|
Survey of Estrogenic Activity in the Danish Aquatic Environment 4 Pre-monitoring tests and quality assurance4.1 Sampling procedure This chapter will briefly discuss the reasons to carry out the special studies and the results developed. It also contains the assessment of the quality assurance programmes. 4.1 Sampling procedureThe ideal sampling procedure for wastewater (and streams/rivers) is flow-proportional sampling. In the Danish national aquatic monitoring programme (NOVANA), flow-proportional sampling is applied when influents and effluents are sampled at the WWTPs. However, for reasons of economy and practicality the flow-proportional sampling was not used in this survey. Therefore, a special investigation was undertaken partly to study the variation in effluent composition during 24 hours and partly to document the possible error introduced by the use of the alternative sampling method applied, the so-called qualified spot sampling (see Section 3.1). By this method, a sample is produced by mixing of five sub-samples of equal volume, taken with intervals of at least 2 minutes within a total period of half an hour. The investigation was concentrated on effluents from three WWTPs representing different treatment technologies and already (from previously sampling rounds in this study) known to contain quantifiable estrogenicity. Effluent samples to describe the variation in composition during one day were taken by spot sampling at different hours during the day. Also the temporal variations between day, evening and night during 24 hours was examined by taking flow-proportional samples covering three consecutive periods of 8 hours, 6 hours and 10 hours. The three WWTP's were:
On these treatment plants it was possible to obtain flow-proportional samples either with the stationary sampler or a portable waste-water sampler. After sampling the samples were treated according to the general procedure in the survey. The samples were subjected to biological measurements and the total and the free estrogenic activity was determined. The results are presented in Table 4.1 for the flow-proportional samples and in Table 4.2 for the spot samples. Some explanations are given:
Table 4.1 Results from the special investigation of the sampling procedure. Upper panel are Flow-proportional samples and lower panel spot samples.
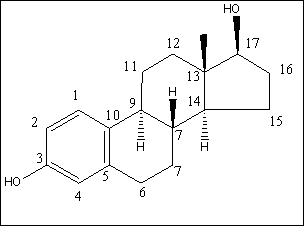
For the flow-proportional samples in Table 4.1 the variation measured as RSD is low, between 6% and 22%, except for results near the limit of detection for the method (approx. 0.1 ng/L). This implies that there is no significant change in the mean of the estrogenic activity during day, evening and night. For the spot samples taken during the day, a RSD between 19% and 28% was found, except for results near the limit of detection. The variations are within a factor of two, when the highest activity is compared with the lowest activity from a certain WWTP. This shows that the variation during the day is relatively low. A comparison between the activities found in the flow-proportional samples and the activities found in the spot samples shows very small and not significant differences. The investigation confirms that the chosen sampling strategy with the use of qualified spot samples instead of flow-proportional samples has not resulted in a significantly increased error from the sampling process. 4.2 Enzymatic treatment of conjugated estrogens4.2.1 BackgroundEstrogens are primarily excreted from humans and animals in conjugated form. The conjugants are either sulphate or glucuronide and each of them can bind to the estrogens on either the 3 or the 17 positions (see Figure 4.1) or on both positions. The di-conjugated estrogens are, however, chemically unstable and are readily cleaved to mono-conjugates. This cleavage occurs almost instantly (D'Ascenzo et al 2003), and the di-conjugated estrogens will therefore not occur in sewage and are therefore irrelevant in the current context. The current section discusses the importance of the conjugated estrogens with regard to interpretation of the results and to the assessment of the potential “delayed” release of estrogens to the environment when conjugated estrogens are cleaved in the environment.
Figure 4.1. Structural formula of 17β-estradiol 4.2.2 Investigation of loss of conjugated estrogensIn the project, the conjugated estrogens have only been measured indirectly, by measuring the total estrogen concentration after enzymatic de-conjugation and subsequently subtracting the observed level of free estrogens. As will be discussed in the following, there is in principle a risk that this determination of conjugated estrogens underestimates the actual amount of these substances. Two factors may have impact on the result (though it turned out that this was not the case here): The first occur if the conjugated estrogens not are cleaved completely during the enzymatic deconjugation procedure. Here, it was shown that the cleavage was quantitative with the exception of E2-17S of which only approximately 9% was cleaved (Appendix 1.4). A number of studies has however, shown that E2-17S is not excreted from humans and therefore this insufficient cleavage is unimportant in the current context (Andreolini et al 1987; D'Ascenzo et al 2003; Zhang and Henion 1999). The second reason for a potential underestimation of the concentration of conjugated estrogens is if the loss of conjugated estrogens during the analytical steps is significantly different than that of the parent compounds. This reduction is not taken into account in the calculation of the concentrations which is based on the use of deuterated internal standards (E1, E2 and EE2). Investigations have therefore been made in order to quantify any on loss of conjugated estrogens due to:
Each of these experiments is described in detail in Appendix 1. The results show that there is no reduction in the amount of conjugated estrogens in the samples during storage. The loss during solid phase extraction is independent of the compound in question and range from 2 to 27%. These findings are consistent with the observations made for the non-conjugated estrogens (the parent compounds) and consequently it can be assumed that the amount of conjugated estrogens is determined with a precision, which is close to that of the parent compounds. 4.2.3 Losses during transport and storageConjugated estrogens were analysed indirectly by using the procedure for enzymatic cleavage of the conjugates and subsequently measuring the concentration of the parent compounds. The stability of the parent compounds during transport is well documented in the scientific literature. In contrast, such data are sparse with regard to the conjugated estrogens. As the conjugated estrogens were measured indirectly in the current project, we therefore performed a study with the aim of assessing the stability of conjugated estrogens during transport and storage. The experimental details as well as the results of this study are described in Appendix 1.1. The study revealed that within 7 days the conjugated estrogens are not significantly degraded in sewage effluent that has been acidified with H2SO4 (pH=3). In conclusion the study revealed that similarly to the free estrogens, the conjugated estrogens are stable during transport and storage. During handling of samples to be analysed for chemicals at very low concentrations, the binding of the analytes to glass equipment may be a significant factor leading to loss of the analyte. Documentation exists, showing that the free estrogens do not bind to glass equipment (Fürhacker et al., 1999). It is characteristic for the conjugated forms of the steroid estrogens that they are more hydrophilic and therefore are more likely to bind to glass equipment during handling and treatment of the samples. A study was therefore conducted to assess whether there was a significant loss of these substances during transport and storage due to their binding to glass equipment. Briefly, the study was conducted by incubating conjugated estrogens in mili-Q water for two days and then comparing the concentrations in water when the experiment was initiated and after two day. The experiment and the results are described in more details in Appendix 1.2. The results revealed that no significant loss of conjugated estrogens could be expected during transport and storage. 4.2.4 Losses during sample pre-treatmentThe recovery of free estrogens on SPE columns has been studied by several authors and it is generally accepted that high recovery (> 60%) is achieved when using a method as the current (see e.g., Desbrow et al., 1998; Snyder et al., 2001, Ternes et al., 1999). In a preliminary study, five different cartridges (Varian® C18 Bond Elut® (6 ml/1 g); Isolute® C18 (6 ml/500 mg); Waters Oasis™ HLB (6 cc/200 mg); Isolute® ENV+ (6 ml/1 g); Waters Porapak™ Rdx (6 cc/ mg)) were tested using spiked tap water to 100 ng/L. The highest recovery was obtained using the Varian cartridges, which subsequently has been applied though out the project. This column material has previously been used for analysis of steroid estrogens (Andersen et al., 2004). Additional recovery experiments were conducted with these columns and selected conjugated estrogens. These studies, which are described in details in Appendix 1.3, revealed that the column material gave recoveries similar to those obtained for the free estrogens. The enzymatic cleavage of conjugated estrogens was developed as a part of this project. If the method should be suitable for its purpose, we needed to document the following:
As described in Appendix 1.4, experiments were performed showing that all of these three demands were fulfilled. In order to remove substances with interference on the chemical analyses a clean-up procedure using silica gel was used for all samples for chemical analyses. Briefly, the acetone eluate from the SPE cartridge was evaporated to dryness under N2-gas. The samples were then redissolved in 200 µl of hexane:acetone (65:35 vol:vol). And then loaded onto the silica gel column and eluted with the hexane-acetone mixture until 5 ml eluate were collected. The absolute recoveries of the analyte for the clean up step were evaluated for the free estrogens and it was found that the clean-up is almost quantitative for each analyte (Appendix 1.4). 4.3 Quality controlSeveral different measures have been taken to ensure the quality of the results obtained during this project. The quality assurance elements consisted of (1) an internal quality control scheme and (2) measures to compare results with other laboratories. 4.3.1 Internal quality control schemeIn connection with every series of samples a set of quality control samples was analysed along with the real samples, i.e. taken through the complete analytical procedure. Each set of quality control samples consisted of one blank sample (tap water) and two identical control samples (tap water to which a mixture of the three estrogens (estrone, 17β-estradiol and ethynylestradiol - E1, E2 and EE2) had been added at a level 2.5 ng/L of each analyte). The same set of quality control samples were used for the biological assay and for the chemical analytical method. Results from the control samples were collected in quality control charts, which all are shown in Appendix 4. The control chart displays mean values of the two control samples for each series of samples (X-chart) as well as the difference between duplicate results (R-chart). 4.3.2 Precision and Limit of Detection (LOD)4.3.2.1 Chemical LODsFrom the control charts the precision can be calculated for each parameter measured. The control chart with results from the biological assay gave an overall precision (RSD, reproducibility)) of 29 % from measurement of estrogenic activity. Similarly, the overall precisions from the chemical determination of the three analytes were 30 % (E2), 17 % (E1) and 27 % (EE2), respectively. For the chemical parameters the Limit of Detection (LOD) was used as a measurement of the lowest amount that can be determined by the method used. The chemical LOD is defined by the formula:
where Sw is the standard deviation determined in the same series of samples (repeatability) at concentrations near the LOD, and t0.995(f) is between 3 and 4 with more than 6 repetitions. Based on this definition, an experimentally generated general LOD for the chemical analyses was determined to 0.1 ng/L for each component. In the both of the abovementioned procedures, tap water was used instead of real samples (i.e. sewage effluent or surface waters) though, obviously, more analytical problems due to effects from the matrix could be expected if real samples were analysed. However, by comparing chromatograms from sewage effluent and surface water with those obtained with tap water, matrix effects generally appeared to have only minor importance. There were, however, situations where such problems occurred and resulted in elevated detection limits. 4.3.2.2 Detection limits in YES-assayFormal limits of quantification and detection cannot be defined in a bioassay as the YES-assay the same way it is possible in a chemical analysis. Samples with a detectable estrogenic activity have been defined by the following criteria:
The lowest level of estrogenicity detected in the investigation by these criteria was 0.03 ng E2 equivalents per litre. Since the absorbances of the blind samples throughout the analytical series were fairly constant, a general detection limit in the YES-assay in the present investigation is 0.05 ng E2 equivalents per litre. In the individual cases it ranges between 0.03 and approximately 0.07 ng E2 equivalents per litre. 4.3.3 Inter-laboratory comparisonAs two laboratories were involved in the chemical analysis of the samples, an inter-laboratory comparison between the two laboratories was performed. A total of 12 identical authentic samples were analyzed by both laboratories in order to reveal any disagreement between results from the two laboratories. The results are presented in Appendix 4. Briefly, good correlations between the results produced in the two laboratories were obtained. 4.3.4 Comparison between control sample results from biological and chemical measurementsSince the control samples used for the quality control in both the biological assay and the chemical analysis are identical samples, data from the control charts can be used to compare the overall agreement between the two methods. This can be expressed as the ratio between the biological and chemical measurement including the standard deviation of the ratio. The calculated mean estrogenic activity of the control samples measured by YES assay was 6.8 ng/L, whereas the calculated mean content (expressed as estrogenic activity) from the chemical analysis was 5.2 ng/L. These results indicate that the ratio (biological/chemical) between measurements performed on identical samples is 1.3. From the control chart of the biological assay a RSD of 29 % is calculated. For the chemical results converted to E2-equivalents the RSD can be calculated from the individual RSD's from the control charts giving a RSD of 39% for the chemical analysis. This means that measurements of ratios between biological and chemical results are accompanied by a combined RSD of 49 %. Consequently, a ratio of 1.3 with a RSD of 49% indicates that ratios (biological/chemical measurements) lower than approximately 2.6 are not statistically significant (given a 95% level).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||