Udvikling af metode til testning af udvaskning af organiske stoffer fra jord og restprodukter
3. Desorptionsstyret udvaskning
Dette afsnit vil beskrive de mekanismer og faktorer, der styrer desorptionen af organiske stoffer fra jord. I teksten vil der blive benyttet forskellige ord og begreber, som kort skal defineres her. Ordet sorption er en samlet betegnelse for flere fysisk-kemisk set forskellige processer, nemlig absorption (binding i en anden fase) og adsorption (binding til overflader). Hvis de organiske stoffer kan ioniseres, dvs. blive negativt eller positivt ladet (som organiske syrer og baser), kan denne overfladebinding helt eller delvist bestå af en ionbytning. Bindingen af de organiske stoffer kan være mere eller mindre reversibel, dvs. at sorptionsprocessen kan gå "baglæns", hvorved stoffet overføres fra jordpartiklerne til porevandet. Dette kaldes desorption. I den kemiske litteratur samt også i noget af sorptionslitteraturen benyttes ordet sorbent om det materiale, der kan binde forureningsstoffet (i samme litteratur kaldet sorbatet). Her benyttes ordet jord for det sorberende materiale. Begrebet dækker topjorde, akvifermaterialer, m.m. Frasen det organiske stof benyttes for det organiske forureningsstof, som sorberer/desorberer. Jordenes væsentligste fraktion for sorption af organiske stoffer benævnes her det naturlige organiske stof. Dette svarer til en anden ofte benyttet term, det faste organiske stof. Denne fraktion kan i nogle tilfælde omfatte menneskeskabt organisk stof i form af rester af ikke vandblandbare organiske væsker (NAPL). I gennemgangen vil der også blive benyttet termer som partikler og aggregater. I mange tilfælde udgøres mange af en jords korn af aggregater af grundpartikler, som er sammenkittet af oxider og andre stoffer.
3.1 Den traditionelle beskrivelse og dens forudsætninger
I en lang årrække har man anvendt en forenklet beskrivelse af organiske stoffers binding i jord. Beskrivelsen gjorde det muligt ud fra få parametre at kvantificere sorptionen. Den traditionelle beskrivelse er indgående beskrevet i (Kjeldsen,1996) og skal kort resumeres her.
Den traditionelle beskrivelse baserer sig på en række forenklede forudsætninger:
| Sorptionen for et aktuelt organisk forureningsstof kan beskrives ved en lineær sorptionsisoterm, hvilket betyder, at der altid er et fast forhold mellem stofkoncentrationen i væskefasen, Cw, og koncentrationen i den faste fase, Cs. Forholdet, som kaldes fordelingskoefficienten, Kd (opgivet i l/kg), er således uafhængigt af koncentrationsniveauet for det organiske stof. | |
| Sorptionen sker alene til jordens indhold af naturligt organisk stof, dvs. at sorption til jordens indhold af mineralske bestanddele (ler og diverse metaloxider/hydroxider) kan negligeres. Typen af naturligt organisk stof betragtes også som underordnet. | |
| Fordelingskoefficienten er proportional med jordens indhold af organisk kulstof, foc,
og fordelingskoefficienten, Koc, mellem organisk kulstof og vand: | |
| Der er fuld reversibilitet. Det vil sige, at sorptionsprocessen, hvor stof optages fra vandfasen til den faste fase, forløber fuldstændigt som desorptionsprocessen, hvor stof frigives fra den faste fase til vandfasen. Dette betyder, at man beskriver begge processer med den samme fordelingskoefficient, Kd. | |
| Momentan indstilling af sorptionsligevægt. Dette betyder, at den stof-overførsel, der skal ske fra vandfasen og over i den faste fase (eller omvendt) for at opnå ligevægt (beskrevet ved fordelingskoefficienten, Kd), sker øjeblikkeligt. Dette skal i praktisk henseende tolkes, således at stof-overførelseshastigheden er så høj, at den ingen begrænsende virkning har for procesbeskrivelsen under enhver praktisk situation. | |
| Negligibel indflydelse fra andre opløste eller kolloide stoffers tilstedeværelse. Dette betyder bl.a., at der kan ses bort fra konkurrenceeffekter, hvis flere organiske stoffer er til stede på samme tid. |
Hvis man, udover de ovennævnte forudsætninger, forudsætter, at det jordvolumen, som betragtes, kan forudsættes homogent med hensyn til rumvægt (r b), porøsitet, (e ), vandindhold, (e w) og indhold af organisk kulstof (foc), kan sorptions-/desorptionsprocessen beskrives ved retardationsfaktoren, R:
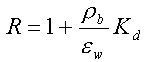
For et homogent jordlag med tykkelsen, L, som er styret af de forenklede forhold beskrevet ovenfor, vil man opnå et udvaskningsforløb som vist på figur 3.1. Man ville således opnå en konstant koncentration, Cw, i udløbet fra jordlaget. Koncentrationen ville holde sig konstant, indtil det organiske stof er blevet udvasket fra hele jordlaget. Tiden, der ville gå til koncentrationen, ville falde til nul, Tg, er:
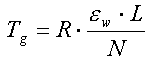
hvor N er nedsivningen gennem jordlaget (f.eks. i mm/år).
Da R er uafhængig af tid og koncentration (under de benyttede forudsætninger), vil R kunne bestemmes ud fra en Kd-måling, eventuelt via et simpelt batchudvaskningsforsøg, hvor et sammenhørende sæt af væske- og faststofkoncentrationer bestemmes, hvorfra Kd beregnes.

Figur 3.1
Ideelt udvaskningsforløb fra homogent jordlag, forudsat at den traditionelle
sorptionsbeskrivelse er gældende er angivet ved den stiplede linie, mens den fuldt
optrukne kurve illustrerer en mere realistisk udvaskning.
3.2 Begrænsninger i den traditionelle beskrivelse for desorptionsstyret udvaskning
Især inden for de seneste fem år har en meget omfattende forskningsindsats vist, at der er flere ting, som gør en udvaskningsbeskrivelse baseret på den traditionelle beskrivelse meget tvivlsom. Et yderligere komplicerende aspekt er, at der stadig gives enkelte eksempler på, at den traditionelle beskrivelse godt kan benyttes, og samtidig flere eksempler på det modsatte.
Der er flere af forudsætningerne i den traditionelle beskrivelse, som der har været fokuseret på. Disse observationer vil kort blive opridset i det følgende:
3.2 1 Isotermtype
Som alternativ til den lineære isoterm benyttes oftest enten Freundlich-isotermen eller Langmuir-isotermen i de tilfælde, hvor der opnås "krumme" sammenhænge mellem koncentrationen på den faste fase og væskekoncentrationen (se Bilag A). På figur 3.2 ses der et eksempel, hvor der blev benyttet en Freundlich-isoterm for at beskrive sorptionssammenhængen. Der er dog også flere nyere eksempler, hvor en lineær beskrivelse er benyttet med succes.

Figur 3.2
Sammenhæng mellem koncentrationen i væske og koncentrationen i den faste fase
modelleret ved hjælp af Freundlich-isotern.
3.2.2 Fordeling til naturligt organisk stof
Sammenhængen:
![]()
hvor foc er fraktionen af naturligt organisk kulstof i jorden (på vægtbasis), og Koc er fordelingskoefficienten mellem det naturlige organiske kulstof i jorden og vand, har været benyttet i mange undersøgelser. Der er eksempler på, at beskrivelse af "sorptionskapaciteten" alene ved jordens indhold af organisk kulstof er utilstrækkelig. I stedet har det naturlige organiske stofs bestanddele af andre grundstoffer end kulstof (f.eks. ilt og kvælstof) været benyttet – se figur 3.3, som viser, at fordelingskoefficienten til det organiske stof vokser med stigende andel af kulstof. Det faktum, at fordelingskoefficienten til det naturlige organiske stof Koc ikke kan betragtes som en konstant for alle jorde (for et specifikt organisk stof), er formentlig hovedårsagen til, at estimationer af Koc ved hjælp af empiriske korrelationssammenhænge med oktanol-vand-fordelingskoefficienten, Kow (som er baseret på et antal sorptionsforsøg med forskellige jorde og organiske stoffer - se afsnit 7.4.2 i (Kjeldsen 1996)), giver vidt forskellige resultater afhængigt af, hvilken empirisk sammenhæng der benyttes. De seneste forsøg på at inddrage flere forsøgsresultater i udvikling af estimationssammenhænge har ikke ændret meget på den store usikkerhed, som disse estimationsformler afstedkommer (Sabljic et al. 1995, Gawlik et al. 1997, Müller 1997).

Figur 3.3
Fordelingskoefficienten til naturligt organisk stof, Kom, som funktion
af (O+N)/C-forholdet i jorden (Rutherford et al. 1992).
3.2.3 Indstilling af sorptionsligevægt
Tabel 3.1 er en sammenstilling af forskellige forsøg lavet for at belyse, hvor hurtigt sorptions- og desorptionsligevægte indstiller sig. Tabellen viser, at ligevægtsindstillingstiden kan variere meget. Det er også tydeligt, at en væsentlig andel af sorptionen oftest sker ganske hurtigt, men at en signifikant andel sorberer langsomt. Det kan faktisk være yderst vanskeligt ved sådanne forsøg at sikre sig, at ligevægt er nået, idet analyseusikkerheder gør det meget vanskeligt at fastslå, om der sker ændringer i den sorberede stofmængde i den sidste del af forsøget.
Den traditionelle beskrivelse benyttes til både at beskrive sorptionen af stof fra væsken til jorden og desorption af stof tilbage igen. Ved vurdering af udvaskning af organiske stoffer fra jord er det primært desorptionen, som skal betragtes. Som nævnt forudsættes det, at begge processer kan beskrives med den samme Kd-værdi, dvs. at der er fuld reversibilitet. Denne forudsætning har via laboratorieforsøg været undersøgt i en lang række tilfælde. Der er tilfælde, hvor der er fundet fuld reversibilitet, men også mange, hvor en
Tabel 3.1 Se her!
Nylige eksempler på observeret langsom sorption eller desorption i naturlige
sorbenter (baseret på sammenfatning af Pignatello og Xing 1996).
væsentlig del af stoffet ikke desorberede i samme grad eller med samme hastighed. I de senere år er også desorption af almindelige neutrale stoffer med beskeden hydrofobicitet (som TCE, benzen, toluen) blevet undersøgt. Figur 3.4 viser adsorptions- og desorptionsisotermer for naphthalen på et sandet overfladesediment med foc på 0,27% (Kan et al. 1994). Det ses af figuren, at adsorptionsisotermen er lineær, og at desorptionsisotermerne adskiller sig radikalt fra adsorptionsisotermerne. Desorptionsisotermerne viser, selv ved adskillige serielle udvaskninger, hvor udvaskningsvandet udskiftes med nyt vand, at der resterer væsentlige stofmængder i jorden. Der opnås således nogle relativt lave udvaskningskoncentrationer, selvom jorden stadig indeholder væsentlige stofmængder. Det er væsentligt at understrege, at det ikke var feltforurenede jordprøver, som benyttedes i forsøget, men jordprøver, hvor de organiske stoffer var tilsat jorden via indledende sorptionsforsøg, hvor stofferne indledningsvist tilsættes til væsken. Den store forskel mellem sorptionen og desorptionen skyldtes således ikke ældningsprocesser (se afsnit 3.6.6). Lignende observationer er gjort i andre forsøg (Miller og Pedit 1992, Harmon og Roberts 1994). Graden af reversibilitet vurderes ikke at have nogen væsentlig indflydelse på selve udførelsen af en udvaskningstest.

Figur 3.4
Sorptions- og desorptionsisotermer for naphthalen på et sediment efter enten 1
døgns (ð ) eller 7 døgns (o) kontakttid i hvert step (Kan et
al. 1994).
3.3 Konceptuel model
Det forrige afsnit viste meget tydeligt, at man stadig er langt fra den fulde forståelse af, hvad der styrer sorptionen og desorptionen af organiske stoffer i jord. En væsentlig del af de seneste års intense forskning har haft til formål at opnå en bedre forståelse af de processer, der styrer stofudvekslingen ved opstilling af konceptuelle modelbeskrivelser af processerne. Disse konceptuelle beskrivelser udvikles til stadighed, efterhånden som nye og flere resultater opnås. Her skal præsenteres en konceptuel model, som i overordnede træk er accepteret af de fleste forskergrupper, der arbejder med dette fagområde.
En beskrivelse af den konceptuelle model kræver, at vi betragter jorden på tre størrelsesskalaer, som vist på figur 3.5. På figur 3.5a ses jorden i forstørrelse med forskellige jordpartikler og porehulrummet imellem partiklerne. På denne skala kaldes porerne makroporer. På partiklerne anes der belægninger, som kan bestå af naturligt organisk stof, evt. i blanding med lermineraler, metaloxider/hydroxider og andet (Pignatello og Xing 1996). Eventuelt indeholder jorden små partikler bestående helt af organisk kulstof eller "ren" kulstof hidrørende fra sod, trækul eller stenkul (Luthy et al. 1997). Jorden kan også indeholde fri fase eller ikke vandblandbar væske (NAPL) i form af olie eller opløsningsmidler, siddende som små "afsnørede" klumper (residual NAPL-fase). Fra disse klumper kan der opløses stoffer, som NAPL’en oprindelig var bestående af (se kapitel 2), men udefra kommende organiske stoffer kan bindes til klumpen – og frigives igen. NAPL kan således virke som en menneskeskabt sorptionskapacitet som supplement til jordens indhold af naturligt organisk stof (Luthy et al. 1997).
I figur 3.5b er en af jordpartiklerne vist i yderligere forstørrelse. Forstørrelsen viser, at partiklen, som det ofte er tilfældet, er et aggregat sammensat af flere forskellige partikler, som er sammenkittet af diverse materialer (organisk stof, metaloxider, m.m.). Figuren viser, at stoffer, som udveksles mellem den faste fase og væskefasen, må passere en stillestående vandfilm. Figuren viser også, at sådanne partikelaggregater ofte er gennemskåret af indre hulrum af forskellig størrelse. Porerne inddeles ofte i to typer, nemlig mesoporer (typisk diameter større end 2 nm) og mikroporer (typisk diameter under 2 nm) (Pignatello og Xing 1996). Disse porer vil oftest være vandfyldte. Transporten i disse porer sker ved diffusion, og diffusionshastigheden kan være kraftigt reduceret, idet porerne kan indeholde naturligt organisk stof, som begrænser stofudvekslingen. Diffusionen begrænses også af porernes tortuositet, som er et mål for, hvor "kringlet" en vej molekylerne skal diffundere (se også afsnit 3.5). I mikroporerne kan tortuositeten være endnu mere udtalt, og der kan samtidig på grund af den begrænsede porediameter være signifikante adsorptionseffekter fra vægge til flere sider og "sterisk hindring" (det faktum, at molekylets størrelse ikke tillader en indtrængning i poren, idet dennes diameter er for lille) (som vist på figur 3.5d) (Pignatello og Xing 1996).
På figur 3.5c er der vist en forstørrelse af et område indeholdende naturligt organisk stof, hvor transporten af det organiske forureningsstof ind i det naturlige organiske stof sker ved diffusionsprocesser i den faste fase. Der har været anvendt analogier til diffusion i polymerer, som der i de seneste år også har været forsket intenst i. Flere forestiller sig således, at det naturlige organiske stof kan være af en "gummiagtig" struktur (fleksibel, amorf ) (A på figur 3.5c) eller af en mere "glasagtig" struktur (kondenseret, hård) (B på figur 3.5c). (Carroll et al. 1994, Young og Weber 1995, Weber et al. 1992, Xing et al. 1996). Herudover har man i polymerer set tegn på adsorptionsprocesser af Langmuir-typen (se Bilag A) til små mikroskopiske hulrum i den glasagtige struktur (C på figur 3.5c) . Man forestiller sig i øvrigt, at alle tre typer kan være til stede samtidig i et område af naturligt organisk stof.
Jorde kan således indeholde forskellige sorptionssites, som enten kan være placeret tæt på den mobile væskefase eller være "gemt" inden i aggregater, hvortil stofferne først skal transporteres via diffusionsprocesser, som kan være yderst langsommelige på grund af forskellige tilbageholdelsesprocesser.
Tabel 3.2 er en sammenstilling af de forskellige sorptionsmekanismer og deres forskellige karakteristika med hensyn til isotermtype, kinetik og konkurrence (Luthy et al. 1997). I det følgende vil de forskellige sorptionsmekanismer blive beskrevet yderligere.
Figur 3.5 Se her!
Konceptuel model for sorption/desorption i jord (efter (Luthy et al. 1997) og
(Pignatello og Xing 1996)) - for nærmere forklaring se teksten.
Tabel 3.2
Kvalitativ sammenligning mellem mekanismer og makroskopiske observationer (efter Luthy
et al. 1997).
Absorption ind i amorft (blødt fast organisk stof eller NAPL |
|
| Isoterm | Lineær |
| Kinetik | Hurtig (<minutter hvis partiklerne ikke er sammensat i aggregater) |
| Hysterese | Nej |
| Konkurrence | Nej |
Absorption ind i kondenseret (hårdt fast organisk stof |
|
| Isoterm | Ofte ikke lineær. Kan være lineær efter ligevægt |
| Kinetik | Langsom (> dage) |
| Hysterese | Ja |
| Konkurrence | Ja |
Adsorption på overflader af fast organisk stof |
|
| Isoterm | ikke lineær |
| Kinetik | Hurtig (<minutter hvis partiklerne ikke er sammensat i aggregater) |
| Hysterese | ? |
| Konkurrence | Ja |
Sorption ind i meso- og mikroporer til stede i jordpartiklerne |
|
| Isoterm | ikke lineær (hvis porerne er af variabel størrelse) |
| Kinetik | Langsom (> dage ) |
| Hysterese | Ja |
| Konkurrence | Ja (pga. begrænset antal sorption sites) |
Adsorption på hulrum i kondenseret fast organisk stof |
|
| Isoterm | ikke lineær |
| Kinetik | Hurtig (<minutter (efter langsom diffusion gennem det omgivende organiske stof)) |
| Hysterese | Ja |
| Konkurrence | Ja |
3.4 Sorptions-/desorptionsprocesser
Som illustreret i forrige afsnit er der flere fysisk-kemisk forskellige processer, som giver anledning til sorption/desorption af organiske stoffer i jord, og som bidrager til, at sorptionsbeskrivelsen adskiller sig fra den traditionelle beskrivelse (se afsnit 3.1). Et organisk molekyle kan således undergå flere diffusionsprocesser, før der sker en endelig sorption, eller før stoffet frigives (desorberer) fra partiklen til det mobile vand. Stoffet må (ved sorptionsprocessen) først transporteres fra den mobile væskefase gennem den stillestående vandfilm, som omgiver jordaggregatet (film-diffusion), eventuelt gennem porer i aggregatet (pore-diffusion) og endelig diffusion gennem det naturlige organiske stof (matrix-diffusion) (Pignatello og Xing 1996).
Diffusionsprocesserne er, som det fremgår af den efterfølgende gennemgang, af forskellig hastighed og vil afhænge af stoffets hydrofobicitet samt af jordtypen. Dette betyder at udvaskningen af de organiske stoffer kan ske meget hurtigt i nogle tilfælde og meget langsomt i andre. Det er p.t. ikke muligt at noget præcist om hvornår man har det ene eller det andet tilfælde. En udvaskningstest hvor væskekoncentrationen måles til forskellige tidspunkter kan afsløre den enkelte jords udvaskningshastighed (se i øvrigt kapitel 9).
3.4.1 Film-diffusion
I de fleste tilfælde vil film-diffusionen ikke være hastighedsbegrænsende for den langsigtede proces (Weber og Miller 1988), men kan formentlig godt være det for den initiale hurtige stofudveksling (Pignatello og Xing 1996). De afgørende parametre for diffusiv stoftransport er dels diffusionskoeffienten, dels den længdeskala, hvorover den diffusive transport foregår (se afsnit 3.5). Diffusionskoefficienten i væskefilmen er langt større end diffusionskoeffienter gældende for transport internt i partikler/aggregaters porer eller i selve det naturlige organiske stof. Ved en batchudvaskningstest, hvor prøven agiteres, kunne det godt tænkes, at tykkelsen eller sammensætningen af væskefilmen bliver påvirket, således at den diffusive transport gennem denne bliver anderledes (formentlig hurtigere), end den ville være i et naturligt system eller i et kolonneudvaskningssystem.
3.4.2 Pore-diffusion
Studier af de hastighedsbegrænsende processer, der er afgørende for den langsigtede udvaskning af organiske stoffer fra jorden, er krævende at udføre, idet de foregår på en mikroskopisk skala. Generelt er det nødvendigt at udvikle nye analyseteknikker, før man kan opnå mere direkte beviser på, hvilke processer der er hastighedsbegrænsende i et konkret tilfælde. Nogle af de indicier, der indtil videre er præsenteret, på at pore-diffusion er en væsentlig proces, er, at man i flere forsøg har opnået hurtigere rater, efter at jorden er blevet pulveriseret (Steinberg et al. 1987, Ball og Roberts 1991, Pignatello 1990). En syrebehandling af jordene har også givet hurtigere rater, hvilket er tolket som et resultat af opløsning af uorganiske oxider, der har virket som "cement" for mindre partikler, og deraf følgende mindre aggregatstørrelser (dvs. mindre diffusionsafstande) (Pignatello 1990).
Et enkelt jordaggregat kan – som vist på figur 3.5b – indeholde porer af helt forskellige størrelser, og porerne kan indeholde mere eller mindre naturligt organisk stof, hvilket resulterer i, at der inden for det enkelte aggregat kan være områder med en relativt høj diffusiv transport, hvor stofafgivelsen i en udvaskningssituation vil ske inden for minutter til timer, og andre områder, hvor den diffusive transport går ekstremt langsomt, og hvor stofafgivelsen vil ske over dage til uger (eller måske over endnu længere tid) (Luthy et al. 1997).
3.4.3 Matrix-diffusion i naturligt organisk stof
Flere forskere har i de seneste år set resultater, som tyder på, at diffusionsprocesser på meget lille skala i belægninger eller diskrete korn af naturligt organisk stof er hastighedsbegrænsende for stofudvekslingen (Brusseau og Rao 1989, Brusseau et al. 1991). Man har bl.a. set eksempler på lavere stofudvekslingsrater ved stigende indhold af naturligt organisk stof (Pignatello et al. 1993, Carroll et al. 1994, Pignatello 1990, Nkedi-Kizza et al. 1989) samt højere udvekslingsrater, når der tilsættes organiske cosolventer, som kan få det naturlige organiske stof til at svelle og blive mere åbent (Brusseau et al. 1991).
Som allerede beskrevet og vist på figur 3.5c kan det naturlige organiske stof bestå af en "blød" og en "hård" del. Der har været benyttet forskellige oxidationsmetoder til at skelne mellem de to typer. Den "bløde" del har været karakteriseret som den andel af det naturlige organiske stof, som kan fjernes ved vådoxidation med persulfat ved lav temperatur. Den "hårde" del er herefter blevet karakteriseret som den andel, der fjernes ved en forbrænding ved høj temperatur, men ikke ved den ovennævnte vådoxidation (Huang et al. 1998).
Som vist i tabel 3.2 betragtes sorptionen til den "bløde" del som en traditionel fasefordeling (lineær isoterm, hurtig diffusiv transport og dermed hurtig indstilling af ligevægt samt ingen konkurrenceeffekter eller forskel mellem sorption og desorption (hysterese). Derimod er sorptionen/desorptionen i den "hårde" del mere kompleks. Isotermerne menes typisk at være ikke-lineære, hvilket betyder, at den effektive diffusionskoefficient bliver mindre med faldende koncentrationer. Der er også set udtalt hystereseeffekt samt konkurrence mellem organiske stoffer til stede på samme tid. Den observerede ikke linearitet har været tolket som et resultat af en "two-site-sorptionsproces", idet den egentlige fasefordeling til det "hårde" organiske stof betragtes som traditionel lineær. Ud over denne kapacitet er det foreslået, at der findes diskrete sorptionssites i mikroskopiske huller i det organiske stof (type C på figur 3.5c), og hvor sorptionen er ikke-lineær af Langmuir typen (som forudsætter, at der er en maksimal sorptionskapacitet til stede). Den samlede sorptionsproces udviser derfor ikke linearitet (af Freundlich-typen) (Luthy et al. 1997).
3.4.4 Adsorption på overflader af naturligt organisk stof
De forudgående beskrevne sorptions-/desorptionsprocesser har generelt været absorptionsprocesser, hvor sorbenten (makroskopisk set) optages i de faste partikler. Herudover kan der også være en sorptionskapacitet i form af en adsorption til heterogene organiske overflader. Udvekslingen vil gå meget hurtigt (som de fleste adsorptionsprocesser), men isotermen vil være ikke-lineær (formentlig af Langmuir-typen på grund af den begrænsede mængde sorptionssites) (Luthy et al. 1997). Det betyder blandt andet, at man for materialer, hvor dette spiller en rolle, skal være forsigtig ved ekstrapolation eller omregning af resultater af udvaskningsforsøg udført ved ét koncentrationsniveau til andre koncentrationsniveauer.
3.4.5 Adsorption til mineraloverflader
Jorde indeholder primært uorganiske partikler som i størrelse kan spænde fra fine partikler i lerfraktionen til grovsand og grus. De fine partikler har meget store specifikke overfladearealer og kan samtidig være elektrisk ladede. Lermineraler er således overvejende negativt ladede og kan derfor have betydning for organiske baser, som i et vist pH-område er positivt ladede; derfor tiltrækkes de af negativt ladede lerpartikler, hvor de ionbytter med naturlige kationer. For neutrale organiske stoffer er sorptionen til mineraloverflader under mættede forhold oftest ikke af betydning (se Kjeldsen 1996, for flere detaljer). Under umættede forhold har de senere års forskning vist, at mineraloverfladerne kan spille en langt større rolle i sorptionen, end det er tilfældet under mættede forhold. Sorption til mineraloverflader er dog kun signifikant for jorde med et meget lavt indhold af naturligt organisk stof (foc < 0,001, Miljøstyrelsen 1996). Hvis jordpartiklerne er dækket af mindre end 5-10 lag vandmolekyler, vil de flygtige organiske stoffer indeholdt i poreluften begynde at konkurrere med vandmolekylerne om at adsorbere direkte til mineraloverfladerne (Ong og Lion 1991, Valsara og Thibodeaux 1992) – se også afsnit 3.6.2.
3.4.6 Sorptions-/desorptionshysterese
Der er flere eksempler på, at stoffer, som er sorberet til jorde, ikke frigives i samme grad, når stofferne forsøges desorberet igen, et begreb, der kaldes hysterese. Fænomenet resulterer i, at sorptions- og desorptionsisotermen ikke er sammenfaldende. Der kan være flere årsager til, at der observeres hysterese i udførte laboratorieforsøg (Huang et al. 1998):
- eksperimentelle artifacter, som giver en øjensynlig hysterese
- irreversibel binding til specifikke sorptionssites
- langsomme desorptionsrater
- "indespærring" af sorberede molekyler.
ad 1. En vigtig artifact er, at der ikke opnås ligevægt i det indledende sorptionsforsøg (hvis det naturlige organiske stof indeholder væsentlige hårde bestanddele, kan det tage op til måneder at opnå ligevægt) (Weber et al. 1998). I det efterfølgende desorptionsstep kan diffusionen foregå både længere inde i aggregatet og ude i bulkvæsken. Den observerede hysterese kan også skyldes tab af stof til forsøgsudstyret eller tilstedeværelse af partikler, som ikke separeres ud ved centrifugering.
ad 2. Der er flere eksempler på, at visse stofgrupper (bl.a. phenoler) bindes kemisk til det naturlige organiske stof og derefter kun meget vanskeligt frigives igen (Burgos et al. 1996, Sabbah og Rebhun 1997). En sådan reaktion, også kaldet kemisorption, adskiller sig fra den almindelige definition på sorption, idet den indbefatter covalent binding. En forskergruppe fra Rice University har i flere artikler beskrevet irreversibel sorption af neutrale organiske stoffer (Kan et al. 1994, Kan et al. 1997, Kan et al. 1998). For alle de jorde, som de studerede, fandtes én fraktion af sorptionen at ske som reversibel sorption. For hver kombination af jord/organisk stof fandt man en maksimal koncentration af irreversibelt sorberet stof, qirrmax, som enten kunne fyldes op på en gang eller ved flere sorptionsstep. En typisk værdi for qirrmax angives til at være (Kan et al. 1998):
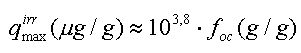
Man fandt også, at fordelingskoefficienten for den irreversible andel næsten er konstant for de ret forskellige organiske stoffer, der blev benyttet, idet man finder Kocirr=105,53± 0,48 l/kg. Efter at den maksimale irreversible sorptionskapacitet, qirrmax, er nået, er der fuld reversibilitet mellem sorption og desorption (hældningerne på sorptions- og desorptionsisotermerne er ens). Generelt findes det, at alle sorptions- og desorptionsdata kan modelleres ved hjælp af én enkel sorptionsisoterm. Isotermligningen består af to led, et lineært led, som repræsenterer den reversible sorption, og et led af Langmuir-typen, som repræsenterer den irreversible del (Kan et al. 1998).
ad. 3+4. En anden forskergruppe omkring Dr. Weber på University of Michigan har også studeret sorptionshysterese og sætter fænomenet i forbindelse med tilstedeværelse af "hårde" bestanddele i det naturlige organiske stof (se afsnit 3.4.3). De hårde bestanddele har en tre-dimensional struktur, som gør, at organiske stoffer kan blive "fanget" (Weber et al. 1998). Ud fra energibetragtninger (Gibbs frie energi) vil det være – energimæssigt – mindre fordelagtigt for sorberede molekyler at desorbere igen, hvilket giver hystereseeffekten med de meget langsomt desorberende stoffer.
Generelt må det konkluderes, at man på nuværende tidspunkt ikke har den fulde forståelse af, hvilke processer og faktorer der er styrende for sorptionshysterese. Det er dog åbenlyst, at der er mange eksempler på, at væsentlige fraktioner af sorberede stoffer er bundet på en form, som gør desorptionsprocessen meget langsommelig. Dette betyder også, at estimerede udvaskningskoncentrationer baseret på måling af totalindholdet i jorden og en fordelingskoefficient baseret på en hurtig udveksling følgende den lineære isoterm i mange tilfælde groft vil overestimere den reelle udvaskning (Kan et al. 1998), hvilket netop nødvendiggør udførelse af udvaskningstests med feltrealistiske opholdstider (se 3.6.8).
3.5 Modellering af langsom desorption
Ovenstående gennemgang af mekanismer for langsom desorption viser med stor tydelighed, at diffusionsprocesser på forskellig skala er afgørende for, hvorledes diffusionen forløber. Diffusionsprocesser modelleres oftest ved hjælp af Ficks lov:
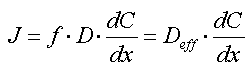
hvor
J er den diffusive flux (i f.eks. g m-2 s-1)
D er stoffets diffusionskoefficient i fasen (som oftest vand)
f er en reduktionsfaktor, som tager højde for geometriske og sorptionsmæssige forhold,
som medvirker til en reduktion af diffusionskoefficienten til den effektive
diffusionskoefficient, Deff
dC/dx er koncentrationsgradienten, således at store koncentrationsforskelle over små
afstande giver større diffusiv transport.
Denne diffusionsbeskrivelse har dannet grundlag for modellering af desorptionen af organiske stoffer fra jord, men vanskeliggøres af, at man på mikroskopiske skala skal kende typiske diffusionsafstande samt effektive diffusionskoefficienter. Den grundlæggende model af denne type er udviklet af Gschwend og medarbejdere (Wu og Gschwend 1986) og er i kortfattet udgave beskrevet i box 3.1 (for mere detaljeret beskrivelse se Kjeldsen (1996).
Som alternativ til diffusionsmodellen beskrevet i box 3.1 blev der af flere foreslået benyttet en mere empirisk tilgang til beskrivelse af desorptionsforløbet. Flere havde observeret, at en del af stoffet blev frigivet nærmest øjeblikkeligt, mens restfraktionen blev frigivet med en tilnærmet eksponentielt aftagende rate. Denne model, også kaldet bicontinuummodellen, har tidligere været benyttet i den kemiske industrier til modellering af lignende problematikker. Figur 3.6 viser koncentrationen i vandet som funktion af tiden for bicontinuummodellen. Til sammenligning er der vist en tilsvarende kurve for diffusionsmodellen. Den store forskel ses især for små tider. De grundlæggende ligninger for bicontinuummodellen er vist i box 3.2 (van Genuchten og Wieranga 1976, Brusseau og Rao 1989). Her er der også udregnet nøgletal, som fremkommer, når bicontinuummodellen indbygges i den én-dimensionelle strømningsligning. Den meget udbredte én-dimensionelle model, CXT-fit, indeholder som én af mulighederne bicontinuummodellen. CXT-fit vil kunne bruges til at vurdere effekten af desorptionskinetikken for udvaskningskoncentrationer i feltrealistiske scenarier, hvor de indgående parametre (F og k2) er bestemt via serier af batchforsøg.
En simpel sammenhæng mellem diffusions- og bicontinuummodellen kan opnås ved at udtrykke hastighedskonstanten som funktion af diffusionskoefficienten (Gong og Depinto 1998):
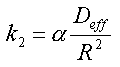
hvor
a er en korrektionsfaktor, som afhænger af tiden og
partikelstørrelsen
Deff er den effektive diffusionskoefficient i partiklen
R er retardationsfaktoren.
Box 3.1
Model for desorption baseret på diffusionsteori
| Forudsætninger De sorberende partikler forudsættes at være kugleformede, homogene partikler med samme radius. En opstilling af en massebalance for en partikel leder frem til følgende differentialligning:
hvor
hvor For et simpelt batchsystem bestående af en fast mængde jord (bestående af partikler med radius R) og en fast mængde vand, kan koncentrationen i vandet som funktion af tiden udregnes ved hjælp af ovenstående differentialligning. Løsningen opgives som et funktionsudtryk, der angiver den totale stofmængde i udvaskningsvandet til tiden t, w(t) i forhold til den endelige stofmængde ved ligevægt,w,¥ . Dette forhold vil afhænge af den dimensionsløse parameter, (D*t)/R2:
hvor F er en monotont voksende funktion med værdier mellem 0 og 1 Løsningen er vist grafisk i figur 3.6. Figuren viser funktionen F. Udover den dimensionsløse tid (D*t/R2) afhænger F af størrelsen Kdrsw,, hvor rsw, er forholdet mellem fast stof (i kg) og vand (i liter) i batchen. Benævnelsen "infinite bath" på figuren er det tilfælde, hvor mængden af partikler er så lille, at Cw kan betragtes som nul, dvs. det tilfælde, hvor den desorberede stofmængde ikke kan detekteres i udvaskningsvandet. Differentialligningen kan også danne grundlag for en model af udvaskningen fra et jordlag, idet diffusionsbeskrivelsen indbygges i en én-dimensional strømningsmodel. Ligningssystemet kan kun løses numerisk. |
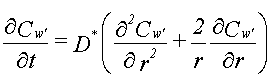
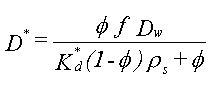
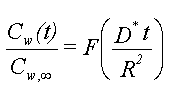
Box 3.2
Model for desorption baseret på bicontinuummodellen.
| Følgende ligninger opstilles:
hvor
Ved indbygning af ovenstående beskrivelse i den én-dimensionelle stoftransportligning fås følgende nøgletal: Damköhler tallet, w :
hvor
Damköhler tallet, w beskriver effekten af kinetik i sorptionsprocessen under givne strømningsbetingelser. De vides fra undersøgelser, at effekten af sorptionskinetik er negligibel, hvis Damköhler tallet, w , er større end 10. |
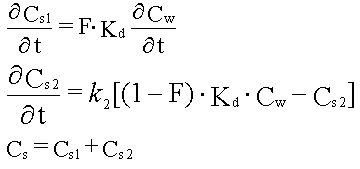
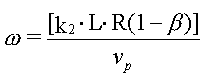
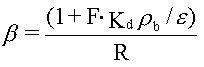
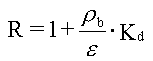

Figur 3.6
Desorptionsforløb beskrevet som væskekoncentrationen i batch som funktion af
tiden. Cw,¥ er ligevægtskoncentrationen. Forløb
både for diffusionsmodel (stiplet) og bicontinuummodel (ikke-stiplet) er vist.
Det er således tydeligt, at hastighedkoefficienten, k2, ikke er konstant over hele desorptionsforløbet. Dette forklarer, at man i flere kinetiske udvaskningsforsøg (hvor udvaskningskoncentrationens tidsafhængighed bestemmes) har haft vanskeligheder med at fitte hele kurveforløbet ved hjælp af bicontinuummodellen, (Wu og Gschwend 1986). Dette gælder også i de tilfælde, hvor diffusionsmodellen har været benyttet. Da desorptionen ofte er styret af forskellige diffusionsprocesser på forskellig skala (se figur 3.5 og tilhørende diskussion) hver med sine vidt forskellige beskrivende parametre, er det forståeligt nok, at diffusionsmodellen benyttende én partikelstørrelse og én diffusionskoefficient ikke kan modellere udvaskningen eksakt. Flere har prøvet at råde bod på dette ved at udbygge og forfine en af de to modeller. Tabel 3.3 viser et sammendrag af de forskellige modeller, som er udviklet med henblik på en mere sammenhængende beskrivelse af desorptionsforløbet. Mange af de beskrevne modeller indeholder flere parametre end de basale modeller, hvilket i sig selv ofte forbedrer en fitting til forsøgsresultater, idet der er flere parametre at "skrue" på. Den sidstnævnte modeltype i tabel 3.3 virker dog som en interessant nyudvikling. Grundtanken bag denne model er, at jorden indeholder en lang række sorptionssites, som har forskellige hastighedskonstanter. Man forestiller sig, at hastighedskonstanterne følger en fordelingsfunktion, idet man har arbejdet med modeller, hvor konstanterne følger en log-normalfordeling (Pedit og Miller 1994, Culver et al., 1997) eller en gammafordeling (Connaughton et al. 1993, Pedit og Miller 1994, Chen og Wagenet 1995, Culver et al. 1997). I begge tilfælde kan fordelingen af hastighedskonstanter beskrives ved to parametre i modsætning til andre multi-compartmentmodeller, som kræver to parametre pr. compartment. Sammenligninger af bicontinuummodellen og multi-compartment-modellen med fordelingsfunktionen faldt ud til den sidstnævnte models fordel, idet den gav en bedre overensstemmelse med forsøgsdata og samtidig var mere robust over for mindre ændringer i inddata (Culver et al. 1997).
Tabel 3.3 Se her!
Sammendrag af forskellige modeller til beskrivelse af desorptionsforløbet for
organiske stoffer fra jord (baseret delvis på Gong og Depinto 1998).
Figur 3.7 viser udløbskoncentrationen i et søjleforsøg som funktion af gennemstrømmede antal porevolumener. Forsøgsdata er med stor succes blevet simuleret med multi-compartmentmodel.

Figur 3.7
Udløbskoncentration fra søjleforsøg som funktion af gennemstrømmet antal
porevolumener simuleret med multi-compartmentmodel.
3.6 Faktorer af betydning for desorption
I dette afsnit vil nogle af de faktorer, som har eller kan have indflydelse på desorptionen blive diskuteret. En forbedret forståelse af disse faktorer vil forbedre muligheden for at forudsige, i hvilke tilfælde desorptionskinetikken vil have afgørende betydning for udvaskningen fra jord.
3.6.1 Temperatur
Flere undersøgelser har vist, at desorptionshastigheden generelt stiger med stigende temperatur. Piatt et al. (1996) fandt således en stigning i hastigheden på 1,2 - 2,6 gange ved en stigning på 22° C. Samtidig faldt fordelingskoefficienten 1,1 - 1,6 gange ved den samme temperaturstigning. Cornelissen et al. (1997) og Werth og Reinhard (1997a,b) fandt lignende sammenhænge. Generelt har temperaturen således ikke den store betydning i forhold til andre ukendte faktorer, og hensynet til desorptionsprocesserne giver derfor ikke anledning til at foreskrive en meget præcis temperaturkontrol ved udførelse af udvaskningstests. Måling af desorptionsforhold ved forskellige temperaturer kan dog benyttes til udregning af aktiveringsenergier for sorption-/desorptionsprocessen. Størrelsen af aktiveringsenergier kan belyse, på hvilken skala diffusionen foregår (pore-diffusion i meso- eller mikroporer eller matrix diffusion i naturligt organisk stof) (Cornelissen et al. 1997, Werth og Reinhard 1997b).
Forsøg til belysning af den meget langsomme desorption kan kræve meget lange forsøgstider. Hvis forsøgene gennemføres ved forhøjet temperatur (60° C), og hastighederne omregnes til feltrealistiske forhold, kan forsøgene klares på 5-10 gange kortere tid (Cornelissen et al. 1997), men andre uønskede ændringer er så til gengæld mulige.
3.6.2 Vandindhold
Ved udtørring af jorde stiger sorptionskapaciteten til den mineralske del af jorden ganske drastisk (se også afsnit 3.4.5). Det kræver dog et meget lavt vandindhold, som under danske forhold ikke er realistisk, undtagen i de allerøverste centimeter af jordprofilet i tørre somre. For normale vandindhold har vandindholdet en ringe effekt på sorptions- og desorptionsforhold (se i øvrigt Kjeldsen (1996) for en mere detaljeret diskussion af fænomenet). Vandindholdet kan således tilpasses testen uden at ændre på sorptions- og desorptionsforholdene.
3.6.3 Jordens surhedsgrad, pH
Jordens surhedsgrad, pH, kan påvirke strukturen af det naturlige organiske stof. Ved stigninger i pH ændres det naturlige organiske stof til en mere åben struktur, hvilket giver hurtigere desorptionsrater (Sahoo og Smith 1997). Der skal dog ændringer på flere pH-enheder til for at se en effekt. For mange organiske syrer og baser vil pH-ændringer direkte påvirke stoffernes ioniseringsgrad og dermed fordelingskoefficienten (se afsnit 3.7). I pH-statiske udvaskningstests på jord udført ved høje pH-værdier (f.eks. over pH = 11) ses ofte kraftig forøgelse af indholdet af totalt opløst kulstof. Det opløste organiske kulstof kan ved sorption og kompleksbinding medvirkedels til mobilisering/udvaskning af andre organiske komponenter, som ikke direkte mobiliseres af det høje pH, dels til mobilisering af uorganiske sporelementer, herunder blandt andet Cu.
Der er store forskelle i stoffers hydrofobicitet, et forhold, som har stor betydning for desorptionsrater. Desorptionen er som nævnt flere gange styret af diffusionsprocesser på flere skalaniveauer. Uafhængigt af skala kan processen beskrives som vist i box 3.1. En afgørende parameter for desorptionsraten er den effektive diffusionskoefficient beskrevet ved ligningen:
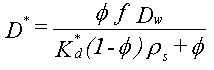
Hydrofobe stoffer har relativt store molekylevægte, hvilket giver lave diffusionskoefficienter i vand, Dw. Den resulterende effekt er meget lave effektive diffusionskoefficienter. Desorptionsrater for PAH’er og lignende er således meget mindre end for benzen eller trichlorethylen for den samme jord, hvilket der skal tages højde for ved fastsættelse af kontakttiden i forbindelse med udførelse af udvaskningstests for disse komponenter.
3.6.5 Konkurrence mellem stoffer til stede på samme tid
Som allerede diskuteret i afsnit 3.4 vil der forekomme konkurrence for nogle af de tilstedeværende sorptionssites. Især for den sorptionskapacitet, som karakteriseres med ikke-lineære isotermer. Dette gælder sorption til den hårde fraktion af det naturlige organiske stof (herunder Langmuir-adsorption til hulrum i det hårde organiske stof) samt adsorption til overflader af naturligt organisk stof (se tabel 3.2). Konkurrerende stoffer kan både være naturligt forekommende organiske stoffer i jorden (Xing og Pignatello 1998) og andre kemikalier (McGinley et al. 1996, Xing et al. 1996). For den del af sorptionen, der sker til den amorfe fraktion af det naturlige organiske stof, og hvor karakteristiske isotermer er lineære, ses der ingen konkurrenceeffekter (McGinley et al. 1996).
3.6.6 Kontakttid ("ældning")
Flere forsøg har i de seneste år vist, at den tid, hvor stofferne har været i kontakt med jorden, er afgørende for desorptionsraterne. Disse observationer er især gjort for hydrofobe stoffer (især PAH’er), hvor desorptionsrater generelt er lave. Carmichael et al. (1997) viste dette ved at tilsætte kulstof-14-mærkede PAH’er til en jord, som i forvejen var forurenet med de samme PAH’er (som naturlige/kulstof-12-stoffer). Jorden var prøvetaget fra en træimprægneringsanstalt, hvor træ var blevet behandlet med kreosot siden slutningen af 1800-tallet. En sammenligning viste, at desorptionsrater for de nyligt tilsatte PAH’er var væsentlig større end desorptionsrater for de oprindelige PAH’er (der sås op til en faktor 1000 til forskel). Lignende observationer er gjort i andre forsøg (Pignatello et al. 1993). Gong og Depinto (1998) så også faldende pore-diffusionskoefficienter med stigende kontakttider for PCB’er og så samtidig, at den øjeblikkelige ligevægtsfraktion (størrelsen F - se box 3.2) ligeledes faldt.
Årsagerne til de observerede ældningsfænomener kan være flere. Som nævnt i afsnit 3.5.4 er PAH’er meget hydrofobe stoffer, hvis diffusionskoefficienter – især i mikroporer eller i den hårde fraktion af det naturlige organiske stof – kan være så små, at det tager årtier at opnå fuldstændig ligevægt. Stoffer, som tilsættes, vil inden for det relativt korte tidsrum ikke have nået at diffundere helt ind i partiklernes indre, hvor diffusionshastighederne er meget små, og vil derfor desorbere hurtigere end det oprindelige stof. En anden årsag kan være, at der sker ændringer i partiklernes indre geometri, som fører til, at stofferne spærres inde.
Observationerne viser klart, at man ikke kan studere udvaskning af hydrofobe stoffer på kunstigt forurenede jorde – undersøgelserne skal ske på prøver udtaget i felten på den konkrete lokalitet.
3.6.7 Jordens alder og sammensætning
Flere har vist, at jordens geologiske alder samt dens oprindelse har stor betydning for sorptions-/desorptionsforhold. Unge jorde udviser generelt lavere fordelingskoefficienter til det naturlige organiske stof (Koc), og isotermerne er generelt lineære. Kile et al. (1995) så således for et stort antal overjorde med et indhold af organisk kulstof på 0,2-6% lineære isotermer og Koc-værdier for tetraklorkulstof fra 45-75 l/kg. De fandt i øvrigt generelt højere Koc-værdier for bundsedimenter. De fandt også meget højere Koc-værdier for jorde forurenet med diverse organiske kemikalier, herunder olie. Huang og Weber (1997) undersøgte 10 forskellige naturlige sorbenter (fra unge tørvejorde til kerogen isoleret fra geologisk gamle skiffer-prøver). Sorbenternes organiske stof blev karakteriseret ved forholdet mellem indholdet af ilt og kulstof, idet kulstoffraktionen i det naturlige organiske stof stiger med den geologiske alder af materialet. Figur 3.8 viser, at der opnås højere Koc-værdier ved faldende O/C-forhold (og højere alder) samt en højere grad af ikke-linearitet (lave Freundlich-eksponenter) ved faldende O/C-forhold.


Figur 3.8
a) Værdier for logKoc som funktion af indholdet af ilt (O) og kulstof (C) i
det naturlige organiske stof repræsenteret ved O/C-forholdet. Forsøgene er gennemført
ved høj koncentration (1000 m g/l) og lav koncentration (1 m g/l) b) Værdier for isotermens ikke-linearitet som funktion af
O/C-forholdet i det naturlige organiske stof (Huang og Weber 1997).
Ligeledes observeredes der udtalt hysterese for de gamle skifferprøver (se figur 3.9). Kleinedam et al. (1999) observerede også udtalt ikke-linearitet (lave Freundlich-eksponenter (1/n)) for geologisk gamle sorbenter. De nævnte observationer passer godt overens med forventningen om, at jorde med stigende geologisk alder får en større andel af hårdt, kondenseret organisk stof, et faktum, der kendes fra organisk stofs diagenetiske ændring til stenkul over en tidshorisont på millioner af år.

Figur 3.9
Phenanthrensorptions- og desorptionsisoterm for Norwood-skiffer. De to isotermer er
ikke sammenfaldende hvilket illustrerer en klar hystereseeffekt (Huang og Weber 1997).
Ud over O/C-forholdet er jorde blevet karakteriseret på forskellig vis med henblik på at bedre kunne forudsige sorptionsprocessen. Xing (1997) benyttede metoder til at karakterisere det naturlige organiske stofs polaritet og aromatiske indhold. Metoderne blev benyttet på fem jorde af forskellig alder og diagenetisk baggrund (omdannelsesstadier) med henblik på karakterisering af jordenes naphthalensorption. Forsøget viste, at fordelingskoefficienten, Koc, steg med stigende aromatisk indhold og faldende polaritet. Kleinedam et al. (1999) målte Koc-værdier for fraktioner af akvifermaterialer, idet materialet opdeltes efter de forskellige partiklers lithologiske oprindelse (sandsten, kalksten, granit, skiffer, m.m.), også kaldet lithokomponenter. Der blev fundet en faktor 590 mellem højeste og laveste Koc-værdi for sorption af phenanthren. De højeste værdier blev fundet i lithokomponenter, hvor det naturlige organiske stof udgjordes af kulpartikler, formentlig stammende fra skovbrande, som er blevet indlejret i de sedimentære bjergarter.
Forskellige fraktioneringsmetoder af jorde har været forsøgt for yderligere at karakterisere sorptionen. Barber (1994) fraktionerede akvifermaterialer efter partikelstørrelse. Undersøgelsen viste, at akvifermaterialernes indhold af naturligt organisk stof især var tilknyttet til fraktionerne med de mindste partikelstørrelser, som derved udviste de største fordelingskoefficienter, Kd.
Holmén, Gschwend (1997) fandt for tre akvifermaterialer, at sorptionskapaciteten var tilknyttet belægninger på akvifermaterialets partikler, som især bestod af kvarts. Belægningerne bestod især af metaloxider, der virkede som "bindemiddel" for underliggende naturligt organisk stof, der normalt antages at sidde yderst. Bestemmelse af fordelingskoefficienter, Kd, ved batch- og søjleforsøg viste divergerende resultater, idet man fandt meget lavere Kd-værdier i søjleforsøget. Dette blev tolket som et resultat af erosion af belægningerne ved batchforsøgene, hvorved naturligt organisk stof, som oprindeligt var utilgængeligt for sorption af stoffer, var blevet frilagt og derved deltog i sorptionsprocessen. Sorptionskinetikken blev med succes modelleret med en pore-diffusionsmodel, hvor det blev forudsat, at diffusionen kun skete i belægningerne, som alene indeholdt sorptionskapacitet.
3.6.8 Strømningshastighed
De meget store ligevægtstider, som er blevet observeret i et stort antal laboratorieforsøg, viser, at der vil være mange tilfælde, hvor den desorptionsbetingede udvaskning i felten må forventes at være betydeligt mindre, end hvis man forudsætter ligevægt mellem jorden og det porevand, som gennemstrømmer det forurenede jordlag. Derfor vil en analyse af typiske opholdstider for porevandet i det forurenede jordlag (herunder en vurdering af fordelingen af typiske opholdstider i et konkret forurenet jordlag) være en nødvendig aktivitet for at kunne vurdere, under hvilke kontakttider eventuelle laboratorieudvaskningstests skal gennemføres, såfremt de præcist skal simulere de pågældende feltforhold. Under alle omstændigheder er det væsentligt at vide, om kontakttiden i et givet naturligt system er større eller mindre end den påkrævede tid for opnåelse af ligevægt for de komponenter, hvis udvaskning ønskes undersøgt. Ved valg eller indretning af testmetode kan der så eventuelt tages hensyn hertil eller oplysningen kan som et minimum indgå i fortolkningen af testresultatet.
I Miljøstyrelsen (1996) angives typiske strømningshastigheder på 10-1000 m/år i den mættede zone og 0,5-2 m/år i den umættede zone (den aktuelle hastighed på en lokaliet kan estimeres udfra kendskab til den aktuelle nedbør samt jordbundsforhold). Opholdstiden i et jordelement på 1 m bliver med disse strømningshastigheder 0,5-2 år i den umættede zone og 0,001-0,1 år i den mættede zone, hvilket indikerer, at opholdstiden i nogle naturlige systemer kan være så lille, at der ikke vil være opnået sorptionsligevægt.
3.7 Specielle forhold for polære/ladede stoffer
De seneste fem års sorptionsforskning har næsten udelukkende beskæftiget sig med neutralt ladede stoffer. Der er således meget få nye erkendelser for ikke-neutrale stoffer (organiske syrer og baser) i det nævnte tidsrum, og der kan derfor henvises til Kjeldsen (1996) – afsnit 7.2.5 og 7.2.6 – for en gennemgang af sorptionen for disse stoffer. Det skal dog nævnes, at man er blevet mere opmærksom på, at phenoler kan indgå en kemisk binding (oxidativ kobling) med det naturlige organiske stof. I dette tilfælde vil stofferne praktisk talt ikke udvaskes på grund af den meget hårde binding (se afsnit 3.4.6).
Desorptionen af organiske syrer og baser styres, udover af de i tidligere afsnit beskrevne faktorer, af pH og stoffets pKa-værdi. Syrekonstanten, pKa (= -logKa), angiver den pH-værdi, hvor halvdelen af stoffet er ioniseret, og den anden halvdel er på neutral form. For en organisk syre vil størstedelen af stoffet være negativt ladet ved højt pH og neutral ved lavt pH. Dette betyder, at desorptionen vil stige ved stigende pH (over pKa-værdien). Mange organiske syrer, som er miljørelevante, har dog relativt høje pKa-værdier, hvorfor disse stoffer praktisk talt vil være fuldt neutrale ved miljørelevante pH-værdier. Dette gælder dog ikke for de fleste chlorphenoler, især de flersubstituerede, som har pKa-værdier i det miljørelevante pH-område - se også (Nielsen et al. 1996).