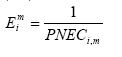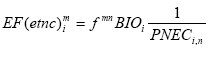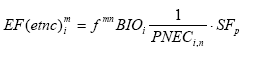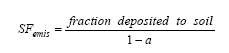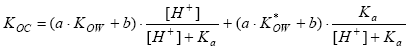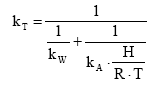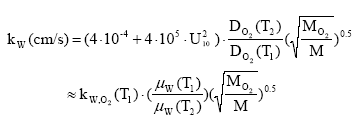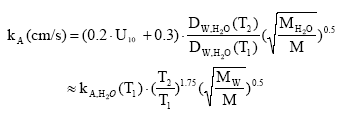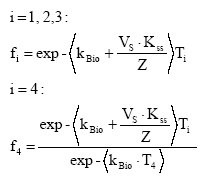|
Background for spatial differentiation in LCA impact assessment - The EDIP2003 methodology 8 Ecotoxicity8.1 Introduction Authors: Jens Tørsløv [77] Michael Hauschild [78]Dorte Rasmussen77 8.1 IntroductionEmissions of chemical substances to the environment may cause effects on organisms as well as on the structure and functioning of the ecosystem through their toxic action. Different types of chemicals act through different toxicological mechanisms, and compared to other environmental impact categories in LCIA, the cause-effect relationship in ecotoxicity is very complex. Also the number of substances in a product system that may contribute to ecotoxicity is higher, and often includes a range of substances with different characteristics. Toxicity is a relative concept, and paraphrasing the ancient Swiss physician Paracelsus, all substances are toxic if the dose ingested is large enough. The ecotoxicological impact of a substance can be regarded as a result of its toxicological properties on the one hand and the exposure to the target on the other hand. The exposure depends not only on the released quantities but also on the fate of the substance in the environment, e.g. distribution between environmental compartments, biodegradation and ability to accumulate in biota. Characterisation modelling for ecotoxic chemicals in LCA seeks its inspiration from environmental risk assessment but there are important differences. Risk assessment is often performed in a legislative context where the purpose can be to help ensure that there is no unacceptable risk to the environment, rather than to provide the best estimate of the actual risk. Therefore, a conservative approach is often followed, whereas a detailed risk assessment is conducted only if a preliminary assessment indicates a risk. Life cycle impact assessment (LCIA), on the other hand, attempts to address all relevant environmental impacts associated with a product, not just the impacts from toxic and persistent chemicals. To avoid an unintentional bias in the treatment of the different impacts, LCIA thus aims at providing the best estimation for each type of impact. Therefore, a conservative estimate of the ecotoxic effect of a substance is unwanted in the context of LCA - we must aim for the best estimate of the actual effect properties of the substance. 8.1.1 Ecotoxicity modelling in LCIAThe SETAC Europe's First Working Group on Life Cycle impact assessment (WIA1) proposed the following framework for characterisation of ecotoxic or human toxic impacts in the environmental impact assessment component of life cycle assessment (LCIA) (Jolliet et al., 1996): (8.1)
The environmental impact potential, EP is presented as the product of an effect factor E, a fate factor F, and the total emitted quantity Q. The index i represents the chemical, n the environmental compartment to which the emission is released, and m the route of exposure of the ecosystem The difference between existing LCIA characterisation models for ecotoxicity lies in the fate factor for which different models have been developed or adopted (e.g. Guinée et al., 1996, Jolliet and Crettaz, 1997, Wenzel et al., 1997, Huijbregts, 1999, Hertwich, 1999). In all these methodologies, the effect factor is the same, taken as the inverse of the predicted environmental no effect concentration, PNEC (8.2)
Assumption of linearity The underlying assumption is that the effect depends on the properties of the substance only, not of the receiving environment. The impact is proportional to the quantity that, upon the reductions and modifications included in the chosen fate modelling, reaches the target system and the proportionality (the slope of the quantity-impact curve) is always the same. This linearity is also assumed in current risk assessment as performed using generic tools like EUSES (RIVM, 1998) or CalTOX (McKone, 1993) but nonetheless it contradicts the experience that thresholds of effects exist and that the curve is not linear in reality (see e.g. Potting and Hauschild, 1997a and b). A novel approach to LCIA, the EcoIndicator99, aiming at damage modelling, attempts in a very generic form to consider the non-linearity by taking into account the background situation of the exposed ecosystem and uses a different effect factor (Goedkoop and Spriensma, 1999). This approach holds a strong potential for spatial characterisation of ecotoxicity since it allows taking the spatial variation in the state of the exposed ecosystems into account. However, sufficient information about background concentrations of manmade chemicals in the different ecosystems is not available today, and in order to make the approach operational for LCIA, the authors generalise their approach by taking out the spatial differentiation. At present, there is thus no alternative to assuming linearity between quantity of substance reaching the ecosystem and impact on the ecosystem in life cycle impact assessment of ecotoxicity. This means that the only fate processes that are relevant to include in spatial characterisation modelling are those processes altering the quantity of substance reaching the different parts of the environment – processes like biological degradation, chemical transformation, evaporation, deposition and sedimentation etc. Processes which influence concentration through dilution and dispersion without altering the total quantity of substance will not influence the characterised ecotoxic impact. When linearity is assumed, exposing 1 m³ of water to a substance concentration of 100 mg/m³ is equivalent to exposing 100 m³ of water to a concentration of 1 mg/m³. 8.1.2 Ecotoxicity modelling in EDIP97In the EDIP97 methodology (Wenzel et al., 1997, Hauschild and Wenzel, 1998), ecotoxicity is considered in aquatic ecosystems (acute and chronic), in terrestrial ecosystems (chronic exposure) and in wastewater treatment plants. For each endpoint, a simplified fate modelling is applied based on a modular approach where redistribution between the environmental compartments and potential for biodegradation are represented as separate factors. The equivalency or impact factor for chronic ecotoxicity in environmental compartment n is determined as: (8.3)
Where the redistribution factor, fmn expresses the fraction that is redistributed from the initial compartment m to the final compartment n, in which the ecotoxicological impact is modelled. BIO represents the potential for biodegradation as determined from standardised tests for ready and inherent biodegradability and the effect factor is the inverse PNEC value for the ecosystems of compartment n as described in expression (8.2). The "site-generic" EDIP methodology is prepared for inclusion of spatial differentiation for all non-global impact categories through site factors SF intended to modify the site-generic characterisation factors: (8.4)
The site-generic impact potential in EDIP97 is interpreted as the largest impact to be expected from the emission and the site factor is seen as the spatially determined probability that the full impact will occur, i.e. SF ranges between 0 and 1. Wenzel and co-authors give guidance on the quantification and use of the SF without making the site factor really operational. The EDIP methodology also involves some other possibilities for spatial differentiation. For the fraction of airborne emissions that deposits, the redistribution factor, fmn is set at "a" when n is the aquatic compartment and 1-a when n is the terrestrial compartment. EDIP97 allows "a" to be chosen according to the conditions of the region where the emission takes place. For Danish conditions, a=0.5 is proposed while a global default is set at a=0.2. Furthermore, in EDIP97, spatial information in the form of initial dilution data for waterborne emissions, is suggested to be included as technical information in the weighting of the potential contribution to acute aquatic ecotoxicity in order to reflect the differences in dilution potential (and hence the probability of acute effects) for different types of aquatic systems. For further background on the EDIP97 methodology, the reader is referred to the description in Wenzel et al. (1997) and Hauschild and Wenzel (1998). 8.1.3 Determining the physical level at which the spatial variation must be quantifiedParameters that influence the fate of substances in the environment often show large spatial variations. This variation depends on the scale, at which they are observed. For soil, the variation in e.g. the content of organic material can vary orders of magnitude on the 10-3 m scale (from rock material to a humus particle). The variation may stay within one order of magnitude between fields (102-103 m scale) and even less between countries or regions (105-106 m scale). At the extreme end, the scale of the whole planet earth (4107 m scale), the spatial variation is zero (which is why spatial characterisation modelling is only discussed for the non-global impact categories). This general relation between spatial variation and physical scale is illustrated in Figure 8.1 a and b Figure 8.1a +b. The degree of spatial variation of different environmental parameters depends on the physical or geographical scale at which it is studied. Examples of physical scales are: 10-2 m between particles of stone, clay and organic material and between water and particles in soil and water systems 10-1 m between small lumps of soil 1 m between patches of soil or water 10-102 m between parts of field, forest or lake 103-104 m between fields/habitats/lakes-streams/estuaries/ocean 105 m between small countries or regions of larger countries, types of climate, lakes, ocean 106 m between larger countries or regions of continents (like Europe), climatic zones 107 m between hemispheres, groups of climatic zones >4·107 m between Earths (only one Earth → no variation) In general, the relevant scale at which to determine the degree of spatial variation must be the scale that is influenced by the substance, i.e. the scale at which the substance is dispersed as part of its transfer and transformation (fate). This means that the relevant scale depends on the lifetime and the dispersion velocity of the substance in the environment. Consequently, removal processes and the type of the receiving environment are important factors (dispersion is quick in air, slow to medium velocity in water and very slow and mainly vertical in soil). The spatial variation is attempted quantified at two scales throughout this chapter
For the further discussion the following levels of spatial variation are defined for the fate modelling of a substance:
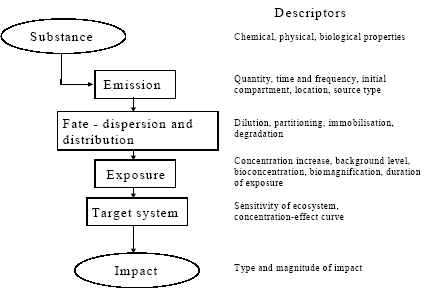
The aim of this chapter on ecotoxicity is to analyse the actual spatial variation for the relevant scale(s) (if possible under consideration of its dependence on the lifetime of the substance), to provide background for the best spatial characterisation modelling deemed possible and discuss how large a part of the actual spatial variation, it covers. Finally, to suggest a framework for calculating site factors to modify the existing site-generic EDIP characterisation factors for ecotoxicity and implement this framework to derive site factors for Europe. 8.2 The cause-impact chain and its descriptorsFigure 8.2 presents the cause-impact chain from the emission of a substance through a number of possible fate and exposure processes to the impact on the target – the part of the environment where the impact is preferably measured for the impact assessment. Each of the links in the chain can be characterised by a set of descriptors, which are important for the processes covered by the link. These descriptors are discussed in Section 8.2.
Figure 8.2. The cause-impact chain. Examples of descriptors for site characterisation are indicated. 8.2.1 The emitted substanceThe inherent physico-chemical and biological properties of a substance have a strong influence on its interaction with the environment. The following physico-chemical and biological properties are examples of parameters that are commonly used in modelling of the environmental fate of chemical substances:
Recommendations The substance-specific properties (descriptors) mentioned above are site-independent and have no direct relevance for the spatial characterisation of the ecotoxicological impact. 8.2.2 The emissionSeveral characteristics of the emission may influence the fate of the emitted substances and therefore also the extent to which an exposure eventually takes place. Important parameters are:
The emitted quantity to the different environmental media Emission to atmosphere, water and soil is given as mass emitted per functional unit in the inventory of the LCA. Generally, no information is included about how large the emission from one functional unit is compared to the total emission output from the process. The characterisation can therefore not be based on the full impact of the process. Emissions to waste treatment facilities are not terminal and the modelling of the relevant waste treatment processes (i.e. their resulting emissions to air, surface water, ground water or soil) is considered part of the inventory analysis. In case of emission to air, the dispersion modelling performed for human exposure reveals that the location of the emission point is not significant since the long transportation distances tends to even out local differences in the fate and exposure except for very short-lived substances. For the ecotoxicity impact category, the target is terrestrial and aquatic ecosystems, and the main descriptors of relevance to spatial characterisation modelling are: the density of ecosystems within the deposition area as described by the fraction that is covered by `natural areas'. Cultivated agricultural land is considered part of the technosphere rather than the environment, and natural ecosystems within urban areas are disregarded due to their insignificant contribution to the total area of natural ecosystems. the distribution of the deposited fraction between land systems and water systems (the relative prevalence of the latter being much lower in continental than in coastal areas) Annex 8.4 shows the relative frequency of natural areas within the countries of Europe. If this information is combined with atmospheric dispersion modelling as performed in e.g. Chapter 3 it is possible to determine the fraction of the emitted substance that deposits to ecosystems. This work has not yet been performed but as a first approximation, Europe is divided into four regions and the relative share of natural areas within each region calculated. Table 8.1. Relative share of natural areas within the four regions of Europe. More information on definition of the regions and the calculations is given in Annex 8.4.
If it is assumed that the major part of an atmospheric emission deposits within the region where it is emitted, Table 8.1. allows introducing a spatial differentiation for airborne emissions in the fraction deposited to ecosystems on land. The sea is considered entirely to be a `natural area' and all emissions deposited to sea are regarded as exposure of a natural area. Annex 8.5 suggests average factors for the distribution between sea and land for deposition originating from the same European regions. The data are based on calculations of the relative fractions of emissions of NH3 and NOx that deposit to land and sea for air-borne emissions from different European countries. Table 8.2. Relative share of emissions depositing to sea and land. For definition of the regions and calculations see Annex 8.5.
* The share of emissions deposited to freshwater systems is considered irrelevant here, due to the relative uncertainty of the deposition factor Used in combination, Table 8.1 and 8.2 allow deriving spatially differentiated factors for the share of emissions deposited from air that exposes aquatic or terrestrial ecosystems. Table 8.3. Relative share of emissions potentially contributing to aquatic and terrestrial ecotoxicity for different regions of Europe.
The temporal course of the emission The emission flux may be continuous or discontinuous, depending on the type of the source. For continuous emissions, there may be a broad variation in the duration from normal industrial processes (lasting from hours to days) to emissions from landfills (lasting from years to millennia). The temporal pattern will have an influence on the environmental impact as organisms respond to the concentrations they are exposed to rather than to the emitted amounts. Intermitted emissions with long intervals where no emission occurs may allow the exposed organisms to overcome the stress from sub-lethal exposures. On the other hand, emissions that come in pulses, e.g. from a batch production, may reach much higher transient environmental concentrations compared to a situation where the same amount of substances is discharged continuously over a longer period of time. A main entrance route for chemical substances to the aquatic environment is via municipal wastewater treatment facilities. Due to the mixing and the hydraulic retention time, emissions from municipal treatment facilities can be regarded as relatively continuous regardless the temporal variation of the original source. Temporal changes in the composition of wastewater from industries with own wastewater treatment facility may, however, be considerable due to variations in production activities. The temporal course of release may thus be important for the environmental concentrations, but tends to lose its importance with increasing distance from the source, as dilution, different fate related processes as well as contributions from other sources contribute to level out temporal variations. Substances emitted to air, that reach the aquatic or terrestrial environment through deposition, can be considered as coming from a continuous source regardless the nature of the emission, with the possible exclusion of targets in the immediate vicinity of the source. This is due to the efficient dispersion in the atmosphere. For characterisation modelling of ecotoxicity, it is therefore assumed that the contribution to atmospheric deposition from a functional unit is infinitesimal and that temporal variations can be disregarded. Apart from atmospheric deposition, the main route for most substances to reach the terrestrial environment are through organic waste products, e.g. sewage sludge, which may be used as an agricultural fertiliser and applied once a year or with longer intervals. The ecotoxicity impact categories are concerned with impacts on natural ecosystems, and the farm fields would normally be considered part of the technosphere, in which case, emission via sewage sludge is without relevance to ecotoxicity (but still a relevant exposure route for human toxicity). As a special group of substances, pesticides, are unintentionally emitted directly to the natural environments in the immediate surroundings of the sprayed field through wind drift and surface run-off. Like the application of the pesticides, this form of emission to the terrestrial environment is highly discontinuous. Pesticides may also reach the surrounding environment through evaporation followed by atmospheric deposition or through leaching to surface water (Hauschild, 2000). Both of these emission routes are considered to be continuous. The extreme end of the spectrum of continuous emissions is represented by leaching of persistent substances, notably metals, from landfills. Here, the duration of the emission may be centuries. Recommendations The emitted quantity to the different environmental media For emissions to air spatial variation is taken into account using the deposition factors in Table 8.3 for that fraction of the emission which deposits. The temporal course of the emission As discussed above, the temporal course of the emission may at the local level have a considerable influence on the environmental concentrations of toxic substances and thus the impact on biota. Consequently, a discontinuous or very quick emission of a substance will be more severe than a slow, continuous emission of the same quantity. Temporal variations in the emission pattern can, however, be disregarded for impacts occurring at a regional level, i.e. at distances from the emission point where the temporal variation are insignificant due to mixing and fate processes. In the aquatic environment temporal variations are relevant only for assessment of the local effects, i.e. acute aquatic ecotoxicity. Here,
Consequently, only in cases of emission with highly temporal variations the emission pattern has relevance for spatial characterisation, and only in the assessment of the acute aquatic ecotoxicity in the area near the emission point. This is not given further treatment here but it is suggested that such emissions be flagged for a more detailed evaluation if relevant. In the terrestrial environment we are only concerned with impacts at the regional level where temporal variations are considered of little importance. Situations with discontinuous emissions are emission through wind drift or surface run-off of pesticides to environment surrounding agricultural areas (other emission routes for pesticides can be regarded as continuous) and emission through adsorption to sludge and application to agricultural areas. 8.2.3 Fate - dispersion and distributionA substance's environmental fate includes transport, distribution between compartments and transformation from the emission point to the target system, a biological community, population or an organism, where it may exert its toxicity. The following fate-descriptors may be relevant for site characterisation and are therefore discussed in details:
8.2.3.1 Dilution and evaporationIn the vicinity of the discharge point to the aquatic environment, dilution is the most important process determining the level of exposure. Locally, acute as well as chronic effects can be observed, but normally, only chronic effects are expected at larger distances from the source where other processes than dilution determine the exposure. Dilution is an important descriptor in the spatial differentiation for acute ecotoxicity effects whereas it has no significance for the spatial differentiation for chronic ecotoxicity. For the present purpose, the "initial dilution zone" of an emission is encompassing the medium from the discharge point to the point where the emission leaves the technosphere and enters the ecosphere (or in other words where it becomes an emission). Particularly for waterborne emissions, the immediate surroundings of the discharge point are often considered to be part of the technosphere in the sense that even acute ecotoxic effects are accepted here. Outside the initial dilution zone, the further dilution is determined by diffusion and mixing due to the turbulence of the receiving water. Examples of the dilution capacity of different types of water courses are presented in Annex 8.1 and summarised in Table 8.4. Table 8.4. Typical dilution factors obtained 1000 m from the discharge point in different types of aquatic environments.
For emissions to soil, the initial dilution depends on the type of the emission, e.g. an application of pesticides through spraying or granules, an application of sewage sludge or a deposition of air-borne pollutant. Regardless the emission type, no spatial differentiation is expected in the dilution or dispersion pattern for emissions to soil. Some substances emitted to the soil or water environment will eventually evaporate. Substance-specific properties as well as environmental factors determine the evaporation rate. The evaporation is represented in the site-generic fate model but will show a spatial variability, which depends among other factors on the temperature. Calculations have been performed in Annex 8.8 for different values of Henrys constant and at three temperatures: 10oC (reference temperature), 6 oC (Northern European countries) and 15 oC (Southern European countries). A site factor is expressed as the ratio between the evaporation in the reference situation and the evaporation in the Southern European and the Northern European situation respectively in Table 8.5. For East and Western European countries, the reference temperature is assumed most descriptive and the correction factor assumes the value 1. Table 8.5. Site factors, SFevap for volatilisation from water for two European regions
Recommendations For the impact categories chronic aquatic ecotoxicity and chronic terrestrial ecotoxicity, dilution is not considered relevant for inclusion in spatial characterisation modelling at the spatial scale considered.
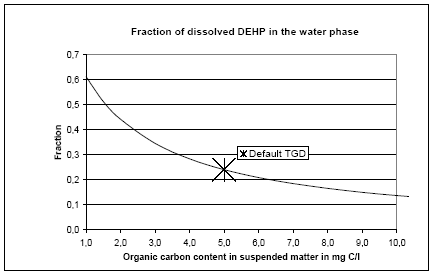
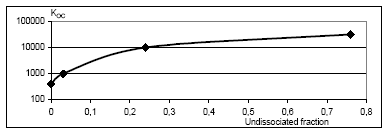
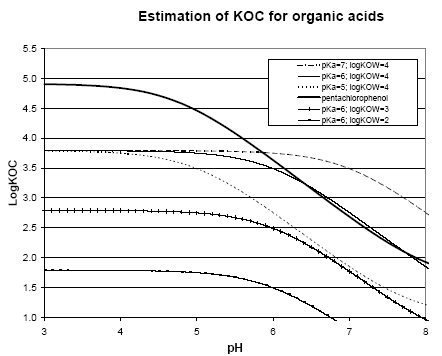
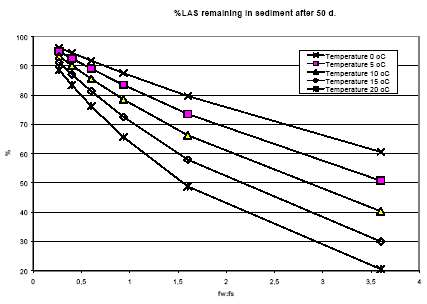
The spatial variation in equilibrium partitioning to air from water or soil for semi-volatile chemicals can be modelled based on knowledge of the annual average temperature as made operational by the site factors in Table 8.5. 8.2.3.2 DispersionIn addition to lowering the concentration to which the environment is exposed, the dispersion that leads to the dilution discussed under 8.2.3.1 also has as a consequence that the emitted substance is spread over a much larger area down the river or in the lake, estuary or ocean as well as potentially allowing different types of ecosystems to be exposed to the substance when the emission takes place to an early part of the hydrological cycle (e.g. a stream). The dispersion is primarily caused by the advective flow of the water and hence by the residence time of the substance in the free water mass as determined by removal processes like degradation, evaporation and immobilisation. Persistent toxicants like heavy metals and chlorinated organic compounds may thus be transported between different environmental compartments and have an impact on several types of organisms and ecosystems before they are degraded or irreversibly incorporated into an organic or inorganic matrix. This mechanism is determined by the ability of the substance to persist in the environment rather than by the point of discharge into the environment. On the effect side, all aquatic ecosystems are generally represented by just one PNEC value for chronic aquatic effects. Within life cycle impact assessment, the impact of a toxic substance can not be modelled as more severe when the release point is at the `upper' part of the hydrological cycle compared to a release directly to the marine environment. The ability to persist in the environment, which strongly influences dispersion potential, is represented as a substance-specific characteristic in the site generic impact potential (see expression 8.4) Recommendation For emissions directly to soil, dispersion is very small and it is considered that the spatial variation at the actual level of the substance's fate as well as at the level relevant for spatial characterisation modelling is negligible. For emissions to aquatic systems the dispersion will depend on the type of receiving system – river, lake, estuary or ocean. As discussed in Section 8.1 and in Section 8.2.3.1 under Dilution, processes that influence exposure without reducing the total quantity of substance available for exposure can not be taken into account in current life cycle impact assessment because the impact factors are the same regardless of the concentration at which exposure occurs. Furthermore, no distinction is made between different types of aquatic systems in calculating the effect factor – the same PNEC is used for all aquatic systems. The ability of a substance to act on several targets from the release into the environment until it is irreversible trapped or degraded is mainly governed by the lifetime of the substance rather than the point of release. Consequently, this issue is addressed through the inclusion of biodegradation and other removal mechanisms in the site-generic characterisation factor. This means that there is no possibility for representing the effects of dispersion and no need to represent the position of the release point in the hydrological cycle for waterborne emissions in the spatial characterisation model. 8.2.3.3 Adsorption and immobilisationAdsorption is an important process that strongly influences the bioavailability and toxic impact of many chemical substances. In Annex 8.2 it is substantiated, that only the dissolved and freely available fraction of a substance will interact (bioaccumulate and exert toxicity) with biological organisms in the environment. Sorption processes (which include adsorption and its reverse process de-sorption) determine the distribution of a chemical between a liquid and a solid phase like pore water and soil or water and particulate material. Adsorption is primarily governed by the fraction of particles that represents the largest surface area. In the water column this is often the small organic particles below 0.2 m, which are regarded as dissolved organic carbon (DOC). The adsorption efficiency indicated by the sorption coefficient Kd depends on the affinity of the substance to these particles. For non-polar organic substances, the octanol-water partition coefficient, (Kow) is often used to indicate the potential for adsorption to organic matter. For substances that dissociate, including most metals, the adsorption efficiency depends strongly on the pH in the receiving environment and on the presence of other types of sorbents, e.g. ionic sites of organic matter and minerals. A review of sorption processes and mechanisms is given in Annex 8.2 while Annex 8.3 exemplifies with the influence of sorption on biodegradation of LAS in sediments. In aquatic systems, sorption may lead to a more permanent removal of the substance from the aquatic compartment when particles become integrated in the sediment. In sedimentation areas, the organic content in of the sediment is often high and may act as a trap for adsorbed substances, e.g. metals and lipophilic organic compounds. Sedimentation is the net result of deposition and re-suspension of particles. The rate of sedimentation depends on the hydraulic conditions of the aquatic system and on the nature of its particulate material. The net sedimentation can be regarded as the product of the net sedimentation in the different types of water environments which the substances pass from the emission point to the sea. In Annex 8.9 the removal of substances through the combined effect of biodegradation and sedimentation is calculated for a standard river, lake, estuary and sea for a readily biodegradable, an inherently biodegradable and a not biodegradable substance with log Kow varying in the interval from -3 to 6. By combination of the fractions left, a site factor SFsed for the removal through adsorption and sedimentation can be calculated for three standard scenarios reflecting different positions of the emission point in the hydro-geological cycle:
For each scenario, the SFsed factor is calculated by multiplication of the substance fractions fi remaining after passing the water systems involved in the scenario. For scenario 1) SFsed is thus calculated as the f-values for river, lake, estuary and sea. The f-values are calculated and shown in Annex 8.9 and the resulting SFsed-values are shown in Table 8.6. Table 8.6. Site factors (SFsed) representing removal by the combined effect of sedimentation and biodegradation for readily biodegradable, inherently biodegradable and not biodegradable organic substances of different lipophilicity for the three emission scenarios: emission to river and from there through lake to estuary and sea, emission through estuary to sea and emission directly to sea.
N.B.: Not biodegradable Immobilisation of metals may take place through chemical precipitation of compounds with low water solubility. In particular, anaerobic sediments act as a trap for some metals e.g. through formation of sulphides. For most metals, the adsorption and precipitation are strongly influenced by the pH of the medium as shown in Annex 8.2. pH also influences the function of micro-organisms and thereby the biodegradation of organic substances in the environment as discussed in the section on biodegradation below. Recommendations Sorption is relevant for characterisation modelling because it facilitates removal of the substance through sedimentation. Sorption is a reversible process and the decreased bioavailability of a substance that sorbs can be seen as a "dilution of the substance in time" – the concentration at any given time will be lower if the substance sorbs. This is not reflected in the characterisation modelling. For the spatial characterisation modelling, it seems reasonable to assume that the variation in sorption and sedimentation among different types of water recipients is more important than the geographically determined variation within one type of recipient across Europe. It is thus the former that should be addressed in the model, i.e. for the aquatic environment the differences between streams, lakes, estuaries and ocean in sedimentation velocity (and hence removal due to adsorption). The removal from the water column through the combination of sedimentation and biodegradation can be represented by the site factors in Table 8.6 For metals that form insoluble salts under environmental conditions, this will be the process that determines the actual propagation of the metal in the environment. This means that the metal will disperse until it interacts with anions and precipitates. The conditions allowing immobilisation of metals through precipitation will, however, always be represented within the actual fate area, and there will thus be no spatial variation in this aspect. Spatial variation of the precipitation of metals will therefore not be considered in the spatial characterisation model. Annex 8.2. gives a more detailed discussion of the mechanisms of adsorption and immobilisation. 8.2.3.4 BiodegradationThe dominating transformation process for organic substance in soil and aquatic systems is biodegradation. Given the right conditions, most organic compounds can be transformed to other substances and ultimately mineralised through microbial degradation. Hereby, the potential for exposure to the mother compound is reduced or eliminated. The potential for biodegradability is one of the most important factors when the environmental impact of substances is assessed, and it is already represented in the site-generic characterisation factor developed under EDIP. Here, the biodegradation potential is treated as a purely substance-specific property derived from the data on the behaviour in standardised biodegradability test according to OECD or EU guidelines (OECD, 1992, EEC, 1987). Results from such standardised screening tests for biodegradability are obtained under laboratory conditions and used as indicator for the potential of a substance to degrade in the environment as discussed further below. A number of factors may have a significant influence on the degradation of substances in the environment:
Organism-related factors Biodegradation of organic substances depends on the presence of competent micro-organisms in sufficient numbers. Natural microbial communities consist of a very diverse biomass and when a `new' substance is introduced in a sufficient high concentration the biomass may adapt to degrade the substance. The mechanism of adaptation includes growth of specific degraders that by nature are competent to degrade the substance, but also enzyme induction, exchange of genetic material and development of tolerance to toxicity of the substances may be involved. Both readily degradable substances and inherently degradable substances may require an adaptation. Biodegradation studies often show an initial lag phase where no or only little mineralisation of the substances is observed, but the environmental significance of adaptation is probably more pronounced for inherently biodegradable substances than for readily biodegradable substances. The adaptation of a microbial community to degrade a substance thus depends on the history of the community, i.e. whether it has been exposed to the substance or a structurally related substance previously. This means that when a xenobiotic substance has been used and emitted for a period of time, the likelihood of finding competent degraders will increase. There is thus a higher chance of adaptation in environments that are exposed to chemicals more or less continuously. Substrate-related factors In the laboratory tests that are used for evaluation of the degradation of chemical substances the substances are applied in high concentrations (mg/l - range) compared to the levels that are expected to be found in the environment (lower g/l range). In general, growth of micro-organisms is not supported when the substrate is present at concentration below about 10 g/l. The reason for this threshold is possibly a lack of sufficient stimulus to initiate an enzymatic response. Consequently, the environmentally relevant concentrations of chemicals are mostly at levels where the chemicals can not be the primary substrate for degrading micro-organisms. In standard tests, the substrate (the tested chemical) is applied as the sole carbon source, while in the environment a large number of other substrates are available. In aquatic environments, the concentration of dissolved organic carbon is often found in the range 1 – 10 mg/l, i.e. around a factor of 1000 higher than the chemical pollutants. Although only a fraction of this pool of organic substance is readily degradable by micro-organisms, it is likely to be more attractive as a carbon source than the much lower concentrations of chemicals present. On the other hand, the natural carbon sources may facilitate a degradation of the chemicals present at lower concentrations by co-metabolism. The co-metabolised substrates may then be available for further degradation. Environmental factors The redox potential, i.e. the availability of oxygen is probably one of the most important environment-related factors. Under normal circumstances aerobic conditions are prevailing in soil, in the water phase of aquatic systems and in top sediments. Anaerobic conditions are present in sediments, soil micro-environments and in some treatment steps in biological treatment plants. Low oxygen concentrations may moreover be present in eutrophic (nutrient rich) lakes and sediments. Thus anaerobic conditions are found in many natural (and technical) sub-compartments. Another important parameter for degradation is the temperature. Most laboratory tests are performed at room temperature (20-25 °C). Under environmental conditions, the temperatures typically range from below 0 to 50 °C and in rare cases even higher. Considering, that the optimum range is probably from 20 to 30 °C for most micro-organisms and that degradation rates on average increase roughly by a factor of 2 for every 10 °C increase of the average ambient temperature within this range, it is clear that the temperature plays an important role in degradation processes. The effect of the temperature on degradation rates is discussed further in Annex 8.3. Annex 8.7 presents annual average temperatures for the European countries and for the four European regions introduced in Section 8.2.2. For the four European regions, the estimated annual average temperatures are given in Table 8.7. Table 8.7. Estimated annual average temperatures for the four European regions.
The biodegradation half-life can be assumed to increase roughly by a factor of 2 for every 10oC decrease in temperature. This gives the following correction factors for the influence of the temperature on biodegradation (reference temperature = 10°C):
The pH in different types of environments may range from 5 – 8 and microbial organisms can be active within this range. However, for bacteria as a group, slightly alkaline conditions favour the activity and the optimum pH range is 6-8, and at pH levels below 5 the activity is decreased significantly. Fungi have in general a lower pH-optimum in the range 5-6. The bioavailability of the substances is critically important also to degradation, which primarily takes place in the aquatic phase of the ecosystems, i.e. the pore water in soil and sediments and in the aquatic compartment. The bioavailability is determined by sorption processes and is discussed under 8.2.3.3. An illustration of the influence that some of the environmental descriptors discussed above may have on the environmental biodegradation is given in Annex 8.3 for the detergent LAS. Recommendations Biodegradation has a major influence on the impact of chemicals in the environment. It is influenced by several factors related to the microbial organisms performing it, the availability of substrate and the environmental conditions. Most of these factors show no variation in their distribution at the spatial scale at which the substance's fate occurs, except for very short-lived substances. This is the case for the redox potential (occurrence of anaerobic conditions) and the pH, except for emissions to lakes where conditions may vary strongly from lake to lake within a region. For temperature, there is a clear geographical trend going from north to south in Europe and therefore temperature-dependence shall be included in the spatial characterisation model. An operational approach may be based on the division of Europe into three to four regions and the assumption that the substance stays within the region that it is emitted in. Site factors for the temperature influence on biodegradation in northern, mid-, and southern Europe are proposed. Also for adaptation of micro-organisms there is a differentiation according to whether the ecosystem is frequently exposed to industrial or municipal wastewater discharges or not. This differentiation will not be included in the spatial characterisation model because of a lack of concrete knowledge of the significance of this parameter. 8.2.3.5 Other transformationFor some substances, transformation may also occur through physical or chemical interactions like photolysis, hydrolysis or chemical reactions. Photolysis Photolysis is the cleavage of light-sensitive molecules under the influence of light. Photolysis may be an important degradation process for some chemicals in water. The rate of photolysis in the water depends on a number of environmental conditions such as latitude, cloudiness, time of day and year, concentration of suspended matter in the water column, and to some extend on the temperature. Apart from the concentration of suspended matter, all of these parameters are related to the geographical location and it is thus possible to include their influence on the photolysis rate in a spatial characterisation model. Since the extinction of light occurs rather quickly down through the water column (and even more quickly in soil), it is in the present context only considered relevant only for substances emitted to air. For air-borne emissions the dispersion will be on a scale that for practical purposes averages out geographically determined differences in light intensity. Photolysis is thus not included in spatial characterisation. Hydrolysis Hydrolysis is the cleavage of bonds in the molecule of many organic substances due to reaction with water molecules. It is only considered relevant for substances present in water, where it may be the predominant degradation route for some organic substances. The rate of hydrolysis depends on the pH and the temperature. Hydrolysis is normally represented in the biodegradation potential since normal test conditions for biodegradability also facilitate hydrolysis. Its temperature dependency is hence considered included under the temperature dependence of biodegradation (8.2.3.4). As for biodegradation, the pH dependence of hydrolysis is not considered relevant to include in the spatial characterisation model. Chemical interaction Once emitted, the substance may interact with the multitude of other chemical substances present in the environment as pollutants or natural compounds. One of the most important reactions with relevance for nearly all volatile organic compounds is the photochemically driven oxidation by hydroxyl radical in the atmosphere under the influence of light and in the presence of nitrogen oxides, NOx. Photochemical oxidation will vary with the intensity of sun light (like photolysis) and the background concentration of hydroxyl radical. The latter will vary locally with the occurrence of organic substances e.g. from other pollution sources, i.e. it may be lower in industrialised and urban areas. For air-borne emissions, however, it is anticipated that the dispersion will be on a scale that averages out such differences so variations in the hydroxyl radical concentration can be disregarded in site characterisation. The highest rate of transformation through reaction with other substances is expected to occur where emissions of man-made pollutants are frequent. Recommendations Apart from hydrolysis, none of the transformation mechanisms show important variations at the actual spatial scale of the fate of the substance except for very short-lived substances. For hydrolysis, the rate will be temperature-dependent and thus follow the geographical temperature gradient mentioned earlier. It is however expected that data on the removal through biodegradation also include removal through hydrolysis and no separate treatment is therefore given to hydrolysis. 8.2.4 ExposureFollowing the transport and transformations of the emitted substance as discussed in 8.2.3, an exposure of one or more target systems occur. The effect elicited by the exposure is a response to the overall exposure experienced by the target system and it can be difficult to isolate the contribution from the product system alone. Concentration increase caused by the emission is in principle modelled or at least represented by the fate model. Some of the fate models used in LCIA actually attempt to estimate a concentration increase in the environment caused by the product system using modified environmental models like EUSES or CalTOX as discussed in Section 8.1.1. Given the constraints of life cycle assessment where the inventory result is an emitted mass corresponding to the functional unit, the value of such concentration estimates is debatable. In EDIP it was chosen to represent the most important fate properties in a transparent way in the fate model rather than adopt already existing models that were developed for something else to arrive at a quite intransparent fate model of questionable relevance. The background level of chemical exposure of the ecosystem is crucial to the quantification of the impact from the concentration increase caused by the product system. Normally, the contribution from a product system to the mixture of chemicals present in the environment is small. Since different chemicals often interact in their toxicity, it is the overall level of exposure that is relevant here, not just the background concentration of a specific substance. As discussed in Section 8.1.1, it is a general assumption in characterisation modelling of ecotoxicity that the concentration-impact curve of the ecosystem is linear. The background level of pollution is thus not taken into account. Even though a high degree of spatial variation in the background level can occur at the relevant scale, this is also ignored in chemical risk assessment today except in very site-specific studies. EU is currently implementing environmental monitoring programmes, which in the future may enable assessment of background levels of toxic pollutants. Bioconcentration of the chemical is attempted represented in a generic way in the PNEC when this is estimated from chronic data generated in tests with a duration that allows the bioconcentration to occur and influence the test results. In the EDIP97 method (in contrast to other LCIA methods for ecotoxicity), a correction for bioconcentration is introduced when the PNEC value is based on acute test data. No spatial variation is anticipated in the bioconcentration potential. Biomagnification, i.e. food chain concentration is not represented in the EDIP97 methodology where exposure is considered through contact to the surrounding medium and there is no specific representation of the top predators that are susceptible to biomagnification. This aspect is not included in the other models for ecotoxicity characterisation modelling that are known to the authors but it should be included in future revisions of the EDIP method. For the current context, it is not likely to show spatial variation at the relevant scale. Duration of exposure depends on the overall removal rate for the substance in that environmental compartment and it should in principle be represented in the site generic fate modelling, i.e. the spatial variation should be taken into account through the considerations presented in 8.2.3. Recommendations The background level of pollution of the exposed ecosystem is expected to be an important site-dependent descriptor, since the species of the ecosystem are exposed to the combination of the chemicals from the product system and the chemicals emitted by other activities exposing the same ecosystem. It is, however, at present not possible to incorporate site-dependent information about the background level of pollutants in the exposed ecosystems in characterisation of ecotoxicity. 8.2.5 Target systemThe target system is the ecosystem or ecosystems that are exposed to the emission from the product system. The relevant descriptor for this link of the cause-impact chain is the sensitivity of the exposed ecosystem as expressed by the concentration-effect curve of the substance. Also the aspect of potential exposure of several consecutive aquatic ecosystems is relevant here. 8.2.5.1 Sensitivity of ecosystemDifferent aquatic ecosystems show different sensitivities depending on their species composition. Ecosystems that are already exposed to man-made pollutants may have lost the most sensitive species so the species sensitivity distribution curve is shifted towards higher values. This means that the already exposed ecosystems are less sensitive and hence that the concentration that protects the function of pristine ecosystems is lower than the concentration protecting already exposed systems. Normally, only one PNEC is derived representing all aquatic (or terrestrial) ecosystems. Only for very few substances, there will be adequate toxicity data to allow estimating a PNEC value based on information for species that are actually representing the ecosystem that is exposed. A review and statistical evaluation of published aquatic mesocosm studies (Petersen 2000) showed a variation in sensitivity of 2.5 orders of magnitude between different zooplankton communities and that the variation among individual groups of macroinvertebrates is 1- 2 orders of magnitude. Such studies indicate that there is a high variability among individual aquatic communities and ecosystems and that the size of this variation may be comparable to the fate based variation in exposure. The results were estimated from actual measured concentrations and can therefore be attributed to biological differences among the exposed organisms. In generic LCA as well as in risk assessments, it is however not possible to include the variation in sensitivities among the individual aquatic systems and in general, the estimated PNEC values are believed to represent a level where the ecotoxicological impact on the system is acceptably low. Recommendations The sensitivity distribution curves and the ecosystem no effect concentrations will vary between aquatic ecosystems, but this is a variation not taken into account in current methods for generic risk assessment. Development in risk assessment tools aims at an incorporation of site-dependent information on background exposure and possibly also ecosystem sensitivity (Feijtel et al., 1997, ECETOC, 1999) but until such tools become available it is not possible to include these aspects in site-dependent characterisation modelling of ecotoxicity. 8.2.6 ImpactIn the characterisation of ecotoxicity, no distinction is made between the different types of effect caused by the chemical (lethality, endocrine disruption or other reproductive effects, avoidance or other). The effect parameter PNEC is determined as the concentration at which no effects are observed in a test system. The effect that is modelled is thus the effect expected to occur if the PNEC value is exceeded. No spatial variation is expected for this part of the cause-impact chain. Recommendations No spatial differentiation is relevant. 8.2.7 Descriptors in the spatial characterisationSummarising Sections 8.2.1-8.2.5, the following descriptors have been identified as potentially relevant to the spatial characterisation modelling in LCIA 8.2.7.1 Emissions to airThe air compartment is treated exclusively as a dispersion medium and therefore, the issue is the spatial variation in exposure of aquatic and terrestrial systems through deposition from the atmosphere. Table 8.8. Descriptors of relevance to the spatial characterisation of emissions to air
8.2.8 Emissions to waterFor emissions to the water compartment, a distinction is made between acute and chronic ecotoxic effects. Acute aquatic ecotoxicity The only descriptors considered of relevance are related to the emission. They concern the temporal course of the emission (pulse or continuous) and the dilution immediately after discharge. Due to the assumption of linearity between concentration and impact according to the current ecotoxicity characterisation modelling including the EDIP97, these descriptors can not be represented in the spatial characterisation modelling (see Section 8.1.1). Chronic aquatic ecotoxicity For chronic aquatic ecotoxicity, several descriptors are identified as relevant to spatial characterisation modelling. These are presented in Table 8.9. Table 8.9. Descriptors of relevance to the spatial characterisation of emissions to water
8.2.9 Emissions to soilFor chronic terrestrial ecotoxicity several descriptors are identified as relevant to spatial characterisation modelling. They are presented in Table 8.10.
Table 8.10. Descriptors of relevance to the spatial characterisation of emissions to soil 8.3 Development of spatial characterisation factorsThree levels of spatial differentiation have been defined:
The existing characterisation factors for ecotoxicity from EDIP97 or similar are considered to represent the full site-generic impact potential and are in principle not modified in the present context. The ambition is to develop site-factors that represent the extent to which exposure allows this full potential to be reached as discussed in Section 8.1.2. 8.3.1 Inclusion of spatial variation of descriptorsFrom the analysis of the causality chain for ecotoxicity in soil and water in Sections 8.2.1-8.2.5, the descriptors relevant to spatial characterisation modelling were identified in Tables 8.8-8.10. Among these, the following are judged possible to include in life cycle impact assessment: Emissions to air: Included:
Not included:
Emissions to water: Included:
Not included:
Emissions to soil: Included:
Not included:
8.4 A framework for spatial characterisation modellingThe already existing site-generic characterisation factor is interpreted as representing the full (in the case of EDIP97, but indeed, for some other LCIA methodologies, it is rather the average) impact from the substance. The spatial characterisation factor is the product of the site-generic characterisation factor and a site factor, which is seen as a modifier expressing the degree to which the full impact comes through:
... as already expressed in equation (3). In contrast to the expression in (8.3), the site factor, SF depends both of the substance properties and the spatial characteristics of the process. Hence, in equation (4), SF has been given the index "I" in addition to the index "p". The modelling of SF must represent the spatial variation in the descriptors listed in Section 8.3.1, and this is attempted through expressing SF as a product of the following variables:
(8.5)
8.4.1 The emission componentFor emissions to air or emissions to air or soil which are found to evaporate, the SFemis factor reflects the fraction of the deposited part of the emission that will expose water or soil ecosystems. In connection to EDIP97, SFemis is defined as (8.6) Chronic aquatic ecotoxicity: (8.7) Chronic terrestrial ecotoxicity: (8.8.) Acute aquatic ecotoxicity: The fractions deposited to water and soil natural areas are found for the four European regions in Table 8.3. The approach may later be sophisticated to allow country-specific modelling by combining the information on the relative frequency of natural areas within the countries of Europe (Annex 8.4) with atmospheric dispersion modelling as performed in e.g. Chapter 3. Based on Table 8.3, SFemis is calculated assuming a global default value of 0.2 for a: Table 8.11 SFemis for emissions occurring in different regions of Europe.
8.4.2 The evaporation componentThe temperature dependency of evaporation is substance specific. In Section 8.2.3.1 the influence of the temperature on evaporation is expressed in generic formulas. Based on some rough assumptions regarding the average temperature in different European regions values for SFevap are proposed in Table 8.5 which is repeated below: Table 8.5. Site factors, SFevap for volatilisation from water for two European regions
SFevap was developed for evaporation from water but it is proposed used also for evaporation from soil. 8.4.3 The biodegradation and transformation componentThe SFbio factor reflects the variation of biodegradability with the average temperature of the region where the fate of the substance takes place. It is relevant for both aquatic and terrestrial systems. The annual average temperature over Europe varies around 10 C between the Nordic region and the Southern region with the Western and Eastern regions in-between. If it is assumed, that the current site-generic fate modelling (in EDIP97 or other LCIA methodology) corresponds to an average mid-European situation, the factor is determined in Section 8.2.3.4 as:
The approach may later be sophisticated to allow country-specific modelling through combining the information on the annual average temperature of the countries of Europe (Annex 8.7) with atmospheric dispersion modelling as performed in e.g. Chapter 3 In a future development of the site-generic EDIP97, it would be relevant to modify the BIO factor representing the substance-specific biodegradation potential based on results of standardised biodegradability tests. In EDIP97, the ratio between BIO for a not biodegradable substance and BIO for a readily biodegradable substance is only a factor 5. This should be increased e.g. seeking inspiration in the suggested biodegradability rates in the EU TGD (1996) for natural terrestrial environments and natural waters based on the same test results. 8.4.4 The sorption and sedimentation componentThe SFsed factor must reflect the spatial variation in the relative importance of sedimentation as a removal process for substances adsorbing to particulate material in different aquatic systems. The SFsed factor is only relevant for substances emitted to or ending in the aquatic compartment of the environment. In EDIP97, there is no consideration of removal due to sedimentation. This is equivalent to operating with a removal factor with the value 1 (just like no potential for biodegradation is represented by a BIO factor value of 1). Site factors for sorption and sedimentation of organic substances are developed in section 8.2.3.3. They include the biodegradability of the substance in the calculation of the fraction, which is removed from the water phase of the aquatic system by sedimentation. Thus, the removal by sedimentation depends on:
These parameters are included in the values for SFsed presented in Table 8.6 and repeated below. Table 8.6. Site factors (SFsed) representing removal by the combined effect of sedimentation and biodegradation for readily biodegradable, inherently biodegradable and not biodegradable organic substances of different lipophilicity for the three emission scenarios: emission to river and from there through lake to estuary and sea, emission through estuary to sea and emission directly to sea.
N.B.: Not biodegradable 8.5 Spatial characterisation factors for ecotoxicityIn the framework laid out for calculation of site factors, SF in Section 8.4 and expressed in Expression (8.5), the determining parameters are Spatial
Using Expression (8.5) and the values given for the factors of SF in the previous sections, the ranges (min-max value) of SF are calculated and shown in Table 8.12. Table 8.12. Ranges (min-max value) of the site factor SF
8.6 Discussion and recommendationsAnalysing the extreme values of the site factors shown in Table 8.12, it is found that the largest variation which can be introduced by using this framework for spatial characterisation of aquatic ecotoxicity is a factor 33 (between the value for a semi volatile and not biodegradable substance emitted directly to the sea in Northern Europe and a strongly lipophilic substance emitted to a river in Western Europe). For substances of less extreme lipophilicity(logKow<4), the largest variation is a factor 7.8, and it is found between the same two situations. For terrestrial ecotoxicity, the largest variation is a factor 5.6 (between the value for a semi volatile and not biodegradable substance emitted to soil in Northern Europe and any substance emitted to soil in Southern Europe). Except for extremely lipophilic substances, this is a rather modest spatial variation, and it is indeed expected to represent only a minor part of the actual spatially determined variation in the fate and exposure of ecosystems to chemicals within Europe. Several reasons can be given for this:
It is the authors' judgement that the site factors developed for ecotoxicity are accompanied by considerable uncertainties, and that these uncertainties may well exceed the variation given by the factors. Application of the factors in life cycle impact assessment may thus introduce a model uncertainty which is larger than the reduction of the spatially determined uncertainty. On this background, the authors do not find it recommendable to apply the developed site factors in an attempt to perform spatial characterisation of ecotoxicity in LCIA. Currently, work is underway in the OMNIITOX project under the fifth Frame Programme of EU on development of a European consensus method for characterisation of ecotoxicity in LCA (http://www.omniitox.net). This method involves a comprehensive multimedia fate model with the option of spatial differentiation at the level of countries for Europe. The reader with interest in spatial characterisation of ecotoxicity is referred to the results of this work which will be available towards the end of 2004. 8.7 ReferencesAnkley, G. T., D. A. Benoit, J. C. Balogh, T. B. Reynoldson, K. E. Day & R. A. Hoke: Evaluation of potential confounding factors in sediment toxicity tests with three freshwater benthic invertebrates. Envir. Toxicol. Chem. 13: 627–635, 1994. Borkar, M.D. and Pillia, K.C.: Sedimentation rate in the Trombay bay, west coast of India. Indian Journal of Marine Sciences, 20(2), 115-117, 1991. Brugmann, L.: Metals in sediments and suspended matter of the river Elbe. Sci.Tot.Env., 159(1), 53-65, 1995. Chakrapani, G.J. and Subramanian, V.: Rates of erosion and sedimentation in the Mahanadi river basin, India. J. of Hydrology, 149(1-4), 39-48, 1993. Chenhall, B.E.: Anthropogenic marker evidence for the accelerated sedimentation in Lake Illawarra, New South Wales, Australia. Oceanographic Literature Review, 43(3), 305, 1996. Christensen T.H.: Cadmium Soil sorption at low concentrations: VIII. Correlation with soil parameters. Water, Air and Soil Pollution 44, 71-82, 1989. Das B.K., and Singh, M.: Rate of sediment accumulation in lakes of Udaipur, Rajasthan, India using the 210Pb method. Environmental Geology (New York), 24(1), 28-33, 1994. ECETOC: Environmental Exposure Assessment. Technical Report No. 61. ECETOC, 1994. ECETOC: GREAT-ER User Manual. Special Report no. 16, European Centre for Ecotoxicology and Toxicology of Chemicals, Brussels, 1999. EEC: 87/302/EEC: Test for potential biodegradability, equivalent to OECD 302 B-C (1992/1981) , 1987. Egebart, C.L.: Modelling aquatic fate of neutral organic pollutants for LCIA. KPD-20-99, Department of Manufacturing Engineering, Technical University of Denmark, 1999. EU-TGD: Technical Guidance Document in support of the commission directive 93/67/EEC on risk assessment of chemical substancesBrussels, 1996. Farmer, J.G.: The determination of sedimentation rates in Lake Ontario using 210Pb dating method. Canadian Journal of Earth Sciences, 15(3), 431-437, 1978. Feijtel, T., Boeije, G., Matthies, M., Young, A., Morris, G., Gandolfi, C., Hansen, B., Fox, K., Holt, M., Koch, V., Schroder, R., Cassani, G., Schowanek, D., Rosenblom, J. and Niessen, H.: Development of a geography-referenced regional exposure assessment tool for European rivers – GREAT_ER, Contribution to GREAT_ER1. Chemosphere, 34(11), 2351-2373, 1997 Georgiyevski, A.Ye. and Kuptsov, V.M.: Determination of sedimentation rates in the Gulf of Ria by the non-equilibrium 210Pb method. Okeanologiya, 26(2), 1986. Goedkoop, M and Spriensma, R: The Eco-indicator 99 A damage oriented method for life cycle impact assessment. Methodology report, preliminary internet version 5 October 1999, PRé Consultants B.V., Amersfoort, the Netherlands, 1999. Guinée, J., Heijungs, R., van Oers, L., van de Meent, D., Vermeire, T. and Rikken, M.: LCA impact assessment of toxic releases. Generic modelling of fate, exposure and effect for ecosystems and human beings with data for about 100 chemicals. VROM, no. 1996/21, The Hague, 1996. Haitzer M., S. Hoss, W. Traunspurger, C. Steinberg: Effects of dissolved organic matter (DOM) on the bioconcentration of organic chemicals in aquatic organisms - a review -. Chemosphere 37(7), 1335-1362, 1998. Harremöes P. & A. Malmgren-Hansen: Lærebog i Vandforurening. Polyteknisk Forlag, 1989. Hatzinger, P.B., M. Alexander: Effect of aging of chemicals in soil on their biodegradability and extractability. Environmental Science and Technology 29(2), 537-545, 1995. Hauschild, M.: Estimating pesticide emissions for LCA of agricultural products. In Weidema, B.P. and Meeusen, M.J.G. (Eds.): Agricultural data for life cycle assessments, volume 2, pp. 64-79, ISBN 90-5242-563-9, Report no. 2.00.01, Agricultural Economics Research Institute (LEI), The Hague, 2000. Hauschild, M.Z. and Wenzel, H.: Environmental assessment of products. Vol. 2 - Scientific background, 565 pp. Chapman & Hall, United Kingdom, 1998, Kluwer Academic Publishers, Hingham, MA. USA. ISBN 0412 80810 2 He, Q., Walling, D.E. and Owens, P.N.: Interpreting the 137Cs profiles in several small lakes and reservoirs in the Southern England. Chemical Geology, 29(1-2), 115-131, 1996. Hertwich E.G.: Toxic Equivalency: Accounting for Human Health in Life-Cycle Impact Assessment. University of California, Berkeley, 1999. Houx N.W.H. & W.J.M. Aben: Bioavailability of pollutants to soil organisms via the soil solution. Sci. Total Environ. Suppl., 387-395, 1993. Huijbregts, M. and J. Seppälä: Towards region-specific, European fate factors for airborne nitrogen compounds causing aquatic eutrophication. Int. Journal of Life Cycle Assessment, 5(2), 65-67, 2000. Huijbregts, M.A.J.: Priority assessment of toxic substances in the frame of LCA. Development and application of the multi-media fate, exposure and effect model USES-LCA. Milieukunde, Universiteit van Amsterdam, Amsterdam, 1999. Jolliet, O. and Crettaz, P.: Critical surface-time 95. A life cycle impact assessment methodology including fate and exposure. École Polytechnique Fédérale de Lausanne, Switzerland, 1997. Kleier, D.A.: Phloem mobility of xenobiotics. Plant Physiol. 86, 803-810, 1988. Knaebel, D.B., T.W Federle, D.C. McAvoy & J.R. Vestal: Effects of Mineral and Organic Soil Constituents on Microbial Mineralization of Organic Compounds in Natural Soil. Appl. Environ. Microbiol. 60(12) 4500-4508, 1994 Kristensen, P. and Hansen, H.O.: European rivers and lakes. Assessment of their environmental state. EEA Environmental Monographs 1, European Environmental Agency, Copenhagen, 1994 Lesuers, P., Weber, O., Marambat, L., Tastat, J.-P., Jouanneau, J.-M. And Turon, J.-L. : Age of an Atlantic shelf mud path supplied by an estuary : the west Gironde example (France) was deposited in historic times. Comptes Rendus de l'Academie des Sciences, Seri II (Mecanique, Physique, Chimie, Sciences de l'Univers, Sciences de la Terre), 308(10), 935-940, 1989. Lyman W.J., W.F. Reehl W.F. & D.H. Rosenblatt : Handbook of chemical properties estimation methods; Environmental behaviour of organic compounds. McGraw-Hill, 1982. Madsen, T., M. Winther-Nielsen, & D. Rasmussen: Studies on the fate of linear alkyl benzene sulfonates (LAS) in sludge and sludge-amended soil. The CLER Review, 5(1), 14-19, 1999. Matthijs E. & H. De Henau: Adsorption and Desorption of LAS. Tenside Detergents 22(6), 299-304(1988). McKone T.E.: CalTOX, A Multimedia Total Exposure Model for Hazardous-Waste Sites UCRL-CR-111456PtI-IV. Lawrence Livermore National Laboratory, Livermore, CA, http://www.cwo.com/~herd1/caltox.htm, 1993. OECD: OECD 302 B-C: Test for potential biodegradability, equivalent to 87/302/EEC, 1992. Pedersen F., A. Damborg & P. Kristensen: Industrispildevands miljøfarlighed. Miljøprojekt, 260, 1994. Petersen S.: Model based tool for evaluation of exposure and effects of pesticides in surface water. Draft report to the Danish Environmental Protection Agency, 2000. Potting, J. and Hauschild, M.: Predicted environmental impact and expected occurrence of actual environmental impact. Part I: The linear nature of environmental impact from emissions in life cycle assessment. International Journal of Life Cycle Assessment 2(3), 171-177, 1997a. Potting, J. and Hauschild, M.: Predicted environmental impact and expected occurrence of actual environmental impact. Part II: Spatial differentiation in life cycle assessment via the site-dependent characterisation of environmental impact from emissions. International Journal of Life Cycle Assessment 2(4), 209-216, 1997b. R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental organic chemistry. Wiley Interscience, New York, 1993. Ravichandran, M., Baskaran, M, Santschi, P.H. and Bianchi, T.S.: Geochronology of sediments in the Sabine-Neches estuary, Texas, USA. Chemical Geology, 125, 291-306, 1995. RIVM: Uniform System for the Evaluation of Substances 2.0 (USES 2.0) 679102044. National Institute of Public Health and the Environment (RIVM), Ministry of Housing, Spatial Planning and the Environment (VROM), Ministry of Health, Welfare, Sport (VWS), The Hague (NL). Rostan, J.C., Juget, J. and Brun, A.M.: Sediment rate measurements in the former channels of upper Rhône river using Chernobyl 137Cs and 134Cs as tracers. Sci.Tot.Env., 193(3), 251-262, 1997. Santschi, P.H., Allison. M.A., Asbill, S. and Perlet, B.: Sediment transport and Hg recovery in Lavaca Bay as evaluated from radionuclide and Hg distribution. ES&T, 33, 378-391, 1999. Stober, J.C. and Thompson, R.: Palaeomagnetic secular variation studies of Finnish lake sediment and the carriers of remannence. Earth and Planetary Science Letters, 37(1), 139-149, 1977. van Leeuwen C,H, & J.L.M. Hermens: Risk Assessment of Chemicals: An Introduction, 1995. Veerkamp and Berge: Hazard assessment of chemical contaminants in soil. Revised appendix 3. ECETOC Technical Report No. 40, 1992. Verschueren K.: Handbook of Environmental Data on Organic Chemicals. Environmental Sciences. Electronic Resources. 3rd edition, 1998. Vogel, R.L., Kjerfve, B. and Gardner, L.R.: Inorganic sediment budget for the North Inlet saltmarsh, South Carolina, USA. Oceanographic Literature Review, 44(6), 579, 1997. Wenzel, H., Hauschild, M.Z., and Alting, L.: Environmental assessment of products. Volume 1. Methodology, Tools and Case Studies in Product Development. Chapman and Hall, London, 1997. Yu, K.N., Young, E.C.M., Stokes, M.J. and Guan, Z.J.: Determination of sedimentation rates in eastern sea areas of Hong Kong with gamma-ray spectrometry. Nuclear Geophysics, 9(1), 73-81, 1995. Zuo, Z., Eisma, D, Gieles, R. And Beks, J.: Accumulation rates and sediment deposition in the Northwestern Mediterranean. Deep-Sea Research II, 44(3-4), 597-609, 1997. Annexes: Annex 8.1: Initial dilution of waterborne emissions Annex 8.2: Adsorption and immobilisation Annex 8.3: Biodegradation of LAS under different temperature and sorptive conditions Annex 8.4: Relative share of natural areas within the regions of Europe Annex 8.5: Relative share of emissions depositing to sea and land within the regions of Europe Annex 8.6: Sedimentation velocities in different aquatic systems Annex 8.7: Annual average temperatures of European countries Annex 8.8: Rate constants for evaporation of substances from water Annex 8.9: Removal of substances from water through the combined effect of biodegradation and sedimentation Annex 8.1 Initial dilution of waterborne emissionsThe initial dilution of waterborne emissions is taken as the average dilution in the cross sections of the wastewater plume at the point where the impulse of the plume equals the impulse of the surrounding water masses or where the plume reaches the water surface. The initial dilution is independent of the substance characteristics and governed by the following parameters: The type of discharge The characteristics of the receiving water body (water exchange rate, depth, difference in salinity of the outlet and the receiving water body) The position of the discharge point in the water column (at the bottom or the surface of the receiving water) The geometry of the discharge pipe The discharge flow velocity (both the volume and the linear flow velocity) Each of these parameters may vary substantially within Europe. Examples of resulting and applied dilution factors are shown in Figure 8.3.
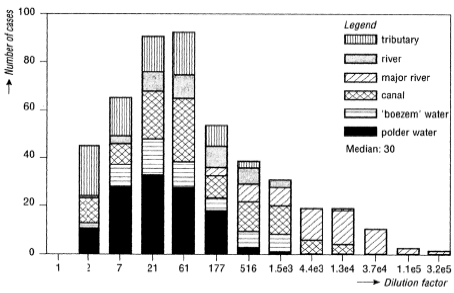
Figure 8.3. Distribution of the dilution factors for Dutch waste water emissions 1000 m downstream of the treatment plants. The type of receiving water is indicated - boezem and polder water refers to flooded marsh areas (Leeuwen and Hermens, 1995). Figure 8.3. shows the estimated dilution factors for municipal wastewater treatment plants in the Netherlands 1000 m downstream from the emission point. The data shows a considerable variation with a median dilution factor of 30. A dilution factor of this size at a distance of 1000 meters indicates a small initial dilution at the discharge point. Examples of default values applied as initial dilution factors in the environmental administration are:
The overall average dilution of wastewater emitted to rivers can be estimated roughly for an area by dividing the wastewater emission per capita (representing all wastewater within a specific region) by the excess precipitation. This approach will underestimate the true average value in countries that discharge wastewater directly to the sea or which receive less polluted river water from neighbouring countries. Assuming an average wastewater volume of 200 l per capita per day (person equivalent) this approach gives an average dilution factor for wastewater discharged to aquatic environments excluding marine waters ranging from 12 (the Netherlands), 15 (Belgium and Luxembourg), and 17 in the UK to 134 in Ireland (ECETOC, 1994). Annex 8.2 Adsorption and immobilisationAdsorption is an important parameter that often has a major influence on the bioavailability and toxic impact of chemical substances. Because of the importance of adsorption to environmental fate and impact of toxicants it is discussed in details below. Sorption (adsorption and desorption) are processes that determine the distribution of a chemical between a liquid and a solid phase in a multiphase system like pore water - soil or sediment - water. Adsorption includes interaction with small organic particles below 0.2 µm, which normally is regarded as dissolved organic matter. Sorption processes involve different types of chemical binding from weak and reversible interactions like hydrogen bonds or van der Waals forces to stronger and less reversible ionic or covalent bonds. Sorption is an equilibrium process, the position of which reflects the relative affinities of the substance to the solid and liquid phases. The weaker the bonds to the solid phase, the more the substance will desorb and be present in the liquid phase. The position of the equilibrium is described through the adsorption coefficient, Kd, defined as the ratio between the concentration on the solid phase and the concentration in the liquid phase:
Kd can be determined experimentally but often, it is estimated from knowledge about the substance's molecular characteristics (through structure-activity relationships). Using Kd, the concentration in the liquid phase (Cl) can be determined from the total concentration of the substance in the compartment. Relevance of sorption In general it can be assumed that only the dissolved and freely available fraction of a substance will interact (bioaccumulate and exert toxicity) with biological organisms in the environment. Several studies support this theory. Ankley et al. (1994) showed that the effect of the pesticide chlorpyrifos on the midge Chironomus tentans was highly dependent on dissolved organic carbon (DOC) in the pore water of sediments. DOC is normally defined as the fraction passing a 0.20 or 0.45 µm filter and includes colloids as well as true dissolved matter. LC50 value in pore water corresponded with the LC50 value obtained in pure water only if the fraction adsorbed to DOC was considered to be unavailable for the animals. The nominal pore water concentration was about 10 times higher than the LC50 value. Houx & Aben (1993) exposed nematodes to chlorpyrifos and pentachlorophenol in different soil types. The toxicity of the spiked soil samples was negatively correlated to the soil organic content and the results indicated that the toxicity could be explained by the dissolved fraction of the substances alone. Knaebel and co-workers measured the degradation over time for the detergent LAS adsorbed to different solid materials. The initial rates of degradation were negatively correlated with the adsorption coefficient indicating that the initial degradation rate decreases with increasing strength of the adsorption (Knaebel et al., 1994). It should, however, be mentioned that in dilute media like the water phase of natural aquatic ecosystems, bacterial growth preferentially takes place on solid surfaces. This observation has inspired the theory that growth may be enhanced by the adsorption of organic matter at liquid-solid interfaces. In addition, there is some evidence that the microbial degradation of certain surfactants in environmental samples is faster in the adsorbed state than in solution. Painter et al. 1992 reported the half-life of LAS in river water without sediment to be 1.4 d and 0.7 d in the presence of sediment. In a recent review Haitzer and co-workers studied the effects of dissolved organic carbon (DOC) on the bioconcentration of different organic substances in aquatic organisms. Hydrophobic organic substances like polychlorinated dioxins, PAHs, pyrethroid insecticides, tributyltin and selected surfactants showed a reduced bioconcentration at DOC levels, which can be found in freshwater environments (0- 10 mg/l). For example a 51% reduction of the uptake of benz(a)pyrene was observed going from 0 to 6 mg DOC/l, and a 23% reduction of the uptake of trichlorobenzene by increasing DOC from 0 to 10 mg/l (natural DOC was used, isolated from a stream water). The reduction in the bioconcentration at low DOC levels (0-10 mg/l) was from 0 - 50% for different hydrophobic substances, although higher levels were reported for cationic tensides (Haitzer et al., 1998). Although the general trend is a reduced uptake at increasing levels of DOC, a stimulated uptake has been reported in specific cases where an increased uptake was observed for highly lipophilic compounds as DDT and polychlorinated biphenyls. The mechanism may be explained by an increased uptake via food. It can be concluded from a large number of experiments, including the reports cited above, that DOC has a considerable influence on the availability of hydrophobic substances in the aquatic environment. As mentioned earlier, dissolved organic carbon includes colloids as well as true dissolved matter. DOC thus includes organic substances with a high molecular weight, which act as a sorbent for lipophilic substances. Because of the size of the sorbent-substance complex the uptake and hence the bioavailability is reduced. Some general conclusions can be drawn from the discussion above regarding site characterisation in LCIA:
Mechanism of sorption Depending on their properties, substances sorb through different types of interactions with the solid phase. Non-ionic organic substances are rather hydrophobic and they sorb through weak van der Waals forces and hydrogen bonds to the organic fraction of the solid phase of soil or sediments. The adsorption is often described by Koc, which expresses the adsorption to the organic carbon content of the solid phase. Koc is defined as:
Where foc is the relative content of organic carbon in the solid phase. The degree of sorption of non-ionic organic substances to soil and sediment matrices (Kd) is thus proportional to the content of organic carbon. The sorption capacity is normally not regarded as a limiting factor because of the high concentrations of organic material in soils and sediments and the relatively low environmental concentrations of the released organic toxicants. The frequently used plasticizer DEHP (di(ethylhexyl)phthalate) is an example of a non-ionic hydrophobic organic substance that sorb readily to organic material in aquatic and terrestrial environments. Estimates of the dissolved fraction of DEHP in an aquatic system show a strong dependency on the amounts of suspended organic matter. The dissolved (available) fraction of DEHP may vary by a factor of 5-6, corresponding to a range of suspended organic carbon from 1 – 10 mg C/l observed in aquatic ecosystems. The relationships between dissolved DEHP and the concentration of suspended organic carbon is shown in Figure 8.4.
Figure 8.4. Estimated fractions of DEHP dissolved in the water phase as a function of the organic carbon content in suspended matter. The default value used in the EU-risk assessment guideline (TGD) is indicated with an asterisk. Ionic substances interact with electrically charged groups on the solid phase. The sorption of cationic substances like metals occurs as ion exchange on negatively charged sites on the solid phase of soils and sediments. Negative charges occur predominantly on the surface of clay minerals and on organic material containing dissociated acid and phenolic hydroxyl groups. The pool of dissolved ionic substances competes about these sites. Hydrogen ion has the strongest positive surface charge, and is naturally present in all systems containing water. Consequently, the sorption of cationic substances is strongly pH dependent. As an example, the sorption of cadmium in soils is suggested to depend on the pH according to the following general expression (Christensen, 1989):
The equation predicts a variation in Kd of cadmium in soil between 73 and 2600 (corresponding to a factor of 35), when the pH of the soil is varied within values naturally occurring in different soils (pH 5-8). A similar pH-dependency can be expected in sediments in the aquatic environment although the natural pH variation here is smaller (6.5-8.3) corresponding to a variation of Kd by a factor of 9 using the same expression. A correction for that the bioavailability of chemicals in soils depends on the soil type is introduced; a standard soil containing 3.4% organic matter is introduced, and the bioavailability of non-ionic organic compounds is assumed only to depend on the organic matter content, only. Ministry of Housing, Spatial Planning and the Environment (1994) (the Netherlands) has suggested a correction term for adsorption of heavy metals in soils, which includes the content of clay and organic matter. The correction term defines a standard soil or sediment containing 25% clay and 10% organic matter is defined here. The correction term reads:
Where a, b and c are constants depending on the compound in question. CStandard is the concentration in standard soil or sediment CMeasured is the measured concentration in soil or sediment fCorrection is the correction term The above expression can in principle be extended to any compound, which adsorbs to the clay and organic matter in the soil and sediment. The coefficient a, b and c should be found from experimental investigations. In Danish soils the clay content varies between 0 and 40% and the organic matter content varies between 1 – 8%. In the table 8.13 below, the equation is used to illustrate the importance of the soil type. For a metal like nickel the composition of the soil has a dramatic effects on the bioavailability, whereas the effect is less significant for other heavy metal as mercury, lead and cadmium. Table 8.13 Influence of soil type on fCorrection for a number of metals
Adsorption of anionic organic substances occur through ion-exchange with positively charged sites on the solid phase as hydrous oxides and partial hydroxides of Al, Fe and Mn –ions (e.g. Al(OH)2(H2O)4+). Anionic adsorption is of particular importance to the sorption of dissociated organic acids. This is illustrated by the adsorption of anionic surfactants, which may be described by the Freundlich equation as: Cs = Kd Cl nf, where `Cs' is the concentration of substance in the solid phase and `Cl' the concentration in the pore water, `Kd' is the distribution coefficient and `nf' the slope of the adsorption isotherm. For dissociating organic substances, the adsorption of the ionic form is normally much smaller than the (non-ionic) adsorption of the undissociated form. pH therefore strongly influences the adsorption of such substances. This is demonstrated in Figure 8.5 where empirically determined Kocs for pentachlorophenol is presented as a function of the undissociated fraction of pentachlorophenol (at different pH values). The correlation between the undissociated fraction and the observed Koc is linear starting at a value around 100 l/kg for the fully dissociated pentachlorophenol (predominantly anionic adsorption). A similar type of linear correlation between the dissociated fraction and the Koc value can be expected for other anionic compounds.
Figure 8.5. Measured Koc for pentachlorophenol as a function of the degree of dissociation (Koc values from Verschueren, 1998) In general, an overall Koc for a dissociating compound may be expressed by:
where, The organic carbon partitioning function of the undissociated part of the compound in expressed by:
K*OW is the octanol-water coefficient for the dissociated part of the compound. This coefficient is approximated with (Kleier, 1988):
The estimated KOC is shown as a function of pH for different values of logKOW and pKa in figure 8.6.
Figure 8.6. Estimation of KOC for organic acids. Immobilisation Sediments often contain high concentrations of organic material, especially in sedimentation areas, where they may act as a trap for pollutant sedimenting together with matter. The sedimentation and binding of pollutants in sediments are governed by the mechanisms discussed above and adsorbing substances (e.g. metals and lipophilic organic compounds) may therefore be present in the sediments in high concentrations. Although the distribution of pollutants between the dissolved and adsorbed fraction is normally regarded as a reversible equilibrium, there is often a net transport of pollutants bound to sedimenting material into the sediment. The initial adsorption is a relatively fast process, which may be followed by a slower process where the fraction of adsorbed substance is increased continuously in periods of months or even longer. From experiments performed in soil it is known that availability of a substance is reduced with increasing incubation time indicating an increasing strength of the chemical bonding involved in the adsorption processes (Hatzinger & Alexander 1995). The behaviour of metals in soils and sediments can be characterised by complex chemical reactions. Heavy metal ions present at trace levels in solution are adsorbed highly selectively in soils. Heavy metal adsorption in soil includes different type of adsorption sites (organic constituents, oxides, clay minerals, etc). The organic constituents determine the mobility of heavy metals in the soil matrix mainly because of the size of the organic constituents involved. The interaction of heavy metals with clay minerals includes adsorption, cation exchange as well as irreversible adsorption mechanisms (i.e. non-extractable with neutral salts or weakly acidic solutions), and will in general result in immobilisation of the metals. The inorganic constituents themselves can be considered as immobile apart from resuspension of particles in turbulent water. In soil the mobility of heavy metals tend to be low. Inclusion of adsorption and immobilisation in site characterisation. It is evident that adsorption and immobilisation of chemical substances in the environment have a strong influence on their bioavailability and hence on the potential for exposure. To a large extent, the sorptive properties and possibility of precipitation depends on inherent substance-specific properties and should as such be considered in the calculation of the site-generic characterisation factor. In the EDIP method adsorption is only considered in the modelling of terrestrial ecotoxicity, while no modification of the laboratory data is performed in modelling the aquatic ecotoxicity potential. This aspect should be considered in future revisions of the model for site-generic characterisation factors. As seen throughout Section 1.3.3, the adsorption and immobilisation processes are strongly influenced by environmental parameters among which the most important are :
Annex 8.3 Biodegradation of LAS under different temperature and sorptive conditionsFigure 8.7 shows the degradation of LAS in a surface sediment, which is assumed to be aerobic. The degradation is modelled as a function of temperature and the water content of the sediment (introducing the influence of bioavailability since LAS adsorbs to the solid phase), using a degradation half-life of 1 day for LAS in water at 15C and assuming that the degradation of LAS takes place only in the water phase. 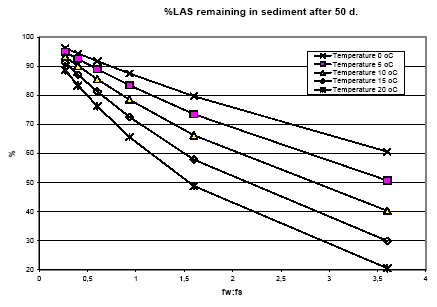
Figure 8.7. Model simulation of the degradation of LAS in aerobic sediment at different temperatures assuming a t½ at 1 day at 15 C. The figure shows moreover the effects of the water content of the sediment (fw:fs: fraction of water : fraction of solids in sediment). Temperature dependency It is well-known that the biodegradation rate increases with increasing temperature within the range tolerated by the degrading micro organisms. The influence of the temperature on the different kinds of micro organisms varies. Investigations on the biodegradation processes in the soil have shown that changes in temperature changes the composition of the microflora. A sudden change in temperature can therefore changes the biodegradation rate immediately, where after a certain adaptation period for the new flora, the biodegradation rate is increases again to a new level, which may not be so far below the initial level. Normally, the rates of biodegradation are described by the Arrhenius equation, which also is used to describe chemicals reaction rates dependency of temperature:
where k(T) is the degradation rate at temperature T k(T0) is a reference degradation rate R is the gas constant T is the absolute temperature Ea an activation energy In Table 8.14 values for Ea/R is given for the degradation of different compounds in water. Table 8.14. Estimated values for Ea/R. In K-1. Data from Verschueren 1998 and Lyman 1982.
It is seen that the dependency of the degradation rate on the temperature varies for the different compounds. With a temperature decrease from 20 oC to 10 oC, the degradation rate may decrease to around 50-75% of the degradation rate at 20 oC. A default value for the temperature dependency on the biodegradation rate in water could be Ez/R = 5000 K-1. With this value the biodegradation rate will be lowered with approximately 50% for every 10 oC decrease in temperature. If a degradation rate (kRef) at a temperature (TRef) is known, the degradation rate at temperature T can be expressed by:
where = Ea/(R•T•T•Ref) For the degradation of pesticides in soil, an value of = 0.08 K-1 have been suggested (Boesten & Van der Linden, 1991). This is the average value of some 50 experiments with a range of pesticides and soils. This coefficient is suggested to be suggested for the description of the temperature influence on the degradation rate in soil. An increase in temperature of 20 oC gives an approximately 5 times higher degradation rate in soil. Bioavailability. Normally it is assumed that the biodegradation primarily takes place in the water phase, meaning that only the dissolved part of the substance in question will biodegrade. Several studies have been carried out to support this theory. Knaebel et al. (1994) measured the degradation (by measuring CO2 evaluation) over time for LAS adsorbed to different solid materials. The desorption coefficient was negatively correlated with the initial rates of degradation – indicating that the initial degradation rate decreases with increasing strength of the adsorption. It should however be mentioned that a general observation is the bacterial growth in dilute media preferentially occurs on solid surfaces. This observation has given rise to the concept that growth may be enhanced by the adsorption of organic matter at liquid-solid interfaces. In addition, there is some evidence that the microbial degradation of certain surfactants in environmental samples occurs faster in the adsorbed state than in solution. It can for example be mentioned that the half-life of LAS in river water without sediment has been measured to be 1.4 d and with sediment the half-life was decreased to 0.7 d (Painter, 19xx). In Figure 8.8 the calculated degradation of LAS in a surface sediment, which is assumed to be aerobic, is shown as a function of temperature and the fw:fs ratio (to illustrate the influence of bioavailability).A degradation half-life in water of LAS is set to 1 day at 15 oC. Even though it may not the case for the degradation for LAS in sediment systems (see the discussion above) the degradation of LAS is in this example is assumed only to take place in the water phase. As seen from this figure the degradation is strongly dependent on the temperature and the fw:fs ratio in the sediment.
Figure 8.8. Degradation of LAS in sediment. Annex 8.4 Relative share of natural areas within the regions of EuropeBased on information from Table 8.15 the relative shares of natural areas and hence the share of air deposited material that exposes natural ecosystems is calculated for the four regions of Europe, the Nordic, the western, eastern and southern countries. The borders of the four regions is taken from Kristensen and Hansen, 1994 (except for Denmark which is considered a Nordic country here). Table 8.15. Frequency of natural areas per European country and for the four European regions
Annex 8.5 Relative share of emissions depositing to sea and land within the regions of EuropeBased on information from Huijbregts and Seppälä (2000), the relative shares of emissions depositing to sea and land is calculated for NH3 and NOx for individual countries and aggregated for the four regions of Europe, the Nordic, the Western, Eastern and Southern countries. The borders of the four regions is taken from Kristensen and Hansen, 1994 (except for Denmark which is considered a Nordic country here). Table 8.16. Fractions of the emissions of NH3 and NOx that are deposited to sea and land for the different European nations and averaged over European regions (based on information from Huijbregts and Seppälä, 2000)
NH3 is a relatively short-lived substance in the atmosphere while NOx is more long-lived. From the average deposition patterns shown in Table 8.16 it is clear, that the difference between the two substances is of little importance except for the Nordic countries. As an average estimate, the following factors are proposed for deposition to sea and land of air-borne emissions originating within the different regions: Table 8.17. Estimated average fractions of emissions deposited to sea and land for the four European regions
8.8 Annex 8.6 Sedimentation velocities in different aquatic systemsBased on information compiled in Egebart (1999), an overview of sedimentation velocities in different aquatic systems is given in Table 8.18. Table 8.18. Deposition velocities in different aquatic systems.
Annex 8.7 Annual average temperatures of European countriesTable 8.19. Monthly and average temperatures for European countries and averaged for European regions.
Annex 8.8 Rate constants for evaporation of substances from waterThe amount of substance evaporating from a water phase is expressed by MEvaporation = A•kT•CW Where kT is the total mass transfer coefficient:
Where: H is Henrys law constant R is the gas constant T is the absolute temperature kW is the liquid phase transfer coefficient, which can be estimated by (Schwarzenbach et al., 1993):
where DO2(Ti) is the diffusion coefficient of oxygen in water at temperature Ti (K) µW(Ti) is the viscosity of water at temperature Ti (K) U10 is the wind velocity at a height of 10 m. U10 is set to 5 m/s. MO2 is the molecular weight of oxygen = 32 g/mole. M is the molecular weight of the substance in question. M is set to 150 g/mole in the present calculations. kA is the air phase transfer coefficient, which can be estimated by (Schwarzenbach et al., 1993):
Where DH2O(Ti) is the diffusion coefficient of water in air at temperature Ti (K) The temperature dependency of Henry's constant can be estimated from (Veerkamp & Berge): H(T2)=H(T1)•e0.024(T2-T1) Calculations have been performed for different values of Henrys constant and at three temperatures: 10oC (reference temperature), 6 oC (Northern European countries) and 15 oC (Southern European countries). The results are given in Table 8.20 below as a reduction factor determined as the ratio between the reference situation and the Northern or Southern European situation. Table 8.20. Temperature dependent evaporation of semi volatile compounds.
Annex 8.9 Removal of substances from water through the combined effect of biodegradation and sedimentationThe net removal from the water phase depends on the sedimentation rates in the different hydro-geological compartments which the substances pass from the emission point to sea, and the time they spend in each of them (which again is determined by the biodegradation rate and the hydraulic retention time for the different compartments). In other words, the fraction of the substance that ends up in the sediments depends on the location of the emission point, and the hydraulic retention time of water in different sections of the hydrological cycle (river, lake). As a reference situation, the chemical is assumed to be emitted directly to the sea. The retention time in the sea is set at 35 days, corresponding to the degradation time of 80% of a readily biodegradable substance with a half-life of 15 days (according to the EU TGD, 1996). In order to derive at the 50% degradation of an inherent biodegradable substance within 35 days, the half-life of an inherently biodegradable substance is set to 35 days in agreement with the application factors for biodegradation (BIO) used in the EDIP. This is lower than the recommended biodegradation half-life of 150 days defined by EU TGD (1996). The biodegradation half-life of a non-biodegradable substance is set to 2500 days corresponding to 1% degradation within 35 days. Biodegradation Biodegradation is treated as a first-order process with the rate constant kBio. The amount removed by biodegradation (MBio) from a water body during the time interval dt is thus expressed as: MBio = kBio•VCW•dt where V is the volume of water body (m³) kBio is the first order biodegradation constant (d-1) Cw is the concentration of the substance in the water phase (mg/m³) Sedimentation Sedimentation is also treated as a first-order process. The amount removed from the water phase by net-sedimentation (MSed) (i.e. sedimentation minus re-suspension) during the time interval dt is: MSed= Vs•A• KSS•CW•dt where Vs is the linear sedimentation velocity (kg solid/m2/d) KSS is the sediment-water partition coefficient (m³/kg) estimated by: Kss = fOC•KOC KOC is the organic carbon partition coefficient here estimated by KOC = KOW A is the area of the water body (m2) Four types of surface waters are considered: river(1), lake(2), estuary(3) and sea(4). For each type of surface water (i), a correction factor fi is calculated representing the fraction of substance which is left after the combined removal through sedimentation and biodegradation:
... since the rate constant of the sedimentation process can be expressed as Vs •KSS/Z Where Z is the water depth of surface water i (m) Ti is the retention time in surface water i (river, lake, estuary, sea) (days) If the sedimentation rate is neglected then f4 = 1, corresponding to the reference state. Table 8.21. The default characteristics of the surface waters are given in Table 8.22.
Table 8.22. Estimated reduction factors (fi) representing removal by the combined sedimentation and biodegradation for readily biodegradable, inherently biodegradable and not biodegradable organic substances in a river, a lake, an estuary or a sea. It is seen that for biodegradation outweighs sedimentation as removal mechanism for the readily and inherently biodegradable substances, particularly when the residence time is long as it is in lakes. The temperature dependence of biodegradation is treated in the next section. Only for extremely lipophilic substances does removal by sedimentation play a substantial part. Footnotes [77] Danish Hydraulic Institute, DHI [78] Institute of Product Development (IPU) in Denmark
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||