og andet prøven uvedkommende materiale. Opmærksomheden rettes dog på det forhold, at indhold af klumper af tjære i prøven må definere denne som værende forurenet. I tvivlstilfælde homogeniseres tjæreklumperne sammen med jordprøven.
Ved den ovenfor omtalte lufttørring burde restvandindholdet i prøven ligge på omkring 10%. Hvis tørstofbestemmelsen indikerer, at jorden stadig indeholder signifikant større mængder vand, lufttørres prøven yderligere eller blandes grundigt med en passende mængde vandfrit Na2SO4 eller MgSO4 (6.2.1) for at sikre en bedre ekstraktionsoverflade og undgå, at prøvens naturlige indhold af vand nedsætter ekstraktionseffektiviteten (gælder primært for de alternative ekstraktionsmetoder ASE og MAE). Tørstofbestemmelse foretages i alle tilfælde ved 105şC indtil konstant vægt.
I de tilfælde, hvor det kræves, at prøverne analyseres hurtigere, end ovenstående procedure tillader, vil prøveforberedelsen kunne foregå på følgende måde, blot laboratoriet i forvejen har verificeret sammenligneligheden:
Til den udtagne prøve tilsættes vandfrit Na2SO4 eller MgSO4 (6.2.1) i tilstrækkelig mængde til at sikre, at prøven ved en efterfølgende grundig homogenisering fremtræder tør, løs og uden klumper. I modsat fald gentages tilsætningen af tørringmiddel, indtil tilstrækkelig tørhed er opnået.
8.4 Ekstraktion
8.4.1 Traditionel rystemetode
Der afvejes ca. 10 g prøve i en 500 mL red cap-flaske, og der tilsættes surrogatstandard (6.5). Efter mindst 10 min. henstand tilsættes 40 mL H2O (6.2.5), og der justeres til pH 10-12 med 4M NaOH. Der tilsættes 150 ml acetone:pentan (6.2.3), og prøven behandles 5 min. på ultralydsbad fulgt af 2 timer på rystebord (250 ryst/min.). Derefter centrifugeres prøven i 5 min. (1500 RPM). Væskefasen overføres til skilletragt, og den organiske fase filtreres gennem faseseperationsfilter med 25 – 30 g glødet Na2SO4 (6.2.1) til en rundbundet kolbe. Ekstraktet inddampes til ca. 5 mL på rotationsfordamper eller andet egnet opkoncentreringsudstyr. Ekstraktet overføres til 10 mL målekolbe, og der fyldes efter til mærket med acetone (6.2.2). Heraf udtages 1,00 mL ekstrakt, som overføres til en GC-vial og der tilsættes evt. en sprøjtestandard (6.6) før analyse på GC-MS.
8.4.2 Soxtec-ekstraktion
Ekstraktion: "Boiling" (ekstraktionshylster neddyppet i solvent) 1 time
Ekstraktion: "Rinsing" (solvent drypper ned i ekstraktionshylsteret) 1 time
Ekstraktionshylster: 33 x 80 mm
Inddampning: til omkring 10 mL (ca. 5 min.).
Ekstraktionsmiddelmængde: 50 mL acetone:cyclohexan (6.2.4)
Der afvejes ca. 10 g prøve i ekstraktionshylster, som blandes med ca. 10 g vandfrit Na2SO4 (6.2.1), og der tilsættes surrogatstandard (6.5). Derefter tilsættes 50 mL acetone:cyclohexan (6.2.4) og ekstraktionshylstret anbringes i Soxtec-apparatet. Prøverne koges 1 time ved 160°C, hvorefter der skylles 1 time (reflux), og opløsningsmidlet inddampes i ca. 5 min. til omkring 10 mL.
Efter ekstraktion inddampes prøven forsigtigt under påblæsning af nitrogen til ca. 5 mL. Ekstraktet overføres kvantitativt til 10 mL målekolbe, og der fyldes op til mærket med cyclohexan (6.2.2). Heraf udtages 1,00 mL ekstrakt, som overføres til en GC-vial, og der tilsættes evt. en sprøjtestandard (6.6) før analyse på GC-MS.
8.4.3 Accelerated Solvent Extraction
Læg et filter i bunden af en ekstraktionscelle på 11 mL. Cellen fyldes helt med tørret prøve (8-10 g) og tilsættes surrogatstandard (6.5). Cellen lukkes, strammes og anbringes i ASE instrumentets karrusel.
Der ekstraheres med følgende betingelser:
| Solvent: | Acetone:cyclohexan (6.2.4) |
| Ovntemperatur: | 100 °C |
| Tryk: | 2000 psi |
| Static time: | 5 min. |
| Skyllevolumen: | 60 % af celle volumen |
| Nitrogen-purge. | 60 sec. ved 150 psi |
Efter ekstraktion overføres prøven, efter inddampning under nitrogen, til henholdvis 10
mL målekolbe (10 g prøve). Der fyldes efter til mærket med cyclohexan (6.2.2). Heraf
udtages 1,00 mL ekstrakt, som overføres til en GC-vial, og der tilsættes evt. en
sprøjtestandard (6.6) før analyse på GC-MS.
Hvis ekstraktet indeholder H2O, tørres dette med glødet Na2SO4 (6.2.1) inden inddampning.
8.4.4 Mikrobølgeekstraktion
10 g prøve afvejes direkte i ekstraktionsbeholderen og tilsættes surrogatstandard (6.5). Derefter tilsættes 2 mL vand (6.2.5) og 25 mL acetone:pentan (6.2.3), hvorefter beholderen lukkes forsvarligt og ekstraktionen igangsættes.
| Ekstraktionsbetingelser: | Mikrobølgeenergi: 1200 Watt (100%), (14 ekstraktionsbeholdere) |
| Ekstraktionstemperatur: | 110° C |
| Opvarmningstid til | |
| operationstemperatur: | 10 min., temperatur holdes: 10 min. |
| Ekstraktionscelletype: | Bestående af et materiale passende til instrumentet (teflon eller lignende) med et volumen på ca. 100 mL. |
Efter ekstraktion afventes nedkøling af ekstraktionscellerne i ca. 30 min., således at
trykket igen er gået af ekstraktionscellerne, og prøven tørres med ca. 10 g vandfrit Na2SO4
(6.2.1). Herefter overføres prøven kvantitativt, efter inddampning med nitrogen, til en
10 mL målekolbe. Der fyldes efter til mærket med acetone (6.2.2). Heraf udtages 1,00 mL
ekstrakt, som overføres til en GC-vial, og der tilsættes en evt. sprøjtestandard (6.6)
før analyse på GC-MS.
8.5 Gaskromatografiske betingelser
Ekstraktet analyseres ved gaskromatografi med massespektrometrisk detektion ved anvendelse af single ion monitoring (GC-MS-SIM) (VKI, 1996).
For hver komponent moniteres 2 ioner. Primær-ionen anvendes ved beregning, sekundær-ionen anvendes ved verifikation af identiteten.
Eksempel på gaskromatografiske betingelser:
| Gaskromatograf: | Temperaturprogrammerbar og trykstyret eller tilsvarende |
| Injektor: | Pulsed splitless 1µL |
| Injektionspuls: | 35 psi/2 min., ved 280° C |
| Kolonne: | 5% phenyl methyl silicone eller tilsvarende, 30 m, 0,25 mm ID, 0,25 µm film |
| Bæregas: | He, 1,6 ml/min., svarende til en bæregashastighed på 46 cm/s som holdes konstant ved hjælp af elektronisk trykregulering |
| Kolonneindgangstryk: | 12,8 psi ved 40 şC (trykket afpasses efter starttemperaturen) |
| Detektortemperatur: | 280° C |
| Temperaturprogram: | 40-50° C (acetone) eller 65-75 ° C (cyclohexan), 2 min. 10° C/min., til 285° C 30° C/min., til 310° C, 5 min. Total analysetid: ca. 32 min. |
| Detektion: | Karakteristiske masser (m/z), SIM mode (se nedenstående tabel) |
Bemærk: Det er vigtigt, at benytte en starttemperatur der svarer til det solvent, som
benyttes til injektionen, for at opnå de bedst mulige kromatografiske betingelser. Hvis
der ikke benyttes elektronisk trykstyring af bæregassen, er det af afgørende betydning
for separationen af de enkelte PAH-forbindelser at sikre sig, at der holdes et starttryk
ved lav temperatur, som resulterer i en bæregashastighed på over 20 cm/s ved
sluttemperaturen.
Det kan ofte være en fordel at benytte biphenyl d10 som surrogat for naphtalen i stedet for Naphtalen d8, specielt for de alternative ekstraktionsmetoder ASE og MAE.
| Karakteristiske ioner til kvantitativ bestemmelse | ||||
| Forbindelse | RT | Primær ion | Sekundær ion | Surrogat, der anvendes til beregning |
| Naphthalen d8 | 10,38 | 136 | 135 | - |
| Naphthalen | 10,42 | 128 | 102 | Naphthalen d8 |
| Biphenyl d10 | 13,1 | 164 | 162 | - |
| Acenaphthylen | 14,2 | 152 | 151 | Biphenyl d10 |
| Acenaphthen | 14,6 | 153 | 154 | Biphenyl d10 |
| Fluoren | 15,8 | 166 | 165 | Phenanthren d10 |
| Phenanthren d10 | 18,0 | 188 | 94 | - |
| Phenathren | 18,1 | 178 | 176 | Phenanthren d10 |
| Anthracen d10 | 18,2 | 188 | 94 | Sprøjte standard |
| Anthracen | 18,2 | 178 | 176 | Phenanthren d10 |
| Fluoranthen d10 | 20,85 | 212 | 106 | - |
| Fluoranthen | 20,91 | 202 | 101 | Fluoranthen d10 |
| Pyren d10 | 21,38 | 212 | 106 | - |
| Pyren | 21,43 | 202 | 101 | Pyren d10 |
| Benz(a)anthracen | 24,3 | 228 | 226 | Benzo(a)pyren d12 |
| Chrysen/Triphenylen | 24,4 | 228 | 226 | Benzo(a)pyren d12 |
| Benzo(b+j+k)fluoranthen | 26,7 | 252 | 250 | Benzo(a)pyren d12 |
| Benzo(a)pyren d12 | 27,2 | 264 | 266 | - |
| Benzo(a)pyren | 27,3 | 252 | 250 | Benzo(a)pyren d12 |
| Indeno(1,2,3-cd)pyren | 29,1 | 276 | 274 | Dibenz(a,h)anthracen d14 |
| Dibenz(a,h)anthracen d14 | 29,1 | 292 | 291 | - |
| Dibenz(a,h)anthracen | 29,2 | 278 | 274 | Dibenz(a,h)anthracen d14 |
| Benzo(g,h,i)perylen | 29,6 | 276 | 274 | Dibenz(a,h)anthracen d14 |
9 Resultater
| 9.1 | Kalibrering |
| 9.2 | Beregning |
| 9.3 | Resultatangivelse |
| 9.4 | Præcision, nøjagtighed og detektionsgrænse |
9.1 Kalibrering
Der analyseres kalibreringsopløsninger for alle komponenter inkl. surrogatstandarder. Der skal som minimum anvendes 5 niveauer, da standardkurven ofte ikke er lineær ved GC-MS. Hvis linien er ret og går gennem (0,0), kan der anvendes lineær regression ved beregningerne. Ellers anbefales det at bruge ikke-lineær kalibrering.
Efter kalibreringen kontrolleres denne ved genberegning af de brugte standarder. Afvigelsen fra den teoretiske koncentration bør efter genberegning ikke overstige 10% generelt og må ikke overstige 25% i hele det beregnede interval.
Laboratoriet skal sikre sig, at koncentrationerne i de analyserede ekstrakter ligger inden for det valgte kalibreringsinterval. Ellers skal ekstrakterne fortyndes og analyseres på ny.
9.2 Beregning
Stofferne identificeres ved deres retentionstid, ved de karakteristiske ioner for stoffet, samt den relative respons mellem de 2 benyttede ioner. Hovedionen anvendes til kvantificering. De kvantitative beregninger udføres i alle tilfælde ud fra toppenes arealer.
Koncentrationen af PAH i ekstraktet beregnes efter laboratoriets interne beregningsmetoder med korrektion for genfindelsen af surrogat standard og evt. sprøjtestandard, under hensyntagen til instrument- og softwareproducentens retningslinier.
Koncentrationen i prøven beregnes efter følgende formel:
Cext, korr * V * 100
Cpr = -----------------------
gpr
* % TS
Hvis det ved blindprøverne viser sig, at der er et blindbidrag, fratrækkes det ved anvendelse af følgende udtryk:
| Cext, korr | = Cext - Cbl |
| Cpr | :koncentration af stof i jordprøven i mg/kg TS |
| Cext | :koncentration af stof i ekstraktet, mg/L |
| Cext, korr | :koncentration af stof i ekstraktet korrigeret for blindbidrag, mg/L |
| Cbl | :koncentration af stof i ekstraktet fra blindprøven, mg/L |
| V | :volumen af slutekstrakt, mL |
| gpr | :mængde prøve taget i arbejde, g |
| % TS | :procent tørstof |
Hvis ekstrakterne skal fortyndes mere end 5 gange for at bestemme høje indhold af PAH,
kan responset fra surrogatstandarderne ikke benyttes længere, hvorfor genfindelsen af
surrogatstandard skal bestemmes i en ikke-fortyndet eller en mindre end eller 5 ganges
fortynding.
Ved fortynding af ekstrakter vurderes det, om en evt. sprøjtestandard skal tilsættes på ny.
9.3 Resultatangivelse
Resultaterne angives i mg/kg TS med to betydende cifre, og for resultater mindre end 0,10 mg/kg med et betydende ciffer, som f.eks.:
| Fluoranthen | 2,3 mg/kg TS |
| Benz(b+k+j)fluoranthen | 1,9 mg/kg TS |
| Benz(a)pyren | 0,03 mg/kg TS |
| Dibenz(a,h)anthracen | 45 mg/kg TS |
| Indeno(1,2,3-cd)pyren | 0,11 mg/kg TS |
| Sum af 7 PAH | 49 mg/kg TS |
9.4 Præcision, nøjagtighed og detektionsgrænse
Præcision og nøjagtighed afventer en interlaboratorie-metodeafprøvning.
Detektionsgrænser og genfinding for rystemetoden bestemt ud fra seksdobbelt tilsætningsforsøg på lavt niveau (5 x detektionsgrænsen).
| Genfinding og detektions- | Tilsat |
Middel |
Standard |
Gen- |
Detektions- |
| grænse bestemmelse på | mængde |
værdi |
afvigelse |
finding |
grænse |
| baggrund af tilsætningsforsøg | mg/kg TS |
Mg/kg TS |
mg/kg TS |
% |
mg /kg TS |
| Naphthalen | 0,05 |
0,047 |
0,002 |
94 |
0,007 |
| Acenaphthylen | 0,05 |
0,051 |
0,003 |
102 |
0,011 |
| Acenaphthen | 0,05 |
0,044 |
0,002 |
87 |
0,007 |
| Flouren | 0,05 |
0,047 |
0,002 |
94 |
0,007 |
| Phenanthren | 0,05 |
0,047 |
0,002 |
93 |
0,006 |
| Anthracen | 0,05 |
0,045 |
0,001 |
90 |
0,005 |
| Flouranthen | 0,05 |
0,046 |
0,002 |
92 |
0,010 |
| Pyren | 0,05 |
0,047 |
0,001 |
93 |
0,006 |
| Benz(a)anthracen | 0,05 |
0,040 |
0,001 |
81 |
0,006 |
| Chrysen | 0,05 |
0,045 |
0,001 |
90 |
0,005 |
| Benzo(b+j+k)fluoranthen | 0,15 |
0,124 |
0,003 |
82 |
0,011 |
| Benzo(a)pyren | 0,05 |
0,042 |
0,002 |
85 |
0,007 |
| Indeno(1,2,3-cd)pyren | 0,05 |
0,039 |
0,001 |
79 |
0,006 |
| Dibenzo(a,h)anthracen | 0,05 |
0,041 |
0,002 |
82 |
0,007 |
| Benzo(g,h,i)perylen | 0,05 |
0,040 |
0,001 |
81 |
0,005 |
| Sum 16 (17) EPA PAH | 0,85 |
0,745 |
0,019 |
88 |
0,075 |
| Sum 7 PAH | 0,35 |
0,293 |
0,008 |
84 |
0,033 |
I de tilfælde, hvor laboratorierne har problemer med at opnå tilstrækkelig følsomhed,
kan det opnåede ekstrakt inddampes yderligere, blot surrogatstandardmængden tilpasses
den ekstra opkoncentrering eller ligger inden for surrogatstandardernes
kalibreringsområde.
Detektionsgrænsen er desuden foreløbigt bestemt ud fra blindbidraget fra de enkelte metoder og kan ses i nedenstående tabel.
| Detektionsgrænse bestemmelse | Ryste |
Soxtec |
ASE |
MAE |
| på baggrund af blindbidrag | mg/kg |
mg/kg |
mg/kg |
mg/kg |
| Naphthalen | 0,02 |
0,03 |
0,01 |
0,01 |
| Acenaphthylen | 0,02 |
0,02 |
0,01 |
0,01 |
| Acenaphthen | < 0,01 |
0,04 |
0,01 |
< 0,01 |
| Flouren | < 0,01 |
0,01 |
< 0,01 |
0,00 |
| Phenanthren | 0,01 |
0,03 |
0,02 |
0,02 |
| Anthracen | < 0,01 |
0,01 |
< 0,01 |
< 0,01 |
| Flouranthen | 0,03 |
0,02 |
0,02 |
0,04 |
| Pyren | 0,02 |
0,04 |
0,01 |
0,03 |
| Benz(a)anthracen | 0,01 |
< 0,01 |
0,03 |
0,01 |
| Chrysen | 0,03 |
0,01 |
< 0,01 |
0,01 |
| Benzo(b+j+k)fluoranthen | 0,07 |
0,02 |
0,01 |
0,04 |
| Benzo(a)pyren | < 0,01 |
0,01 |
0,01 |
0,01 |
| Indeno(1,2,3-cd)pyren | 0,03 |
0,02 |
0,06 |
0,02 |
| Dibenzo(a,h)anthracen | 0,01 |
0,01 |
0,01 |
0,02 |
| Benzo(g,h,i)perylen | 0,02 |
0,02 |
0,07 |
0,01 |
| Sum 16 EPA PAH | 0,24 |
0,14 |
0,22 |
0,13 |
| Sum 7 PAH | 0,11 |
0,06 |
0,08 |
0,04 |
10 Analyserapport
Analyserapporten bør som minimum indeholde følgende:
| koncentrationer i mg/kg TS af alle enkelt-PAH, der er bestemt, og sum af alle enkelt-PAH, der er bestemt, samt sum af de 7 PAH, som indgår i jordkvalitetskriteriet | |
| nøjagtig identifikation af den anvendte metode (ekstraktions- og GC-MS-betingelser) | |
| nøjagtig prøvemærkning | |
| angivelse af tidspunkt for prøvernes modtagelse på laboratoriet | |
| enhver afvigelse fra denne standard, såsom afvigende opbevaringsbetingelser, skal anføres i analyserapporten | |
| henvisning til denne metode |
11 Litteratur
Bjørseth, A., Ramdahl, T. (editor) (1985). Handbook of Polycyclic Aromatic Hydrocarbons, Volume 2. Marcel Dekker inc., New York,.
Hyver, K.J., Sandra, P. (editor) (1989) High Resolution Gas Chromatography, 3. Ed. Hewlett-Packard Co., Delaware.
Means, J.C. (1998). Compound-Specific Gas Chromatographic/Mass Spectrometric Analysis of Alkylated and Parent Polycyclic Aromatic Hydrocarbons in Waters, Sediments, and Aquatic Organisms. Journal of AOAC International, 81 s. 657- 672.
Schantz, M.M., Nichols, J.J., Wise, S.A. (1997) Evaluation of Pressurized Fluid Extraction for the Extraction of Environmental Matrix Reference Materials. Analytical Chemistry, 69, 1997, s. 4210-4219.
Heemken, O.P., Theobald, N.,. Wenclaviak, B.W (1997). Comparison of ASE and SFE with Soxhlet, Sonication, and Methanolic Saponification Extractions for the Determination of Organic Micropollutants in Marine Particulate Matter. Analytical Chemistry, 69, 1997, s. 2171-2180.
Fisher, J.A., Scarlett, M.J., Stott; A.D. (1997). Accelerated Solvent Extraction: An Evaluation for Screening of Soils for Selected U.S. EPA Semivolatile Organic Priority Pollutants. Environmental Science and Technology, 31, 1997, s. 1120-1127.
Dupeyron, S., Dudermel, P-M., Couturier, D., Guarini, P., Delattre, J-M. (1999). Extraction of Polycyclic Aromatic Hydrocarbons from Soil: A Comparison Between Focused Microwave Assisted Extraction , Supercritical Fluid Extraction, Subcritical Solvent Extraction and Soxhlet Techniques. International Journal of Environmental Analytical Chemistry, 73, 1999, s. 191-210.
Letellier, M., Budzinski; H. (1999). Influence of Sediment Grain Size on the Efficiency of Focused Microwave Extraction of Polycyclic Aromatic Hydrocarbons. The Analyst, 124, 1999, s. 5-14.
Bestemmelse af PAH i jord og vand ved GC-MS, VKI-metode 0-32. 3. udgave, 20/10-96.