Miljøprojekt nr. 1271, 2009
Udvikling og validering af en metode til analyse af kolophonium i kosmetiske produkter
Indholdsfortegnelse
2 Analysemetode til kolofonium
- 2.1 Fase 1: Litteraturstudie af analysemetode
- 2.2 Fase 2: Udvikling af en analysemetode
- 2.3 Fase 3: Validering af den udviklede metode
Bilag B: En metode til kvantificering af harpikssyrer i kosmetik
Forord
Projektet er udført af Göteborgs Universitet (GU), Stockholms Universitet (SU) og Danmarks Miljøundersøgelser (DMU) ved Århus Universitet. DMU er ansvarlig for projektorganisering og afrapportering.
Litteraturstudiet af eksisterende publicerede metoder til analyse af kolofonium er udført af Ann-Theresa Karlberg, GU, og Ulrika Nilsson, SU.
Udviklingen af en analysemetode til analyse af kolofonium i kosmetiske produkter er udført af Ulrika Nilsson og Naghmeh Berglund, SU.
Metodevalideringen er udført af Pia Lassen og Gitte Hellerup Jensen, DMU.
Projektet er udbudt af Miljøstyrelsen (MST) i forbindelse med Udvikling af analysemetoder til kosmetik under Virksomhedsordningen.
Kontaktperson i MST er Dorrit Skals
Projektet startede i januar 2007 og sluttede i december 2007.
Sammenfatning og konklusioner
Kolofonium (harpiks) er en almindelig årsag til kontaktallergi og allergisk kontakt dermatitis. I følge EU’s lovgivning for farlige stoffer – Direktiv 67/548/EEC Annex 1 skal et indhold på >1 % af kolofonium i et produkt deklareres og produktet skal mærkes med risikosætning R 43 (kan give overfølsomhed ved kontakt med huden). Kolofonium er optaget på listen over farlige stoffer under følgende CAS numre: 8050-09-7 (som stofgruppe) samt 8052-10-6 og 73138-82-6. I henhold til kosmetikreguleringen (76/768/EØF) er stoffet optaget på INCI-listen (Index on Cosmetic Ingredients) under navnet ”Colophonium”, (EF. nr. 232-475-7) og der er ingen restriktioner på anvendelsen.
Kolofonium, som er et naturprodukt, er en kompleks blanding, som består af ca. 90 % forskellige organiske syrer og ca. 10 % neutralt organisk materiale. Ved en kemisk analyse er det ikke muligt at bestemme den totale mængde af en sådan blanding, som eksempelvis kolofonium, i et produkt. Årsagen til dette er, at et naturprodukt som oftest består mange enkeltstoffer og det er ikke muligt at kvantificere dem alle. En mulig metode til bestemmelse af indholdet af en kompleks blanding i et forbrugerprodukt er i stedet ved at kvantificere et eller flere udvalgte markørstoffer, som er specifikke for den komplekse naturlige blanding og på baggrund af koncentrationen af disse markørstoffer angive et estimat af indholdet af blandingen i produktet. I dette projekt var målet at udvikle en analysemetode til screening af indhold af kolofonium i kosmetik. På baggrund af den ovenstående argumentation var det nødvendigt, at kvantificeringen blev baseret på kemisk analyse af enkelte specifikke primære komponenter.Til kvantificeringen af kolofonium blev der udvalgt 2 primære harpikssyrer; abietinsyre (CAS-nr 514-10-3) og dehydroabietinsyre (CAS-nr. 1740-19-8) samt et oxidationsprodukt; 7-oxodehydroabietinsyre (CAS-nr. 18684-55-4) Luftoxidation af harpikssyrerne sker let ved kontakt med luft under almindelig opbevaring og håndtering af kolofonium.
Tidligere publicerede analytiske metoder til identifikation og kvantificering af de primære kolofonium komponenter blev undersøgt ud fra forskellige aspekter med henblik på udvælgelse af en analysemetode. De vigtigste aspekter var, at metoden skal kunne bruges af mindre avancerede analyselaboratorier, være robust og detektering af komponenter skal ske uden derivatisering. På baggrund af litteraturstudiet blev det besluttet, at en ny metode var nødvendig til dette projekt. Vi har derfor udviklet en omvendt fase HPLC metode med UV detektion ved brug af diode-array-detektor (DAD). Som referencestoffer blev anvendt rene ikke-oxiderede harpikssyrer og et stabilt oxidationsprodukt (sidstnævnte som markør for en mulig autooxidation og dannelse af allergifremkaldende oxidationsprodukter). Den analytiske separation er god, hvilket ikke tidligere har været opnået med en HPLC metode for kolofonium.
Summary and conclusions
Colophonium (rosin) is a common cause of contact allergy and allergic contact dermatitis. According to the EU legislation on dangerous substances – Annex 1 (Directive 67/548 /EEC) a content of >1% colophonium in a product must be declared and the product must be labelled with risk phrase R 43 (“May cause sensitisation by skin contact”). Colophonium is listed under the following CAS numbers: 8050-09-7, 8052-10-6 and 73138-82-6. According to the Cosmetics Directive (76/768/EØF) the component is listed on the INCI-list (Index on Cosmetic Ingredients) under the name ”Colophonium”, (EF. nr. 232-475-7) and there is no restriction for its application.
However, colophonium is a mixture of many compounds. Quantifying a mixture used as an ingredient of another mixture cannot be achieved by any analytical means. This applies to any natural extracts used as ingredient of a compounded consumer product. To overcome the impossibility of quantifying a complex ingredient in a product, the quantification of tracers (= defined substances) specific of the complex natural sources is feasible. Therefore, quantification must be based on chemical analyses of specific major compounds. The objective of the present study was to develop a suitable method to be used for screening of cosmetics with regard to content of colophonium. We have chosen to base our quantification on its two major recin acids abietic acid (CAS no. 514-10-3) and dehydroabietic acid (CAS no. 1740-19-8) together with a tracer, 7-oxodehydroabietic acid (CAS. 18684-55-4), for air oxidation which readily takes place at storage and handling of colophonium.
Earlier published analytical methods for identification and quantification of the major colophonium components have been thoroughly scrutinized from different aspects e. g. possibility to be used outside very sophisticated analytical laboratories, robustness, and possibility to detect compounds without derivatisation. Based on this, it was concluded that a new method was needed for the purpose of the present study. We have thus developed a reversed phase HPLC method with UV detection using diode-array-detector (DAD). Pure non-oxidized recin acids and a stable major oxidation product as a marker for a possible autoxidation and the formation of allergenic oxidation products are used as reference substances. The analytical separation obtained is good and has never been obtained before using HPLC methods.
1 Baggrund
Eksponering til kolofonium (harpiks) er en kendt årsag til kontaktallergi og kontakt dermatitis (1). Efter EU’s lovgivning for farlige stoffer – Bilag 1 (Direktiv 67/548/EEC) skal et indhold >1 % af kolofonium i et produkt deklareres og produktet skal mærkes med risikosætning R 43 (kan give overfølsomhed ved kontakt med huden). Kolofonium er optaget på listen over farlige stoffer under følgende CAS. numre: 8050-09-7 (som dækker over en stofgruppe), 8052-10-6 og 73138-82-6. . I henhold til kosmetikreguleringen (76/768/EØF) er stoffet optaget på INCI-listen (Index on Cosmetic Ingredients) under navnet ”Colophonium”, (EF. nr. 232-475-7) og der er ingen restriktioner på anvendelsen.
I kliniske studier er der observeret klare sammenfaldende positive reaktioner ved lappe-test for kolofonium og parfumemarkører (fragrance mix og Perubalsam) blandt patienter (2-4). De sammenfaldende testreaktioner kan bedst beskrives som et resultat af kombineret sensitivitet forårsaget af samtidig eksponering til et stort antal duftstoffer og kolofonium komponenter i dagligdags produkter (5). Der har ingen undersøgelser været af indholdet af kolofonium i kosmetik, da der ikke har eksisteret en anvendelig analysemetode, som også medtager de allergene oxiderede harpikssyrer.
Kolofonium er en harpiks (eng. resin), som udvindes fra forskellige arter af nåletræer, og hvorfra terpentinolien er afdestilleret. Det består af en kompleks blanding af harpikssyrer (omkring 90 %) og neutrale stoffer (10 %), hvilket varierer afhængig af art, oprensningsproces og opbevaringsbetingelser. De primære harpikssyrer er abietinsyre (eng. abietic acid) (AbA) og dehydroabietinsyre (eng. dehydroabietic acid) (DeA) (Figur 1) (1). Oxideret materiale er til stede i kolofonium som følge af luftpåvirkning under normal håndtering (dvs. produktfremstilling og efterfølgende almindelig brug) samt opbevaring ved stuetemperatur. Der er således påvist en sammenhæng mellem graden af oxidation og opbevaringstiden. Omfattende studier har vist at oxiderede komponenter, der er dannet ved luftpåvirkning, hovedsagelig dannes fra AbA (6-9). Mens AbA selv er et svagt allergen eller ikke-allergen, er nogle af de dannede oxidationsprodukter allergener. Et af de mest almindeligt forekommende oxidationsprodukter er 15-hydroperoxyabietinsyre (eng. 15-hydroperoxyabietic acid), der er et kraftigt allergen (10).
Kolofonium er ofte modificeret eller derivatiseret for at opnå bedre tekniske egenskaber. Oftest modificeres kolofonium kun delvist indtil de ønskede tekniske egenskaber er opnået, hvilket betyder at umodificeret kolofonium vil være til stede i produktet. Både modificeret og umodificeret kolofonium kan forårsage kontakt allergi. Imidlertid har undersøgelser vist at hvis patienter reagerer på både modificeret og umodificeret kolofonium, skyldes reaktionen oftest indholdet af umodificeret kolofonium og ikke en sensitivitet overfor begge typer. Det vil sige at kontakt allergi forårsaget af modificeret kolofonium kan alternativt derfor også skyldes dets indhold af umodificeret kolofonium (11, 15).
Kolofonium i umodificeret og modificeret form anvendes i en række produkter og materialer f.eks. loddevæske, papir (papir masse) maling, lim, plaster og også i kosmetik. Mængden af umodificeret kolofonium kan variere i forskellige produkter fra 20 % eller mere i visse plastre, maling og loddevæske til små mængder i andre produkter, som hovedsagelig indeholder modificeret kolofonium (12)
Da kolofonium er ikke-toksisk, ikke-irriterende og meget klæbrig, er det et perfekt materiale til at binde kosmetik på huden. Allergisk kontakt dermatitis udløst af kolofonium i forskellige kosmetiske produkter såsom øjenskygge, rouge, læbeprodukter og mascara er rapporteret i litteraturen. Kolofonium er også fundet i mascara, som er mærket som ”hypoallergeniske” og indholdet af kolofonium i disse produkter var højt nok til at udløse reaktioner hos sensitive personer (13). Omfattende reviews omhandlende brugen af kolofonium er blevet publiceret gennem årene (6, 11, 14, 15).
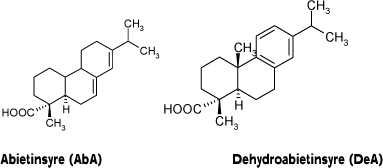
Figur 1. De primære harpikssyrer i umodificeret kolofonium
2 Analysemetode til kolofonium
- 2.1 Fase 1: Litteraturstudie af analysemetode
- 2.2 Fase 2: Udvikling af en analysemetode
- 2.3 Fase 3: Validering af den udviklede metode
Formålet med dette projekt er at udvikle en metode til brug for screening af kosmetik med henblik på bestemmelse af indholdet kolofonium, baseret på de primære komponenter AbA og DeA samt en markør for oxideret materiale. Projektet er opdelt i 3 faser. Fase 1: Et litteraturstudie af publicerede analysemetoder. Fase 2: Udvikling af en analysemetode. Fase 3: Validering af den udviklede analysemetode.
2.1 Fase 1: Litteraturstudie af analysemetode
Publicerede metoder til identifikation og kvantificering af kolofonium komponenter er blevet gennemgået og evalueret for at vurdere, om der eksisterede en metode som kunne anvendes til videre udvikling af en ny metode.
2.1.1 Resultater
De publicerede metoder til identifikation og kvantificering af kolofonium komponenter er beskrevet i Bilag A. Der er korte beskrivelser og evalueringer af de fundne metoder. På baggrund af litteraturstudiet kunne 2 metoder fremhæves. Metode I er en GC-FID metode, som kræver derivatisering. Metode II er en HPLC-fluorescens metode, som dog er uspecifik og derfor kræver videre udvikling for at kunne bruges på komplekse prøver. Da ingen af disse metoder lever op til de fastlagte krav, blev det besluttet, at det var nødvendigt at udvikle en ny metode til dette projekt.
2.2 Fase 2: Udvikling af en analysemetode
I denne fase blev der udviklet en metode til bestemmelse og kvantificering af de vigtigste komponenter i kolofonium, AbA og DeA, samt en markør for oxideret harpikssyre. En HPLC-UV metode med diode array detektor (DAD) blev valgt. Denne metode giver en høj specifik bestemmelse for de udvalgte komponenter uden brug af derivatisering.
Traditionelt set bestemmes de vigtigste komponenter i kolofonium ved brug af gas kromatografi (GC) (16), men HPLC metoder har også været anvendt (12). Analyser, der kun er baseret på ikke-oxiderede harpikssyrer, fortæller ikke noget om de oxiderede harpikssyrer. En lille detekteret koncentration af AbA kan betyde at der kun er en lille mængde kolofonium tilstede i produktet, men det kan også skyldes en kraftig oxidation af kolofonium. Eftersom oxidationsprodukterne i kolofonium også er de primære allergener er det være særlig nødvendigt at udvikle en metode til kvantificering, som også medtager oxidationsprodukterne. Da metoden skal bruges til screening, er det vigtigt at den er robust, billig og simpel samt ikke involverer meget farlige kemikalier. Det er ikke muligt at anvende den kraftigt allergene 15-hydroperoxyabietic som markør for oxidationsprodukter, da det er meget ustabilt og derfor besværlig at håndtere. I stedet for blev det stabile oxidationsprodukt 7-oxodehydroabietinsyre (7-O-deA) valgt som markør for oxideret kolofonium (Figur 2). Dette oxidationsprodukt er stabilt nok til at blive brugt som referencestof og er samtidig ikke så kraftig en allergen som hydroperoxiderne, hvilket gør den nemmere at håndtere.
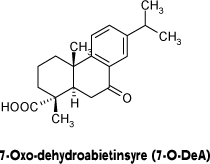
Figur 2: Et af oxidationsprodukterne identificeret i luft–eksponeret kolofonium og udvalgt som markør for den oxidative nedbrydning af AbA
Indholdet af AbA varierer mellem 30-50 % i kolofonium af typen gum rosin og 35-40 % i tallolie kolofonium. Hvis det antages, at indholdet af harpikssyrer er 90 %, vil en detektionsgrænse på 10 mikrogram/g AbA således svare til en detektionsgrænse på cirka 30 mikrogram/g (ppm) for kolofonium. Dette vil blive betragtet som et acceptabelt niveau i forhold til det niveau, der udløser reaktion hos allergi patienter. Følsomme patienters reaktion på kolofonium af typen gum rosin er testet med en serie fortyndinger af gum rosin på 20-0,001 % i vaseline. Ud fra testen reagerede 50 % af forsøgspersonerne på en koncentration ned til 0,1 % (1000 ppm), mens den mest følsomme patient reagerede på et niveau ned til 0,001 % (10 ppm) (18). For andre patienter testet med tallolie kolofonium (20-0,001 % i vaseline) fandt man ingen reaktion lavere end 1 % (10 000 ppm) (7).
2.2.1 Materialer og metoder
2.2.1.1 Kemikalier
Standarder
AbA, DeA og primarinsyre (alle >95 % renhed) er fra Helix Biotech, Vancouver, Canada. Cis-5,8,11,14,17-Eicosapentaenoinsyre (EPA, >98,5 %), der anvendes som intern standard, er fra Fluka, Sigma-Aldrich ChemieGmbH, Steinheim, Germany. 7-O-DeA er syntetiseret som beskrevet nedenfor.
Stamopløsninger af alle komponenterne er fremstillet i 100 % acetonitril i en koncentration på 1 mg/ml. De blev opbevaret i fryser gennem projektets forløb.
Standardopløsninger til kvantificering på HPLC blev lavet i acetonitril:MilliQ vand (i forholdet 9:1) med 0,2 % myresyre. Opløsningerne blev opbevaret på køl og opretholdt stabilitet i mindst 2 uger. Stabiliteten blev kontrolleret overfor friske opløsninger. Der blev ikke kontrolleret for en længere periode end to uger.
2.2.1.2 Solventer og prøver
Methanol med en renhed på 99,8 % er fra Ltd BDH, myresyre (renhed 98-100 %) fra Scharlau og acetonitril af kromatografisk kvalitet fra Sigma-Aldrich.
Gum rosin (kolofonium) prøver (umodificeret og maleinsyre-anhydrid-modificeret fra Soccer, Portugal) og umodificeret kolofonium (Fluka) var opløst i 100 % acetonitril i en koncentration på 1,5 mg/ml.
Kosmetik prøver (1g foundation fra Face, Stockholm) blev spiket med standarder på forskellige koncentrationsniveauer, fra 1 mikrogram/g til 500 mikrogram/g af hver komponent.
2.2.1.3 Syntese af 7-O-DeA
7-O-deA er syntetiseret i to trin som beskrevet i litteraturen (17). En blanding af AbA (5,2 g svarende til 17,19 mmol) og 5 % Pd/C (260 mg, 5 % w/w) blev opvarmet til 245 °C i 1 time under nitrogenflow i en to-halset rundbundet kolbe. Efter afkøling blev det faste stof opløst i ethylacetat (4 x 50 ml eller indtil alt var opløst) og filtreret igennem celite. Den organiske fase blev ekstraheret med 0,25 M natrium hydroxid (2 x 100 ml) efterfulgt af forsuring af den vandige fase med 1 M saltsyre. Råproduktet var derefter isoleret fra den vandige fase ved udrystning med ethylacetat (3 x 50 ml). Den organiske fase indeholdende råproduktet blev tørret med magnesium sulfat og opkoncentreret under reduceret tryk. Oprensning af råproduktet foregik ved hjælp af flash kromatografi (ethylacetat/heman/trifloureddikesyre i forholdet 14/85/1), hvilket gav et udbytte på 2,5 g (46 %) DeA. Dette var i overensstemmelse med tidligere rapporterede data.
Til en blanding af DeA (2,1 g, 6,99 mmol) og kalium hydroxid (0,4 g) i vand (40 ml) blev der drypvis tilsat en opløsning af kalium permanganat (2,8 g, 17,72 mmol) i vand (60 ml) ved stuetemperatur. Slutblandingen blev omrørt i 2,5 time efterfulgt af mætning med svovl dioxid. Et hvidt bundfald blev dannet og centrifugeret fra, vasket med vand og tørret natten over. Stoffet blev oprenset ved hjælp af flash kromatografi (ethylacetat/hexan/ trifloureddikesyre 14/85/1) og gav et udbytte på 1,15 g (52 %) af 7-O-DeA med en renhed på 95 %. Renheden blev bestemt ved Nuclear Magnetic Resonance (NMR) analyse, som er almindelig anvendt til renhedsbestemmelse i forbindelse med synteser. Resultatet var i overensstemmelse med tidligere rapporterede data.
2.2.1.4 Udstyr
Et Agilent 1100 gradient HPLC system blev anvendt til analyse. Der anvendtes et 20 mikroliter injektionsloop, en DAD og en kolonnetermostat sat til 25 °C. HPLC kolonnen var en Prism RP-12 fra Thermo (4,6 mm i.d., længde 150 mm, partikelstørrelse 3 mikrometer). Den stationære fase i kolonnen indeholder både C12-kæder og urea funktioner. Til SPE blev anvendt Oasis MAX kolonner (6cc, 500mg) fra Waters.
2.2.2 Analysemetode
2.2.2.1 Analyse af kosmetikprøver
1 g spiket prøve blev tilsat 20 ml acetonitril og sat i ultralydsbad i 30 min. 500 mikroliter EPA opløsning (928 mikrogram/ml) blev tilsat som intern standard (IS) før ekstraktionen.
Hvert ekstrakt blev overført til plastrør og centrifugeret i 15 min ved 4000 rpm. Supernatanten blev overført til en kolbe, mens bundfaldet (pellet) blev genopløst i acetonitril, ultralydsbehandlet og centrifugeret igen som beskrevet ovenfor. Supernatant fraktionerne blev samlet og inddampet ved hjælp af rotations inddamper til et volumen på cirka 5 ml. Volumenet blev justeret til 7 ml ved tilsætning af acetonitril. MilliQ vand blev derefter tilsat til et slutvolumen på 10 ml indeholdende acetonitril:vand i forholdet 7:3.
Tilsætning af vand gjorde visse af opløsningerne uklare. I de tilfælde blev opløsningen filtreret gennem sprøjtefilter (0,45 mikrometer polypropylene) før fast-fase ekstraktion (SPE).
2.2.2.2 Fast-fase ekstraktion
SPE kolonnen blev konditioneret med 1) 3 ml methanol og 2) 3 ml MilliQ. Prøven blev påsat og kolonnen blev efterfølgende skyllet med 1) 3 ml 50 mM natriumacetat i MilliQ vand for at eliminere meget polære stoffer eller baser, 2) 4 ml methanol for at eliminere hydrofobe stoffer og til sidst 3) 1 ml myresyre (2 %) i methanol. Prøven blev derefter elueret med 2 ml myresyre (2 %) i methanol. Yderligere 1 ml myresyre (2 %) i methanol blev tilsat i tilfælde af gennembrud. Frisk myresyre opløsning blev fremstillet hver dag.
2.2.2.3 Analyse af kolofonium
Intern standard, 200 mikroliter (186 mikrogram) blev tilsat til 2 ml af 1,5 mg/ml gum rosin opløsninger i acetonitril. Gum rosin prøverne blev filtreret og analyseret direkte på HPLC (uden SPE oprensning).
2.2.2.4 HPLC metode
Methanol:vand i forholdet 8:2 med 0,05 % myresyre blev brugt som mobil fase med et flow på 1 ml/min (dette giver et tryk på cirka 240 bar). Eluering foregik ved konstant koncentration og sammensætning af den mobile fase, dvs. isokratisk eluering. Der blev anvendt 4 forskellige bølgelængder 210, 220, 240 og 250 nm. Forholdene mellem absorptionen ved de forskellige bølgelængder blev brugt til identifikation af komponenterne. Der blev anvendt isokratisk eluering for at undgå stigning af basislinjen, hvilket ville have givet anledning til højere LOD værdier. Mængden af myresyre var kritisk for både retentionstid og støj på basislinjen.
Der blev lavet en kalibreringskurve i koncentrationsintervallet fra 0,8 ng/mikroliter til 500 ng/mikroliter for hver komponent. Til kvantificering blev der dagligt lavet et-punkts kalibrering ved tredobbelt injektion af en 250 ng/mikroliter opløsning af komponenterne. Kvantifikationen blev foretaget ved brug af relative respons faktorer analyt/IS.
Specifikke absorptionsforhold (CV mindre end 10 %) blev brugt til identifikation af de forskellige komponenter:
| AbA: | 240nm/250nm: forhold 1,36 |
| DeA: | 220nm/210nm: forhold 0,55 |
| 7-O-DeA: | 220nm/210nm: forhold 0,53; 240nm/210nm: forhold 0,26; 250nm/210nm: forhold 0,45. |
| EPA (IS): | 220nm/210nm: forhold 0,40 |
Til kvantificering af DeA og EPA blev der anvendt arealtoppe ved bølgelængden 210 nm. For AbA og 7-O-DeA anvendtes 240nm.
2.2.3 Resultater
2.2.3.1 Selektivitet
Den udviklede metode bygger på separation og identifikation af de tre udvalgte komponenter i kolofonium (AbA, DeA og 7-O-DeA) (Figur 3). Detektionen af syrerne er foretaget ved de ovenfor angivne bølgelængder. Der blev opnået er basislinje separation af AbA og DeA toppene. Et af problemerne med LC-separation ved UV-detektion er co-eluering mellem pimarinsyre (en harpikssyre, som også findes i kolofonium) og AbA. Den nye metode anvender en LC-kolonne, hvor den stationære fase er mere selektiv overfor molekyleform, hvilket sikrer en separation af pimarinsyre og AbA (dog ikke med total basislinje separation). Med den valgte mobile fase (methanol:vand 8:2 med 0,05 % myresyre) og et flow på 1,0 ml/min, kan de udvalgte harpikssyrer i kolofonium separeres i løbet af 22 min. Det er vigtigt at kontrollere mængden af myresyrer i den mobile fase præcist. For lav koncentration (0,02 %) påvirker separationen i en dårlig retning og ved højt niveau (0,1 %) stiger støj på basislinjen kraftigt. Der blev anvendt isokratisk eluering, da gradient eluering øgede støjen på basislinjen.
2.2.3.2 Separation af komponenter i kolofonium prøver
Basislinje separation af toppe svarende til flere harpikssyrer blev også opnået, hvilket analyse af umodificeret kolofonium viser (Se Figur 4).
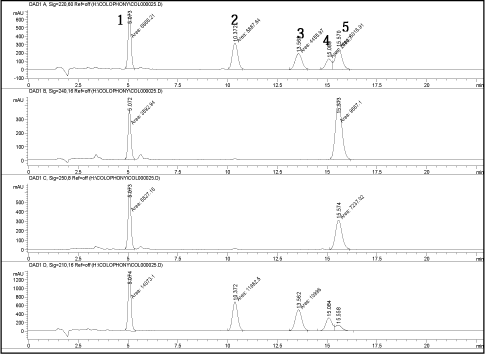
Figur 3. Standard opløsning (500 mikrogram/ml) analyseret med den udviklede metode. 1= 7-O-DeA, 2= DeA, 3= EPA, 4= pimarinsyre, 5= AbA
Krommatogrammer er vist ved 220, 240, 250 og 210 nm
Figur 4: Kolofonium prøve, umodificeret Rosin fra Soccer (1,5 mg/ml) analyseret med den udviklede metode. 2 = DeA, 3 = EPA, 4 = pimarinsyre, 5 = AbA. Den oxiderede harpikssyre 7-O-DeA kunne ikke detekteres. Kromatogrammerne er vist ved 220, 240, 250 and 210 nm
2.2.3.3 Kvantifikation af komponenter i kolofonium prøver
De forskellige analytter er kvantificeret i umodificerede og modificerede kolofonium prøver. Resultaterne er vist i Tabel 1.
Tabel 1. Koncentration af komponenterne i forskellige prøver af umodificeret og modificeret kolofonium (mg/g)
| AbA | DeA | 7-O-DeA | |
| Umodificeret kolofonium af gum rosin type (Soccer) |
255 | 62 | N. D. |
| Umodificeret kolofonium af gum rosin type (Fluka) |
691 | 40 | Not det |
| Malein anhydrid-modificeret gum rosin (Soccer) |
74 | 23 | Not det. |
2.2.3.4 Linearitet
Toparealerne for AbA, DeA og 7-O-DeA varierer lineært med harpikssyre koncentrationen indenfor koncentrationsområdet 10 mikrogram/g til 500 mikrogram/g.
2.2.3.5 Detektionsgrænser
Under anvendelse af den beskrevne metode var instrument detektionsgrænsen (LOD) 4 mikrogram/ml for AbA, 31 mikrogram/ml for DeA og 3 mikrogram for 7-O-DeA.
2.2.3.6 Genfindinger
Genfindingerne blev undersøgt på 3 forskellige koncentrationsniveauer i spiket foundation. De forskellige tilsatte niveauer af de enkelte analytter var 100, 200 og 500 mikrogram/ml. Genfindingen for AbA var 78 % (CV 1 %), DeA var 88 % (CV 5 %), 7-O-DeA var 76 % (CV 2 %) og EPA 97 % (CV 5 %).
2.2.3.7 Præcision
Præcision angiver graden af overensstemmelse mellem uafhængige målinger opnået under fastlagte betingelser. Som oftest udtrykkes præcision som ”mangel på præcision” og beregnes som en spredning mellem måleresultaterrne, hvilket er tilfældet her. Præcision kan bestemmes under både repeterbarheds- og reproducerbarhedsbetingelser. Præcisionen for HPLC metoden var mindre end 10 % (CV) både med hensyn til reproducerbarhed (dag til dag variation, n=6) og repeterbarhed (n=3) beregnet ud fra den relative respons faktor analyt/IS. Præcisionen blev testet med en 250 mikrogram/g standard.
2.3 Fase 3: Validering af den udviklede metode
Metoden udviklet i fase 2 blev valideret for kvantificeringsgrænse, repeterbarhed og genfinding for de udvalgte harpikssyrer og oxidationsprodukt på 2 koncentrations niveauer ved standard addition til kosmetik prøver. En større screening af kolofonium i en række produkter på baggrund af de udvalgte komponenter var ikke mulig på grund af de begrænsede ressourcer i dette projekt. Det anbefales i stedet at indholdet af kolofonium i kosmetikprodukter undersøges i et separat projekt. Enkelte kosmetikprodukter var dog analyseret for indhold af de udvalgte komponenter indenfor dette projekt.
2.3.1.1 Separation af de udvalgte komponenter i kosmetik
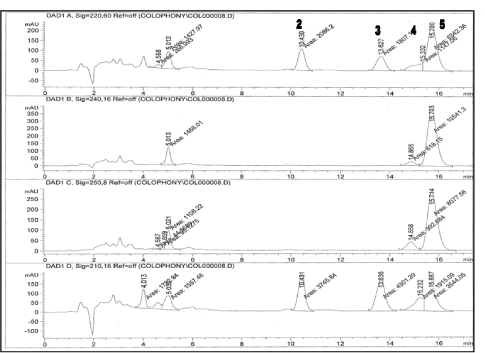
Komponenterne blev kvantificeret i forskellige kosmetikprøver i henhold til den udviklede metode. Separationen var fyldestgørende for identifikation og kvantificering i de testede kosmetikprøver. Dette er illustreret med et kromatogram af et voks-strip produkt til hårfjerning (Figur 5).
Kromatogrammet målt ved 210 nm er vist som eksempel.
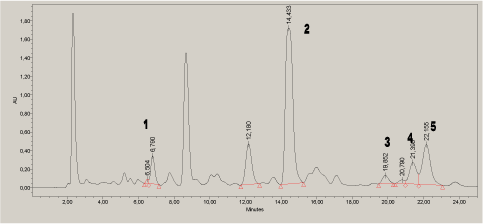
Figur 5. Kromatogram af voks strip ved 210 nm. 1=7-O-DeA, 2=DeA, 3=EPA, 4=pimarinsyre, 5=AbA.
2.3.1.2 Præcision
Repeterbarheden, beregnet som CV, blev bestemt på 2 niveauer baseret på spikede prøver (standard addition). For alle komponenterne var repeterbarheden på lavt niveau (50-90 mikrogram/g for hver komponent) i god overensstemmelse med resultaterne fra fase 2 (se Tabel 2). For det høje niveau (135-250 mikrogram/g) var præcisionen 5-7 % (se Tabel 3).
Tabel 2. Analyser af kosmetikprodukter ved lavt niveau (50 – 90 mikrogram/g) ved standard addition
| komponent | repeterbarhed | % repeterbarhed | % genfinding | LOD | LOQ |
| AbA | 2,4 | 2,4 | 85 | 7,2 | 21,6 |
| DeA | 6,4 | 13 | 84 | 19,2 | 57,6 |
| 7-O-DeA | 4,3 | 11 | 91 | 12,9 | 38,7 |
Tabel 3. Analyser af kosmetikprodukter ved højt niveau (135 – 250 mikrogram/g) ved standard addition
| komponent | repeterbarhed | % repeterbarhed | % genfinding |
| AbA | 13,1 | 6,5 | 94 |
| DeA | 11,2 | 4,8 | 92 |
| 7-O-DeA | 11,8 | 7,5 | 94 |
2.3.1.3 Linearitet og repeterbarhed af HPLC
Lineariteten og repeterbarheden blev testet for den udviklede HPLC metode ved gentagne injektioner (n=10). Metoden er lineær i koncentrationsområdet 1 til 400 mikrogram/g (Korrelationskoeficient, r²: 0,999-1,000). Repeterbarheden af instrumentet var god i området 5-185 mikrogram/ml. AbA: 2-4,5 %, DeA: 0,3-2 %, 7-O-DeA: 0,3-1 % og den interne standard, EPA: 2,5 % ved en koncentration på 125 mikrogram/ml.
2.3.1.4 Detektionsgrænse
Detektionsgrænsen er bestemt som 3 x SD baseret på 6 analyserede prøver spiket på lavt niveau og lå i omkring 7-20 mikrogram/g (se Tabel 2).
2.3.1.5 Kvantifiseringsgrænse
Bestemmelse af metodens kvantifikationsgrænsen var baseret på spikede kosmetikprøver ved brug af ligningen: LOQ = blind + 3 x LOD. Dette svarede til LOQ = 3 x LOD, da der ikke var målbare blindværdier.
2.3.1.6 Genfinding
Genfindingerne, baseret på standard addition, svarede i niveau til de værdier der var fundet i fase 2. Se Tabel 2 og 3. CV for genfindingerne lå på 4-6 %.
2.3.1.7 Identifikation af analytter
De specifikke absorptionsforhold for identifikation af de enkelte analytter blev kontrolleret, da forskellige detektorer blev brugt i fase 2 og fase 3. Der er nogle afvigelser, specielt for DeA og EPA. Det er derfor anbefalet, at de specifikke absorptionsforhold kontrolleres ved hjælp af rene standarder før analysen påbegyndes. CV var mindre end 10 %, hvilket er i overensstemmelse med fase 2 (se Tabel 4).
Tabel 4: Specifikke absorptionsforhold for identifikation angivet for de to forskellige detektorer anvendt i fase 2 og 3.
| AbA | DeA | 7-O-DeA | EPA | |||||
| nm ratio | Fase 2 |
Fase 3 | Fase 2 | Fase 3 | Fase 2 |
Fase 3 | Fase 2 |
Fase 3 |
| 220/210 | 0,55 | 0,83 | 0,53 | 0,53 | 0,4 | 0,06 | ||
| 240/210 | 0,26 | 0,24 | ||||||
| 250/210 | 0,45 | 0,44 | ||||||
| 240/250 | 1,36 | 1,54 | ||||||
2.3.1.8 Kvantifikation af produkter
Forskellige almindelige kosmetik produkter blev analyseret for indhold af de udvalgte harpikssyrer. Baseret på de målte data kan indholdet af kolofonium beregnes (tabel 5). Kun én voks strip havde et signifikant indhold svarende til 7 mg/g kolofonium. For andre produkter kunne indholdet AbA og DeA detekteres (0,5 – 4,5 mikrogram/gram), men koncentrationerne var dog under detektionsgrænsen.
Tabel 5: Kvantifikation af produkter (mikrogram/g)
| Komponenter | Foundation | Lip gloss | Voks strip, normal |
Voks strip, ansigt |
| AbA | <7 | <7 | 2302 | <7 |
| DeA | <19 | <19 | 2517 | <19 |
| 7-O-DeA | <13 | <13 | 27,8 | <13 |
3 Diskussion
En ny metode til bestemmelse og kvantificering af to primære komponenter, AbA og DeA, i kolofonium (harpiks, eng. rosin) er blevet udviklet. Metoden inkluderer også mulighed for bestemmelse og kvantificering af et af de vigtigste oxidationsprodukter, 7-O-DeA, som dannes ved oxidativ nedbrydning af kolofonium, som har været i kontakt med luft. Den udviklede metode involverer en HPLC med en UV-DAD detektor som er i stand til at separere harpikssyrerne. Metoden er hurtig, robust og simpel samt uden anvendelse af giftige kemikalier. Endvidere har metoden høj specificitet i forhold til ordinære UV metoder, da der anvendes absorptionsforhold til identifikation af de udvalgte komponenter. Dette betyder at metoden har en mere sikker identifikation af komponenterne sammenlignet med ordinære UV metoder, hvor identifikation primært er baseret på retentionstid. Den høje specificitet er nødvendig på grund af den komplekse sammensætning i kosmetik. Dette er opnået ved brug af en DAD detektor som muliggør specificitet uden anvendelse af massespektrometri. I forhold til GC metoder er det ikke nødvendigt at anvende derivatisering.
Ifølge EU lovgivning om farlige stoffer – Annex 1(Direktiv 67/548/EEC) skal en koncentration >1 % af kolofonium i produkter deklareres og produktet skal mærkes med R-sætning R 43 (kan give overfølsomhed ved kontakt med huden). Produkter, der ikke skal klassificeres som sensibiliserende, men som indeholder en koncentration af kolofonium på = 0,1 %, skal mærkes ”Indeholder kolofonium. Kan udløse allergisk reaktion”. Dette gælder dog ikke kosmetiske produkter, der mærkes i henhold til forskrifterne i kosmetikbekendtgørelsen (Bek. nr. 422, 2006) og kosmetikreguleringen (76/768/EØF). Når kolofonium anvendes som ingrediens i kosmetik skal det altid fremgå af indholdsdeklarationen uanset koncentrationen i produktet.
Imidlertid er kolofonium en blanding af mange komponenter og det er ikke muligt at kvantificere dem alle. Dette gælder i alle tilfælde, hvor et naturligt ekstrakt, som oftest består af mange enkeltstoffer, anvendes som ingrediens i et forbrugerprodukt. En mulig metode til bestemmelse af indholdet af en kompleks blanding i et produkt er i stedet ved at kvantificere et eller flere udvalgte markørstoffer, som er specifikke for den komplekse naturlige blanding og på baggrund af koncentrationen af disse markørstoffer angive et estimat af indholdet af blandingen i produktet. På baggrund af den ovenstående argumentation, var det derfor nødvendigt at kvantificeringen i dette projekt blev baseret på kemisk analyse af enkelte specifikke komponenter. For kolofonium har vi valgt at basere vores kvantificering på 2 primære harpikssyrer. Det er dog ikke muligt at angive den eksakte koncentration af kolofonium i et produkt på baggrund af analyse af disse syrer, da der er forskel i mængden og sammensætningen af syrerne i forskellige typer af kolofonium på grund af variation i ekstraktionen, håndtering, opbevaring og fabrikation. Dette har specielt betydning for indholdet af DeA. Indholdet af AbA kan variere mellem 30-50 % i gum rosin og 35-40 % for tallolie kolofonium. For DeA er tallene 5-10 % i gum rosin og omkring 30 % i tallolie kolofonium. Endvidere er der ved bølgelængden 220 nm, som anvendes til detektion af DeA, høj risiko for interferens fra matricen. På baggrund af dette anvendes detektionen af AbA som kvantificering af kolofonium i de fleste tilfælde.
Et lavt niveau af AbA kan indikere et lavt indhold af kolofonium. Det er dog vigtigt at kontrollere, at det lave niveau ikke skyldes en oxidativ nedbrydning af AbA, som resulterer i meget allergifremkaldende komponenter. For at undgå fejl i vurdering af tilstedeværelse af kolofonium er der også analyseret for DeA, som er en mere stabil komponent end AbA samt for 7-O-DeA, der er et oxidationsprodukt af AbA. Høje niveauer af disse stoffer sammen med et lavt niveau af AbA vil indikere, at AbA er kraftigt dekomponeret i prøven, og at indholdet af kolofonium er større end estimatet på basis af AbA analysen. Endvidere betyder dette også, at indholdet af allergifremkaldende oxidationsprodukter kan være højt.
To generelle typer af analyser af komponenter i kolofonium er beskrevet i litteraturen. Dels en traditionel GC-FID metode med derivatisering af harpikssyrer for at opnå en god separation. Denne metode er især brugt i f.eks. papirindustrien. På det seneste er LC metoder med UV, fluorescens eller massespektrometriske detektionsmetoder blevet udviklet. De fleste publicerede metoder er udviklet med henblik på at undersøge specifikke produkters indhold af kolofonium (Se Bilag A). Den udviklede metode i projektet er ikke begrænset til en specifik produktgruppe som f.eks. kosmetik. Metoden kan anvendes bredt. Imidlertid kan der være begrænsninger begrundet i metoden for prøvebehandling overfor visse produkttyper, eksempelvis gummi og plastik, hvor stofferne kan være bundet hårdere i materialet end i kosmetikprodukter.
De anvendte SPE kolonner til prøveoprensning i dette projekt består af en blanding af lipofile og svage anion udbytningssites, hvilket ser ud til at passe harpikssyrerne perfekt. Andre lipofile komponenter kan vaskes ud af kolonnen ved brug af methanol, hvorimod harpikssyrerne holdes tilbage indtil syre tilsættes. Den anvendte metode giver meget rene ekstrakter i henhold til LC analyserne.
Prøver fra forskellige produkter er blevet analyseret. Kun en voks strip til fjernelse af hår havde et signifikant indhold af kolofonium. For de andre produkter (lip gloss, foundation, voks strip til ansigt) er der spor af AbA og DeA, men koncentrationerne var under detektionsgrænsen. I følge varedeklarationen for de udvalgte produkter var der kun deklareret indhold af kolofonium i de to typer voks strips.
4 Konklusion
Tidligere publicerede analysemetoder for identifikation og kvantificering af de primære komponenter i kolofonium er blevet gennemgået ud fra forskellige aspekter, herunder f.eks. mulighed for at metode kan blive anvendt af mindre avancerede laboratorier, robusthed, samt mulighed for at detektere stofferne uden brug af derivatisering. På basis af litteraturstudiet blev det konkluderet, at det var nødvendigt at udvikle en ny metode til formålet i dette projekt. Der blev udviklet en omvendt-fase HPLC metode med UV detektion vha. diode-array-detektor. Metoden er simpel, hurtig og anvender ikke farlige kemikalier. Da der anvendes absorptionsforhold til identifikation af markør stofferne er metoden mere specifik sammenlignet med ordinære UV metoder, hvor identifikation primært er baseret på retentionstid. Rene ikke-oxiderede harpikssyrer og et stabilt oxidationsprodukt blev anvendt som referencestoffer. Oxidationsproduktet var medtaget som markør for en mulig autooxidation og dermed også en indikator for dannelse af andre allergene oxidationsprodukter i kolofonium. Den opnåede analytiske separation af harpikssyrerne er god, hvilket ikke tidligere har været opnået med en HPLC metode for kolofonium.
5 Referencer
- Karlberg A-T. Colophony. In: Handbook of Occupational Dermatology. Eds. L Kanerva, P Elsner, J E Wahlberg, H I Maibach. Springer-Verlag, Heidelberg. 2000 pp 509-516.
- Färm G. Contact allergy to colophony and hand eczema. Contact Dermatitis 1996: 34: 93-100.
- Wohrl S, et al. The significance of fragrance mix, balsam of Peru, colophony and propolis as screening tools in the detection of fragrance allergy. Br J Dermatol 2001: 145: 268-273.
- Matura M, et al. Selected oxidized fragrance terpenes are common contact allergens. Contact Dermatitis 2005: 52: 320-328.
- Bråred-Christensson J, et al. Hydroperoxides form specific antigens in contact allergy. Contact Dermatitis 2006: 55: 230-237.
- Karlberg A-T. Contact allergy to colophony. Chemical identifications of allergens, sensitization experiments and clinical experiences. Acta Derm-Venereol 1988; Suppl. 139, 1-43.
- Karlberg A-T. Air oxidation increases the allergenic potential of tall oil rosin. Gum rosin contact allergens also identified in tall oil rosin. Am J Contact Dermatitis, 1991; 2: 43-49.
- Hausen BM, et al. Contact allergy due to colophony (IX). Contact Dermatitis 1993: 29: 234-240.
- Gäfvert E, et al. Contact allergy to resin acid hydroperoxides. Hapten binding via free radicals and epoxides. Chem Res Toxicol 1994; 7: 260-266.
- Karlberg A-T, et al. Identification of 15-hydroperoxyabietic acid as a contact allergen in Portuguese colophony. J Pharm Pharmacol 1988; 40: 42-47.
- Gäfvert E. Allergenic components in modified and unmodified rosin. Acta Derm Venereol (Stockholm)1994: Suppl. 184: 1-36.
- Ehrin E, Karlberg A-T. Detection of rosin (colophony) components in technical products using an HPLC technique. Contact Dermatitis 1990; 23: 359-366.
- Karlberg A-T, Lidén C, Ehrin E. Colophony in mascara as a cause of eyelid dermatitis. Acta Derm-Venereol 1991; 71: 445-447.
- Färm G. Contact allergy to colophony. Clinical and experimental studies with emphasis on clinical relevance. Acta Derm Venereol (Stockholm)1997: Suppl. 201: 1-42.
- Downs AMR, Sansom JE. Colophony allergy: a review. Contact Dermatitis 1999: 41: 305-310.
- Holmbom B, et al. Capillary gas chromatography-mass spectrometry of resin acids in tall oil rosin. J Am Oil Chem Soc 1974: 51: 397-400.
- Ayer, W. A. and Migaj, B. S. Acids from blue-stain diseased lodgepole pine. Can J Bot. 1989: 67: 1426-1428
- Karlberg A-T and Lidén C. Comparison of colophony patch test preparations. Contact Dermatitis 1988: 18: 158-165.
Bilag A: Litteraturstudie: Bestemmelse af primære komponenter i umodificeret kolofonium i produkter – i relation til kontakt allergi
1 Introduktion
1.1 Analytter
De to primære komponenter i kolofonium er diterpenoiderne, abietinsyre (AbA), CAS-nr. 514-10-3 og dehydroabietinsyre (DeA), CAS-nr. 1740-19-8 [1]. Derfor er disse to syrer også de mest almindelige analytter ved bestemmelse af indholdet af umodificeret kolofonium i kommercielle produkter [2-11]. Et stort antal af andre terpener, ofte oxiderede, er ligeledes også fundet i umodificeret kolofonium [8, 12-17]. Flere vigtige allergener bliver dannet gennem autooxidation af AbA og DeA, hvorved koncentrationen af de originale syrer falder. Det er derfor vigtigt at medtage en eller flere at de oxiderede stoffer i analysen ud over AbA og DeA ved bestemmelse af kolofonium i en prøve [8, 12,14-16]. For at være en passende markør for oxideret umodificeret kolofonium, skal oxidationsproduktet være stabilt nok til analysen og helst også være et af hovedstofferne i den oxiderede blanding for at være til stede i så høj mængde som muligt. 7-oxo-dehydroabietinsyre (7-O-DeA), CAS-nr. 18684-55-4, opfylder ovenstående betingelser [8, 12, 14, 15].
1.2 Undersøgte prøver
Forskellige typer af produkter er blevet undersøgt for indhold af kolofonium. Disse inkluderer engangsbleer [2], hygiejnebind [6], urteolier og -salver [4], sulfat sæber [9], mascaraer [11, 18], plastre [3, 5, 7], forskellige former for karton, pap og papir [3, 8, 10, 19, 20], bonevoks [8] og skæreolie, loddevæske og malingsprodukter [3]. Ved opvarmning af harpiks (rosin) i forbindelse med lodning udvikler kolofonium aerosoler, som også er blevet analyseret [21]. Støv [8], træ [9] og papirproduktionsvand [22] er ligeledes undersøgt. Endvidere er DeA målt som biomarkør i urin hos fabriksarbejdere der arbejder med lodning i forbindelse med studier af eksponeringen for kolofonium [23].
1.3 Prøve forberedelse
Før analyse af prøven er analytterne ekstraheret over i et passende opløsningsmiddel. Afhængig af prøvetype er forskellige opløsningsmidler blevet anvendt: acetone [2, 7, 8, 10, 19], methanol [3, 12, 19], ethanol [14], acetonitril [4, 5], dichlormethan [3, 8, 15] ethylacetat [3], methylen chlorid [21], dietylether [4, 9,23] og methyl tert-butylether (MTBE) [22]. Hvis nødvendigt kan den efterfølgende oprensning involvere filtrering [3, 8, 12], centrifugering [4] og/eller fast-fase ekstraktion (SPE) [4, 5].
1.4 Analysemetode, separation og detektion
Til bestemmelse af tilstedeværelsen af kolofonium i prøver kan prøvens komponenter derivatiseres og separeres ved hjælp af gas kromatografi (GC) [2, 6, 8-10, 13, 21-23] eller separeres ved hjælp af højtryks-væske kromatografi (HPLC) i deres umodificerede form [3-5, 8, 12, 13]. Detektion af de separerede komponenter ved GC er blevet bestemt med flamme-ionization (FID) [2, 8-10, 22] eller massespektrometri (MS) [21, 23]. Til detektion ved HPLC er almindeligvis anvendt ultraviolet (UV) [3-5, 8, 12, 13], fluorescens detektion [4, 5] og/eller MS [12, 13]. Analytter, der er følsomme over for den høje temperatur i GC, er blevet sprøjtet ind i massespektrometret gennem en direkte injektionsport [13,14]. En alternativ metode for sådanne analytter er matrix-assisteret laser desorption/ionisering massespektrometri (MALDI-MS), hvor en cellulose coatet tyndlagskromatografi plade (TLC) indgår som prøveopsamler [15].
2 Metodebeskrivelser og evaluering
2.1 GC metoder
2.1.1 GC-FID
Metode [9]
- Beskrivelse: Heptadecanoinsyre som intern standard. Ekstraktion med diethylether. AbA og DeA derivatiseret til metylestere med diazomethan.
- Fordele: Top identifikation baseres både på retentionstid af kendte, rene analytter og massespektre.
- Ulemper: Modificering af analytter. Diazomethan er klassificeret som kræftfremkaldende. Ingen oxidationsprodukter analyseres. Ikke kvantitativ.
- Anvendes til: Træ ekstraktion, sulfat sæber, råekstrakt og destilleret tallolie kolofonium.
Metode [22?]
- Beskrivelse: Heneicosanoinsyre, betulinol, cholesteryl heptadecanoate og 1,3-dipalmitoyl-2-oleoyl glycerol anvendes som fire interne standarder. Ekstraktion med MTBE. AbA og DeA derivatiseres ved silylering med bis(trimethylsilyl)-trifluoro-acetamid (BSTFA) og trimethylchlorosilan (TMCS).
- Fordele: Kvantitativ. Mange andre komponenter analyseres også.
- Ulemper: Detektionsgrænse ikke angivet. Modificering af analytter. Silylerede prøver nedbrydes og bør analyseres indenfor 12 timer. Ingen oxidationsprodukter analyseres. Ikke optimeret specielt til separation af AbA og DeA. Metoden til top identifikation er uspecifik.
- Anvendes til: Papir produktionsvand og udløbsvand.
Metode [2, 8, 10]
- Beskrivelse: Methyl stearat som intern standard. Ekstraktion med acetone. Analytter derivatiseres til metylestre med diazomethan.
- Fordele: Bestemmelse af AbA, DeA og 7-O-DeA. Kvantitativ.
- Ulemper: Modificering af analytter. Diazomethan er kræftfremkaldende. Top identifikation er baseret på retentionstid af kendte, rene analytter; risiko for interferens med eller overlapning af ukendte toppe. Nødvendig at syntetisere 7-O-DeA, da den ikke er kommerciel tilgængelig. Detektionsgrænse ikke angivet.
- Anvendes til: Papir, gulvbelægning til linoleum og bleer.
2.1.2 GC-MS
Metode [21?]
- Beskrivelse: Heptadecanoinsyre som intern standard. Ekstraktion med methylene chloride. Detektion af AbA og DeA; derivatisering to metylestre med kalium karbonat og methyl iodide.
- Fordele: Top identifikation baseres på både massespektre og retentionstid af kendte, rene analytter. Kvantitativ.
- Ulemper: Modificering af analytter. Ingen oxidationsprodukter analyseres. Detektiongrænse er ikke angivet.
- Anvendes til: dampe fra lodning.
Metode [23?]
- Beskrivelse: Heptadecanoinsyre som intern standard. Ekstraktion med diethylether. DHA derivatiseret med dimethylformamide dimethylacetal. Detektionsgrænse: 50 nM DHA.
- Fordele: Top identifikation baseres på både massespektre og retentionstid af kendte, rene analytter. Kvantitativ
- Ulemper: Ingen bestemmelse af AbA eller oxidationsprodukter.
- Anvendes til: Biomarkør i urin.
2.2 HPLC metoder
2.2.1 HPLC-UV
Metode [3]
- Beskrivelse: UV detektion af AbA ved 242nm og DeA ved 265 nm. Kalibrering med rene syrer. Ekstraktion med methanol, ethylacetat eller dichlormethan. Detektionsgrænse; 10 ppm AbA, 150ppm DeA.
- Fordele: Ingen modifikation af analytter. Yderligere top identifikation baseres på UV-spektre ved topspids. Kvantitativ.
- Ulemper: Ingen oxidationsprodukter måles. Top identifikation baseret på retentionstid af kendte, rene analytter; risiko for interferens og overlapning af ukendte toppe. DeA har lav absorption ved 267nm; ved max absorbans 215nm er der høj matrix interferens.
- Anvendes til: plastre, skæreolier, loddevæske og malingsprodukter
Metode [13]
- Beskrivelse: UV detektion af 15-HPDA ved 254nm. 15-HPDA er termisk ustabil; til MS opsamles HPLC-fraktion; DIP.
- Fordele: Ingen modifikation af analytter. Detektion af oxidationsprodukt 15-hydroperoxydehydroabietinsyre (15-HPDA). Yderligere top identifikation med MS.
- Ulemper: AbA og DeA detekteres ikke. Ikke kvantitativ. Syntese af reference stof 15-HPDA er nødvendigt, da dette stof er ustabilt og derfor ikke er kommercielt tilgængeligt. Top identifikation er baseret på retentionstid af kendte, rene analytter; risiko for interferens af eller overlapning af ukendte toppe.
- Anvendes til: Umodificeret kolofonium
Metode [8]
- Beskrivelse: Metode svarende til [3]. Ekstraktion med acetone eller dichlormethan.
- Fordele: Ingen modifikation af analytter. Detektion af oxidationsproduktet 7-O-DeA samt AbA og DeA.
- Ulemper: Syntese af reference stof 7-O-DeA er nødvendigt, da dette stof ikke er kommercielt tilgængeligt. Top identifikation er baseret på retentionstid af kendte, rene analytter; risiko for interferens af eller overlapning af ukendte toppe. DeA har lav absorption ved 267nm; ved max absorbans 215nm er der stor matrix interferens. Ikke kvantitativ.
- Anvendes til: Papir, bonevoks, støv.
Metode [12]:
- Beskrivelse: UV detektion af AbA og DeA ved 245nm. Ekstraktion med methanol. Separation er optimeret til største mulige antal komponenter.
- Fordele: Detektion af 7-O-oxo samt AbA og DeA og en række andre komponenter. Ingen modifikation af analytter. Identifikation af toppe med MS.
- Ulemper: Top identifikation er baseret på retentionstid af kendte, rene analytter; risiko for interferens af eller overlapning af ukendte toppe. Syntese af reference stof 7-O-DeA er nødvendigt, da dette stof ikke er kommercielt tilgængeligt. DeA har lav absorption ved 245nm; ved max absorbans 215nm er der stor matrix interferens. Ikke kvantitativ.
- Anvendes til: Umodificeret kolofonium
2.2.2 Kombination af HPLC-UV og fluorescens detektion
Metode [5]
- Beskrivelse: UV detektion af AbA ved 240nm og fluorescens detektion af DeA, excitation 225nm, emission 285nm. Kalibrering med rene syrer. Ekstraktion med acetonitril. SPE oprensning før HPLC. Grænseværdi: 1,25 ng AbA, 0,5ng DeA.
- Fordele: Ingen modifikation af analytter. SPE begrænser matrix interferens og gør detektionen mere specifik. Detektion af DeA er 100 gange mere følsom med fluorescens end ved UV. Samme kolonne er brugt til begge detektorer. HPLC separation på 10 minutter. Kvantitativ.
- Ulemper; Ingen oxidationsprodukter er detekteret. Top identifikation er baseret på retentionstid af kendte, rene analytter; risiko for interferens af eller overlapning af ukendte toppe.
- Anvendes til: Klæbemiddel i makeup
Metode [4]
- Beskrivelse: UV detektion af AbA ved 200nm og fluorescens detektion af DeA, excitation 225nm, emission 285nm. Kalibrering med rene syrer. Ekstraktion med acetonitril eller diethylether. SPE oprensning før HPLC. Detektionsgrænse: 0,4ppm eller 1 ng for AbA og DeA.
- Fordele: Ingen modifikation af analytter, SPE begrænser matrix interferens og gør detektionen mere specifik. Detektion af DeA er 100 gange mere følsom med fluorescens end ved UV. Samme kolonne brugt til begge detektorer. HPLC separation på 10 minutter. Kvantitativ.
- Ulemper; Ingen oxidationsprodukter detekteret. Top identifikation er baseret på retentions tid af kendte, rene analytter; risiko for interferens af eller overlapning af ukendte toppe.
- Anvendes til: urteolier og -salver
2.2.3 HPLC-MS
Metode [13]
- Beskrivelse: HPLC-UV fraktionering (se tidligere). Fraktioner analyseres ved DIP-MS.
- Fordele: Ingen modifikation af analytter. Massespektre af oxidationsproduktet 15-HPDA, der er termisk ustabilt.
- Ulemper: AbA og DeA ikke detekteret. Ikke kvantitativ. Syntese af reference stof 15-HPDA er nødvendigt, da dette stof ikke er kommercielt tilgængeligt.
- Anvendes til: Umodificeret kolofonium
Metode [12]
- Beskrivelse: HPLC-UV fraktionering (se tidligere). Fraktioner analyseres ved kemisk ionisering-MS.
- Fordele: Ingen modifikation af analytter. Detektion af oxidationsproduktet 7-O-DeA, samt AbA og DeA og en række andre komponenter.
- Ulemper: Ikke kvantitativ. Til 7-O-DeA identifikation; IR og NMR.
- Anvendes til: Umodificeret kolofonium
2.2.4 Direkte indsat probe MS metode
Metode [14]
- Beskrivelse: Ekstraktion med ethanol. Kemisk ionisering- og elektron impact ionisering-MS. Komponent identifikation baseres på reference massespektre.
- Fordele: Detektion af 7-O-DeA samt AbA og DeA og en række andre stoffer inklusiv oxidationsprodukter. Ingen modifikation af analytterne.
- Ulemper: Ingen separation, komplekse massespektre. Ikke kvantitativ.
- Anvendes til: Umodificeret kolofonium
2.2.5 MALDI-MS metode
Metode [15]
- Beskrivelse: Ekstraktion med dichlormethan. TLC plade coatet med cellulose anvendes som prøveopsamler.
- Fordele: Detektion af 7-O-DeA og andre oxidationsprodukter. Ingen modifikation af analytter.
- Ulemper: Ingen separation, komplekse massespektre. Ikke kvantitativ. Kræver mekanisk ændring af instrumentets prøveopsamler
- Bruges til: Umodificeret kolofonium.
2.3 Konklusion
Indholdet af AbA varierer mellem 30-50 % i kolofonium af typen gum rosin og 35-40 % i tallolie kolofonium. Hvis det antages, at indholdet af harpikssyrer er 90 %, vil en detektionsgrænse på 10 mikrogram/g AbA således svare til en detektionsgrænse på cirka 30 mikrogram/g (ppm) for kolofonium. En detektionsgrænse på dette niveau vil være acceptabelt i forhold til det koncentrationsniveau, der udløser reaktion hos allergi patienter [3]. Derfor bør en detektionsgrænse for AbA 10 ppm anvendes som en retningslinje til at vurdere størrelsesordnen af den ønskede detektionsgrænse i den udvalgte analysemetode.
Foreslået metode I, eksisterende
GC-FID metoden [2, 8, 10] inkluderer kvantitativ analyse af AbA, DeA og 7-O-DeA. Målbare mængder ned til 1 ppm AbA [10], 1 ppm DeA [2] og 2 ppm 7-O-DeA er præsenteret, selv om grænseværdierne ikke er specificerede. Det er nødvendigt at derivatisere analytterne, hvilket inkluderer brugen af farlige kemikalier. For at undgå top interferens og overlapning af ukendte toppe, kan det være nødvendig at udvikle og anvende en supplerende GC-MS analyse [12].
Foreslået metode II, metodeudvikling
HPLC-UV-fluoresens metode [4, 5] er kvantitativ for AbA og DeA ned til 0,5-1,25 ng, hvilket svarer til omkring 0,4 ppm. Det er ikke nødvendigt at derivatisere komponenterne. Matrix interferens er begrænset og specificiteten af detektionen er øget ved hjælp af SPE oprensning. Oxidationsproduktet 7-O-DeA er inkluderet i metoden. Denne analyt er undersøgt ved hjælp af HPLC-UV [8], dog ikke kvantitativt. Da HPLC-UV-flourescens metoden er ikke-destruktiv, er fraktionering og efterfølgende analyse en mulighed, f. eks. direkte ved MS for at undgå forveksling med ukendte toppe [12-14].
2.4 Referencer
- Hausen, B.M., et al., Contact Allergy Due to Colophony.3. Sensitizing Potency of Resin Acids and Some Related Products. Contact Dermatitis, 1989. 20(1): p. 41-50.
- Karlberg, A.T. and K. Magnusson, Rosin components identified in diapers. Contact Dermatitis, 1996. 34(3): p. 176-8O.
- Ehrin, E. and A.T. Karlberg, Detection of rosin (colophony) components in technical products using an HPLC technique. Contact Dermatitis, 1990. 23(5): p. 359-66.
- Lee, B.L., et al., High-performance liquid chromatographic determination of dehydroabietic and abietic acids in traditional Chinese medications. Journal of Chromatography A, 1997. 763: p. 221-226.
- Lee, B.L., et al., High-Performance Liquid-Chromatographic Method for Determination of Dehydroabietic and Abietic Acids, the Skin Sensitizers in Bindi Adhesive. Journal of Chromatography A, 1994. 685(2): p. 263-269.
- Kanerva, L., et al., Colophonium in sanitary pads. Contact Dermatitis, 2001. 44(1): p. 59-60.
- Gafvert, E. and G. Farm, Rosin (colophony) and zinc oxide in adhesive bandages - An appropriate combination for rosin-sensitive patients? Contact Dermatitis, 1995. 33(6): p. 396-400.
- Karlberg, A.T., et al., Airborne contact dermatitis from unexpected exposure to rosin (colophony) - Rosin sources revealed with chemical analyses. Contact Dermatitis, 1996. 35(5): p. 272-278.
- Holmbom, B., Improved Gas Chromatographic Analysis of Fatty and Resin Acid Mixtures with Special Reference to Tall Oil. J Am Oil Chem Soc, 1977. 54: p. 289-293.
- Karlberg, A.T., E. Gafvert, and C. Liden, Environmentally Friendly Paper May Increase Risk of Hand Eczema in Rosin-Sensitive Persons. Journal of the American Academy of Dermatology, 1995. 33(3): p. 427-432.
- Karlberg, A.T., C. Liden, and E. Ehrin, Colophony in Mascara as a Cause of Eyelid Dermatitis - Chemical-Analyses and Patch Testing. Acta Dermato-Venereologica, 1991. 71(5): p. 445-447.
- Sadhra, S., C.N. Gray, and I.S. Foulds, High-performance liquid chromatography of unmodified rosin and its applications in contact dermatology. J Chrom B, 1997. 700: p. 101-110.
- Shao, L.P., et al., 15-Hydroperoxydehydroabietic Acid - a Contact Allergen in Colophony from Pinus Species. Phytochemistry, 1995. 38(4): p. 853-857.
- Scalarone, D., et al., Direct-temperature mass spectrometric detection of volatile terpenoids and natural terpenoid polymers in fresh and artificially aged resins. Journal of Mass Spectrometry, 2003. 38(6): p. 607-617.
- Scalarone, D., et al., MALDI-TOF mass spectrometry on cellulosic surfaces of fresh and photo-aged di- and triterpenoid varnish resins. J Mass Spectrom, 2005. 40(12): p. 1527-35.
- Hausen, B.M., et al., Contact Allergy Due to Colophony.9. Sensitization Studies with Further Products Isolated after Oxidative-Degradation of Resin Acids and Colophony. Contact Dermatitis, 1993. 29(5): p. 234-240.
- Karlberg, A.T. and E. Gafvert, Isolated colophony allergens as screening substances for contact allergy. Contact Dermatitis, 1996. 35(4): p. 201-207.
- Sainio, E.L., M.L. HenriksEckerman, and L. Kanerva, Colophony, formaldehyde and mercury in mascaras. Contact Dermatitis, 1996. 34(5): p. 364-365.
- Karlberg, A.T. and C. Liden, Colophony (Rosin) in Newspapers May Contribute to Hand Eczema. British Journal of Dermatology, 1992. 126(2): p. 161-165.
- Bergh, M., T. Menne, and A.T. Karlberg, Colophony in Paper-Based Surgical Clothing. Contact Dermatitis, 1994. 31(5): p. 332-333.
- Smith, P.A., et al., Detection of resin acid compounds in airborne particulate generated from rosin used as a soldering flux. American Industrial Hygiene Association Journal, 1997. 58(12): p. 868-875.
- Örså, F. and B. Holmbom, A convenient method for the detemination of wood extractives in papermaking process waters and effluents. Journal of Pulp and Paper Science, 1994. 20(12): p. 361-365.
- Jones, K., et al., Identification of a possible biomarker for colophony exposure. Occup Med (Lond), 2001. 51(8): p. 507-509.
Bilag B: En metode til kvantificering af harpikssyrer i kosmetik
Kemikalier
Cis-5,8,11,14,17-eicosapenraenoinsyre (EPA, >98,5 %) og abietinsyre (CAS-nr. 514-10-3, teknisk kvalitet, 75 %). Begge fra Fluka.
Pimarinsyre (CAS-nr. 510-39-4)og dehydroabietinsyre (CAS-nr. 1740-19-8) er begge fra HelixBiotech og 7-oxo-dehydroabietinsyre (CAS-nr. 18684-55-4) er syntetiseret i henhold til Ayer og Migaj (1989).
Standardopløsninger til HPLC laves i acetonitril: MilliQ vand 9:1 med 0,2 % myresyre. Opløsningerne opbevares på køl og er stabile i mindst 2 uger.
Kolofonium prøver opløses i 100 % acetonitril i en cirka koncentration på 1,5 mg/ml
Prøver
Metoden er blevet testet på voks strips, foundation, rouge og mascara.
Procedure
Ekstraktion:
1 til 2 g prøve (1 g for voks strips og mascara) ultralysbehandles med 20-100 ml acetonitril (afhængig af prøvetype) i 30 minutter. Voks strips klippes i stykker. Papirerne fjernes for at opnå kontakt mellem voks og solvent.
500 ml EPA fra en 928 mikrogram/ml opløsning tilsættes som intern standard (IS) før ekstraktion.
For alle kosmetik prøver undtagen voks strips: Hvert ekstrakt overføres til plastrør og centrifugeres i 15 min. ved 4000 rpm. Supernatanten overføres til en kolbe, bundfaldet genopløses i acetonitril, ultralydbehandles og centrifugeres efter samme procedure. Supernatant fraktionerne samles og inddampes ved hjælp af rotationsinddamper til et volumen på cirka 5 ml. Volumenet justeres til 7 ml med acetonitril. MilliQ vand tilsættes til et slutvolumen på 10 ml indeholdende Acetonitril:vand i forholdet 7:3.
Tilsætning af vand gør opløsningen uklar. Opløsningen filtreres derfor gennem et sprøjtefilter (0,45 mikrometer polypropylene membranfilter).
Note: Kontroller at filtret kan tåle acetonitril.
Til voks strips: Efter ultralydsbehandling, inddampes prøven ved brug af rotationsinddamper, til ca. 5 ml. Volumenet justeres til 7 ml med acetonitril. MilliQ vand tilsættes til et slutvolumen på 10 ml indeholdende Acetonitril:vand i forholdet 7:3. Prøven overføres til plastrør. Kolben skylles med en lille mængde acetonitril, der også overføres til røret. Prøven centrifugeres ved 4000 rpm i 15 min. Supernatanten overføres direkte til en konditioneret SPE kolonne, uden filtrering.
Fastfase ekstraktion (SPE):
Der anvendes Oasis MAX kolonne (6cc, 500mg) fra Waters til oprensning. Konditionering af SPE kolonne foretages ved eluering med 1) 3 ml methanol og 2) 3 ml MilliQ vand. Prøven påsættes og kolonnen skylles med 1) 3 ml 50 mM NaOAc i MilliQ vand, 2) 4 ml methanol og til sidst 1 ml 2 % myresyre i methanol. Prøven elueres med 2 ml 2 % myresyre i methanol. Der tilsættes yderligere 1 ml myresyre i methanol i tilfælde af gennembrud. 3 fraktioner gemmes: 1) sidste skyllefraktion (1 ml) 2) elueringsfraktionen (2 ml) og 3) den sidste ekstra fraktion (1 ml). Kun elueringsfraktionen analyseres.
Note 1: Forholdet acetonitril:vand 7:3 i prøven er vigtigt for en god genfinding ved oprensning på SPE.
Note 2: Myresyre opløsningen skal laves frisk hver dag. Det er meget vigtigt for genfindingen at opsamle nøjagtig volumen af elueringsfraktionerne.
Til kolofonium prøver: Intern standard (IS), 200 mikroliter (186 mikrogram) tilsættes til en 1,5 mg/ml kolofonium opløsning. Kolofonium prøver analyseres direkte efter filtrering på HPLC (uden oprensning på SPE).
HPLC:
HPLC system skal være udstyret med et 20 mikroliter injektionsloop, en diode array detektor (DAD) og en kolonne termostat sat til 25 °C. HPLC kolonnen er en Rp-12 (4,6mm i.d., længde 150mm, partikelstørrelse 3 mikrometer). Den stationære fase indeholder både C12-kæder og urea funktioner. Methanol:vand 8:2 med 0,05 % myresyre bruges som mobil fase med et flow på 1 ml/min (hvilket giver et tryk på 240 bar). Der anvendes en isokratisk eluering (konstant koncentration og sammensætning af mobil fase). Der måles på fire forskellige bølgelængder 210, 220, 240, og 250 nm. De forskellig absorptions forhold anvendes til identifikation af de enkelte komponenter.
Note: Der anvendes isokratiosk eluering for at undgå en stigende basislinje, som vil medføre højere LOD værdier. Mængden af myresyre er vigtig for både retentionstiden og støjniveau på basislinjen.
Kromatogrammer af en standard opløsning (200 ng/µl) ved de enkelte bølgelænger er vist på næste side.
De forskellig absorptions forhold til brug til identifikation (CV under 10 %).
| 7-OxoDeA: | 220nm/210nm forhold 0.53 240nm/210nm forhold 0.26 250nm/210nm forhold 0.45 |
| DeA: | 220nm/210nm forhold 0.55 |
| EPA: | 220nm/210nm forhold 0.40 |
| AbA | 240nm/250nm forhold 1.36 |
Det anbefales dog at kontrollere de specifikke absorptionsforhold på rene standarder, da disse kan variere afhængig af detektor..
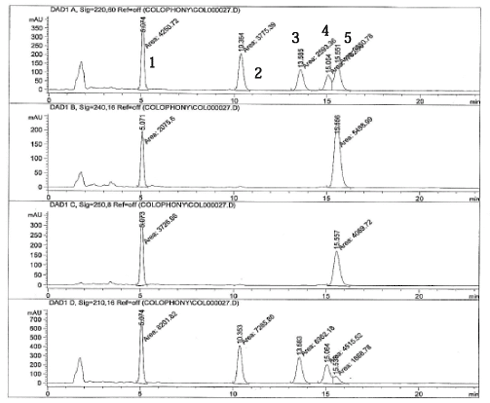
Top 1 = 7-oxo-dehydroabietinsyre
Top 2 = Dehydroabietinsyre
Top 3 = EPA, intern standard
Top 4 = Pimarinsyre
Top 5 = Abietinsyre
Metoden er udviklet af Ulrika Nilsson og Nagmeh Berglund, Afd. for Analytisk Kemi, Stokholm Universitet, Sverige
Reference
Ayer, W. A. and Migaj, B. S. Acids from blue-stain diseased lodgepole pine. Can J Bot. 1989: 67: 1426-1428