Development of an analysis method for quantification of colophonium components in cosmetic products
2 Analytical method for colophonium
- 2.1 Phase 1: Literature study of analytical methods
- 2.2 Phase 2: Development of an analytical method
- 2.3 Phase 3: Validation of the developed method
The objective of the present study was to develop a suitable method to be used for screening of cosmetics with regard to content of colophonium, based on its major components AbA and DeA and including a marker for oxidized material. The project is divided into three phases. Phase 1: A literature study of published analytical methods; Phase 2: Development of an analytical method; and Phase 3: Validation of the developed method.
2.1 Phase 1: Literature study of analytical methods
Published methods for identification and quantification of colophonium components have been scrutinized and evaluated to find out if any method was suitable for further development into a new method.
2.1.1 Results
Published methods for identification and quantification of colophonium components are presented in Appendix A. Brief descriptions and evaluations of the methods are given. From the literature search two methods could be suggested. Method I is a GC-FID method which requires derivatisation. Method II is an HPLC-fluorescence method which is unspecific and therefore requires development to be applied on complex samples. Thus, it was concluded that a new method was needed for the purpose of the present study.
2.2 Phase 2: Development of an analytical method
In this phase a method for detection and quantification of the major colophonium components AbA and DeA, including a marker for oxidized resin acids, was developed. An HPLC – UV method with diode array detection (DAD) was chosen in order to gain high specificity for the target compounds and without need of derivatisation.
Traditionally, the major colophonium components are quantified using gas chromatography (GC) (16), but also HPLC methods have been developed (12). However, analyses based only on the non-oxidized resin acids do not tell anything about the amount of oxidized acids. A small amount of AbA detected might be due to a small amount of colophonium present, but could also be due to an extensive oxidation. As the oxidation products in colophonium are the major allergens, it is necessary to develop a method for quantification that takes the presence of oxidized material into consideration. However, as the method should be used for screening purposes it is important that it is a robust, inexpensive and simple method, not involving highly dangerous materials. Use of the highly allergenic 15-hydroperoxyabietic acid as a marker for oxidation products is not suitable, since it is very unstable and difficult to handle. Instead, the stable oxidation product 7-oxodehydroabietic acid (7-O-DeA) was selected as a marker for oxidized colophonium (Figure 2). This oxidation product is stable enough to be used as reference compound and not as allergenic to handle as the hydroperoxides.
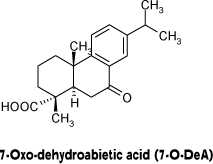
Figure 2: One of the oxidation products identified in air-exposed colophonium chosen as a marker compound for the oxidative degradation of AbA.
For AbA, the content may vary between 30 – 50% for gum rosin and
35 – 40% for tall oil rosin. Assuming that the resin acid content is 90%, a detection limit of 10 microgram/g AbA thus corresponds to a detection limit of approximately 30 microgram/g (ppm) of colophonium. This could be considered acceptable compared to the reactivity in allergic patients. Patients sensitized to colophonium of the gum rosin type were tested with serial dilutions of gum rosin 20 - 0.001% in petrolatum. Of the subjects tested, 50% reacted down to a concentration of 0.1% (1000 ppm), while the most sensitive patient reacted to a level as low as 0.001% (10 ppm) (18). In other patients tested with tall oil rosin (20 – 0.001% in petrolatum), no reactions were found to concentrations lower than 1% (10 000 ppm) (7).
2.2.1 Materials and Methods
2.2.1.1 Chemicals
Standard compounds
AbA, DeA and pimaric acid (all >95% purity) were obtained from Helix Biothech, Vancouver, Canada. cis-5,8,11,14,17-Eicosapentaenoic acid (EPA, >98.5%), used as internal standard, was purchased from Fluka, Sigma-Aldrich ChemieGmbH, Steinheim, Germany. 7-O-DeA was synthesized as described below.
Standard stock solutions of each compound were prepared in 100% of acetonitrile at a concentration of 1mg/ml. They were stored in freezer throughout the study.
Standard solutions for HPLC were made in acetonitrile:MilliQ water 9:1 with 0.2% formic acid. They were stored in refrigerator and were stable for at least 2 weeks (not tested for longer periods).
2.2.1.2 Solvents and samples
Methanol of 99.8% purity was purchased from Ltd BDH, formic acid, 98-100%, from Scharlau and acetonitrile of Chromasolv quality from Sigma-Aldrich.
Gum rosin (colophonium) samples (unmodified and maleic acid anhydride-modified from Soccer, Portugal) and unmodified colophonium (from Fluka) were dissolved in 100% acetonitrile to approximately 1.5 mg/ml.
Cosmetic samples, (1 g foundation from Face Stockholm) were spiked with standard compounds at different concentration levels, from 1 microgram /g up to 500 microgram/g of each compound.
2.2.1.3 Synthesis of 7-O-DeA
7-O-DeA was synthesized in two steps according to the procedure described in literature (17). A mixture of AbA (5.2 g, 17.19 mmol) and 5 % Pd/C (260 mg, 5 % w/w) was heated to 245 °C for 1 hr under nitrogen in a two necked round bottom flask. After cooling, the solid was dissolved with ethyl acetate (4 x 50 ml, or until all was dissolved) and filtered through celite. The organic layer was then extracted with 0.25 M sodium hydroxide (2 x 100 ml) followed by acidifying the aqueous layer with 1 M hydrochloric acid. The crude product was obtained by washing the aqueous layer with ethyl acetate (3 x 50 ml), dried with magnesium sulphate and concentrated under reduced pressure. Purification by flash chromatography (ethyl acetate/hexane/ trifluoroacetic acid: 14/85/1) afforded 2.5 g (46 %) of DeA. This was in agreement with previous reported data.
To a mixture of DeA (2.1 g, 6.99 mmol) and potassium hydroxide (0.4 g) in water (40 ml), was added a solution of potassium permanganate (2.8 g, 17.72 mmol) in water (60 ml) drop-wise at room temperature. The resulting mixture was stirred for 2.5 hr followed by saturating with sulphur dioxide. A white precipitate was formed and filtered off, washed with water and air dried over night. Purification by flash chromatography (ethyl acetate/hexane/ trifluoroacetic acid: 14/85/1) afforded 1.15 g (52 %) of 7-O-DeA in 95% purity determined by Nuclear Magnetic Resonance (NMR). This was in agreement with previous reported data
2.2.1.4 Equipment
An Agilent 1100 gradient system was used for analyses, equipped with an injection loop of 20 microliter, a DAD and a column heater set at 25°C. The HPLC column was a Prism RP-12 from Thermo (4.6mm i.d., length 150mm, particle size 3 micrometer). The stationary phase in the column contains both C12-chains and urea functions. For SPE, Oasis MAX columns (6cc, 500mg) from Waters were used.
2.2.2 Analytical Procedure
2.2.2.1 Analysis of cosmetic samples
Spiked 1 g samples were ultrasonicated in 20 ml of acetonitrile during 30 min. An aliquot of 500 microliter EPA from a 928 microgram/ml solution, was added as internal standard (IS) prior to extraction.
Each extract was transferred to plastic tubes and centrifuged during 15 min at 4000 rpm. The supernatant was transferred to a flask, while the pellet was redissolved in acetonitrile, ultrasonicated and centrifuged using the same procedure. The supernatants were pooled in the flask and evaporated in a rotavapor to a volume of approximately 5 ml. Adjustment of the volume to 7 ml was made by adding acetonitrile. MilliQ water was then added to a final volume of 10 ml containing acetonitrile:water 7:3. Addition of water made some of the solutions opaque. In those cases, filtration through a syringe filter (0.45 micrometer polypropylene membrane) was performed prior to the solid-phase extraction (SPE).
2.2.2.2 Solid phase extraction
Conditioning was performed by percolating 1) 3 ml of methanol and 2) 3 ml of MilliQ water through the SPE column. The sample was loaded and the column was then washed with 1) 3 ml of 50 mM sodium acetate in MilliQ water to eliminate very polar neutrals or bases, 2) 4 ml of methanol to eliminate lipophilic neutrals and finally 3) 1 ml of 2% formic acid in methanol. The sample was then eluted with 2 ml of 2% formic acid in methanol. An additional volume of 1 ml 2% formic acid in methanol was gathered in case of breakthrough. Fresh formic acid solution was made daily.
2.2.2.3 Analysis of gum rosin
Internal standard, 200 microliter (186 microgram) was added to 2 ml of 1.5 mg/ml gum rosin solutions in acetonitrile. The gum rosin samples were analyzed directly by HPLC after syringe filtration (without SPE clean-up).
2.2.2.4 HPLC method
Methanol:water 8:2 with 0.05% formic acid was used as mobile phase at a flow rate of 1 ml/min (yielding a pressure of approx 240 bar). An isocratic elution was performed. Four different wavelengths were used 210, 220, 240 and 250 nm. The different absorption ratios were used for identification of the compounds. Isocratic elution has to be performed to avoid increasing baseline, which would lead to higher LODs. The amount of formic acid was found to be critical for both retention and noise level.
A calibration curve was made from injected concentrations in the interval 0.8 ng/µl to 500 ng/microliter of each compound. For quantification single-point calibration was performed by a daily triplicate injection of a 250ng/microliter standard. Quantification was performed by using relative response factors analyte/IS.
Specific absorption ratios (CV below 10%) were used for identification of the different compounds:
AbA: 240nm/250nm 1.36
DeA: 220nm/210nm ratio 0.55
7-O-DeA: 220nm/210nm ratio 0.53; 240nm/210nm ratio 0.26; 250nm/210nm ratio 0.45.
EPA (is): 220nm/210nm 0.40
Quantification of DeA and EPA was performed by integration of peaks at 210 nm. For both AbA and 7-O-DeA the wavelength at 240 was used.
2.2.3 Results
2.2.3.1 Selectivity
The method developed is able to separate and identify the three reference colophonium compounds selected (AbA, DeA and 7-O-DeA) (Figure 3). Detection of the acids is performed at the wavelengths given above. Baseline separation of the peaks corresponding to AbA and DeA was obtained. One problem with LC-separation based on UV detection is the co-elution of pimaric acid with AbA. The new method with an LC column, which makes the stationary phase more selective to molecular shape, allows for a separation of pimaric acid and AbA (even though not fully baseline-separated). With the chosen mobile phase (MeOH:H2O 8:2 with 0.05% formic acid), and flow rate (1ml/min) the selected resin acids in colophonium can be separated within 22 min. It seems to be important to control the amount of acid thoroughly. To low a concentration (0.02%) affected the separation in a bad way and to a high level (0.1%) increased the noise level severely. There was used isocratic elution, since gradient elution increased the noise.
2.2.3.2 Separation of the target compounds in colophonium samples
Baseline separation of peaks corresponding to other resin acids was also obtained as analysis of unmodified colophonium shows (See Figure 4).
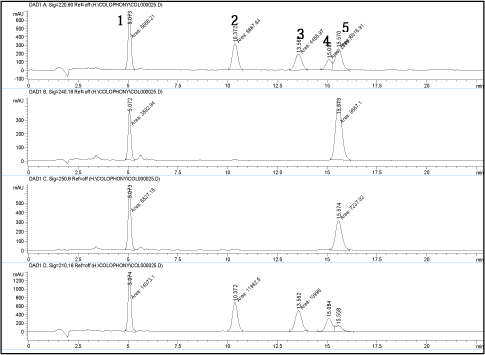
The chromatograms are measured at 220, 240, 250 and 210 nm, respectively
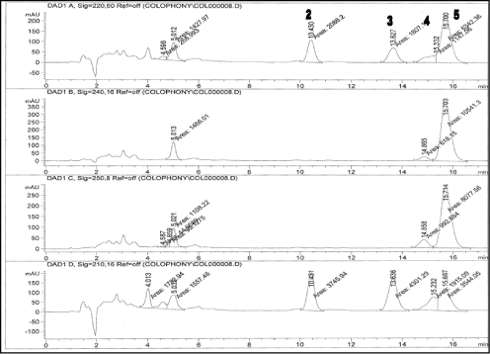
Figure 4: Colophonium sample, unmodified rosin from Soccer (1.5 mg/ml) analyzed with the developed method. 2 = DeA, 3 = EPA, 4 = pimaric acid, 5 = AbA. The oxidized resin acid 7-O-DeA could not be detected. The chromatograms are measured at 220, 240, 250 and 210 nm, respectively
2.2.3.3 Quantification of the target compounds in colophonium samples
The different analytes were quantified in unmodified and modified colophonium samples and the results are shown in Table 1.
Table 1. Concentrations of the target compounds in different samples unmodified and modified colophonium (mg/g)
| AbA | DeA | 7-O-DeA | |
| Unmodified colophonium of the gum rosin type (Soccer) |
255 | 62 | Not det |
| Unmodified colophonium of the gum rosin type (Fluka) |
691 | 40 | Not det |
| Maleic anhydride-modified gum rosin (Soccer) |
74 | 23 | Not det. |
2.2.3.4 Linearity
The peak areas for AbA, DeA and 7-O-DeA varied linearly with resin acid concentrations within the concentration range of 10 microgram/g and 500 microgram/g.
2.2.3.5 Limit of detection
Using the procedure described, the instrumental limit of detection (LOD) was 4 microgram/ml for AbA, 31 microgram/ml for DeA, and 3 microgram/ml for 7-O-DeA.
2.2.3.6 Recovery
The recoveries were investigated at three different levels of spiked foundation. The different spiked levels for each analyte were 100, 200 and 500 microgram/g. The found recovery for AbA was 78% (CV 1%), 88% (CV 5%) for DeA, 76% (CV 2%) for 7-O-DeA and 97% (CV 5%) for EPA.
2.2.3.7 Precision
The precision of the HPLC method in terms of both repeatability (within day variation, n=6) and reproducibility (n=3) of the relative response factor analyte/IS was in both cases below 10% (CV). The precision was tested with a 250 microgram/g standard.
2.3 Phase 3: Validation of the developed method
The method developed in Phase 2 was validated for quantification limit, repeatability and recovery of colophonium and oxidized colophonium marker at two concentration levels by standard addition using cosmetic products. Analysis of colophonium and oxidized colophonium in a series of products was considered, but due to limited resources available for this project, it is proposed that the investigation of colophonium content in the marketed cosmetic products should be performed in a separate project. However, selected cosmetic products were analyzed for the content of colophonium.
2.3.1.1 Separation of the target compounds in cosmetics
The analytes were quantified in different cosmetic samples according to the method developed. The separation was sufficient for identification and quantification in the tested cosmetics samples. This is illustrated by the chromatogram of a wax strip product for hair removal (Figure 5). The chromatogram measured at 210 nm is shown as an example.
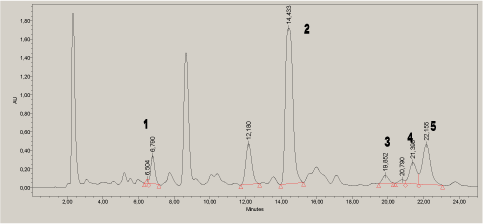
Figure 5. Chromatogram of a wax strip product at 210 nm. 1=7-O-DeA, 2=DeA, 3=EPA, 4=pimaric acid, 5=AbA: .
2.3.1.2 Precision
The repeatability in terms of CV was determined at two levels based on standard addition. For all compounds, the repeatability at low levels (50 – 90 microgram/g for each compound) was in good agreement with the results from phase two (See table 2). At high levels (135 - 250 microgram/g) the precision was in the range of 5-7% (See Table 3).
Table 2. Analysis of cosmetic product at low level (50 – 90 microgram/g) by standard addition
| component | precision | % precision | % recovery | LOD | LOQ |
| AbA | 2,4 | 2,4 | 85 | 7,2 | 21,6 |
| DeA | 6,4 | 13 | 84 | 19,2 | 57,6 |
| 7-O-DeA | 4,3 | 11 | 91 | 12,9 | 38,7 |
Table 3. Analysis of cosmetic products at high level (135 – 250 microgram/g) by standard addition
| component | precision | % precision | % recovery |
| AbA | 13,1 | 6,5 | 94 |
| DeA | 11,2 | 4,8 | 92 |
| 7-O-DeA | 11,8 | 7,5 | 94 |
2.3.1.3 Linearity and Repeatability of HPLC
The linearity and repeatability were tested with regard to the developed HPLC method by multiple injections (n=10). The method was linear in the concentration range of approximately 1 to 400 microgram/g (Correlation, r²: 0.999 – 1.000). The repeatability of the instrument was good in the range of 5 to 185 microgram/ml. AbA: 2-4.5%, DeA: 0.3-2%, 7-O-DeA: 0.3-1% and the internal standard, EPA: 2.5% at a concentration of 125 microgram/ml.
2.3.1.4 Limit of detection
Limit of detection was calculated as 3 x SD based on 6 analyzed samples spiked at low level and ranged from 7-20 microgram/g, (See Table 2).
2.3.1.5 Limit of quantification
The method limit of quantification was calculated based on the spiked cosmetic samples using the equation: LOQ = blank + 3 x LOD. This was equal to LOQ = 3 x LOD as there was no detectable blank values.
2.3.1.6 Recovery
The recoveries based in standard addition were in accordance with those obtained in phase 2. See table 2 and 3. CV of the recoveries was 4-6%.
2.3.1.7 Identification of the analytes
The specific absorption ratios for identification were controlled as different detectors were used in Phase 2 and Phase 3. There are some differences, especially for DeA and EPA. It is therefore recommended that the ratios were verified by standards before analysis. The CV was less than 10% in accordance with phase 2 (see Table 4).
Table 4: Absorption ratios for identification
| AbA | DeA | 7-O-DeA | EPA | |||||
| nm ratio | Phase 2 |
Phase 3 |
Phase 2 |
Phase 3 |
Phase 2 |
Phase 3 |
Phase 2 |
Phase 3 |
| 220/210 | 0,55 | 0,83 | 0,53 | 0,53 | 0,4 | 0,06 | ||
| 240/210 | 0,26 | 0,24 | ||||||
| 250/210 | 0,45 | 0,44 | ||||||
| 240/250 | 1,36 | 1,54 | ||||||
2.3.1.8 Quantification in products
Different common cosmetics products were tested with respect to content of the target resin acids. Based on the obtained data the content of colophonium was estimated (See Table 5). Only one wax strip had significant content corresponding to approximately 7 mg/g colophonium. For the other products AbA and DeA could be detected (0.5 – 4.5 microgram/gram), however, the concentrations were below detection limits.
Table 5: Quantification in products (microgram/g)
| Component | Foundation | Lip gloss | Wax strip, normal |
Wax strip, face |
| AbA | <7 | <7 | 2302 | <7 |
| DeA | <19 | <19 | 2517 | <19 |
| 7-O-DeA | <13 | <13 | 27,8 | <13 |
Version 1.0 Marts 2009, © Danish Environmental Protection Agency