Udvikling og validering af en metode til analyse af kolophonium i kosmetiske produkter
2 Analysemetode til kolofonium
- 2.1 Fase 1: Litteraturstudie af analysemetode
- 2.2 Fase 2: Udvikling af en analysemetode
- 2.3 Fase 3: Validering af den udviklede metode
Formålet med dette projekt er at udvikle en metode til brug for screening af kosmetik med henblik på bestemmelse af indholdet kolofonium, baseret på de primære komponenter AbA og DeA samt en markør for oxideret materiale. Projektet er opdelt i 3 faser. Fase 1: Et litteraturstudie af publicerede analysemetoder. Fase 2: Udvikling af en analysemetode. Fase 3: Validering af den udviklede analysemetode.
2.1 Fase 1: Litteraturstudie af analysemetode
Publicerede metoder til identifikation og kvantificering af kolofonium komponenter er blevet gennemgået og evalueret for at vurdere, om der eksisterede en metode som kunne anvendes til videre udvikling af en ny metode.
2.1.1 Resultater
De publicerede metoder til identifikation og kvantificering af kolofonium komponenter er beskrevet i Bilag A. Der er korte beskrivelser og evalueringer af de fundne metoder. På baggrund af litteraturstudiet kunne 2 metoder fremhæves. Metode I er en GC-FID metode, som kræver derivatisering. Metode II er en HPLC-fluorescens metode, som dog er uspecifik og derfor kræver videre udvikling for at kunne bruges på komplekse prøver. Da ingen af disse metoder lever op til de fastlagte krav, blev det besluttet, at det var nødvendigt at udvikle en ny metode til dette projekt.
2.2 Fase 2: Udvikling af en analysemetode
I denne fase blev der udviklet en metode til bestemmelse og kvantificering af de vigtigste komponenter i kolofonium, AbA og DeA, samt en markør for oxideret harpikssyre. En HPLC-UV metode med diode array detektor (DAD) blev valgt. Denne metode giver en høj specifik bestemmelse for de udvalgte komponenter uden brug af derivatisering.
Traditionelt set bestemmes de vigtigste komponenter i kolofonium ved brug af gas kromatografi (GC) (16), men HPLC metoder har også været anvendt (12). Analyser, der kun er baseret på ikke-oxiderede harpikssyrer, fortæller ikke noget om de oxiderede harpikssyrer. En lille detekteret koncentration af AbA kan betyde at der kun er en lille mængde kolofonium tilstede i produktet, men det kan også skyldes en kraftig oxidation af kolofonium. Eftersom oxidationsprodukterne i kolofonium også er de primære allergener er det være særlig nødvendigt at udvikle en metode til kvantificering, som også medtager oxidationsprodukterne. Da metoden skal bruges til screening, er det vigtigt at den er robust, billig og simpel samt ikke involverer meget farlige kemikalier. Det er ikke muligt at anvende den kraftigt allergene 15-hydroperoxyabietic som markør for oxidationsprodukter, da det er meget ustabilt og derfor besværlig at håndtere. I stedet for blev det stabile oxidationsprodukt 7-oxodehydroabietinsyre (7-O-deA) valgt som markør for oxideret kolofonium (Figur 2). Dette oxidationsprodukt er stabilt nok til at blive brugt som referencestof og er samtidig ikke så kraftig en allergen som hydroperoxiderne, hvilket gør den nemmere at håndtere.
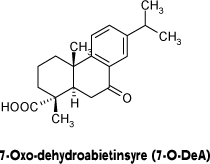
Figur 2: Et af oxidationsprodukterne identificeret i luft–eksponeret kolofonium og udvalgt som markør for den oxidative nedbrydning af AbA
Indholdet af AbA varierer mellem 30-50 % i kolofonium af typen gum rosin og 35-40 % i tallolie kolofonium. Hvis det antages, at indholdet af harpikssyrer er 90 %, vil en detektionsgrænse på 10 mikrogram/g AbA således svare til en detektionsgrænse på cirka 30 mikrogram/g (ppm) for kolofonium. Dette vil blive betragtet som et acceptabelt niveau i forhold til det niveau, der udløser reaktion hos allergi patienter. Følsomme patienters reaktion på kolofonium af typen gum rosin er testet med en serie fortyndinger af gum rosin på 20-0,001 % i vaseline. Ud fra testen reagerede 50 % af forsøgspersonerne på en koncentration ned til 0,1 % (1000 ppm), mens den mest følsomme patient reagerede på et niveau ned til 0,001 % (10 ppm) (18). For andre patienter testet med tallolie kolofonium (20-0,001 % i vaseline) fandt man ingen reaktion lavere end 1 % (10 000 ppm) (7).
2.2.1 Materialer og metoder
2.2.1.1 Kemikalier
Standarder
AbA, DeA og primarinsyre (alle >95 % renhed) er fra Helix Biotech, Vancouver, Canada. Cis-5,8,11,14,17-Eicosapentaenoinsyre (EPA, >98,5 %), der anvendes som intern standard, er fra Fluka, Sigma-Aldrich ChemieGmbH, Steinheim, Germany. 7-O-DeA er syntetiseret som beskrevet nedenfor.
Stamopløsninger af alle komponenterne er fremstillet i 100 % acetonitril i en koncentration på 1 mg/ml. De blev opbevaret i fryser gennem projektets forløb.
Standardopløsninger til kvantificering på HPLC blev lavet i acetonitril:MilliQ vand (i forholdet 9:1) med 0,2 % myresyre. Opløsningerne blev opbevaret på køl og opretholdt stabilitet i mindst 2 uger. Stabiliteten blev kontrolleret overfor friske opløsninger. Der blev ikke kontrolleret for en længere periode end to uger.
2.2.1.2 Solventer og prøver
Methanol med en renhed på 99,8 % er fra Ltd BDH, myresyre (renhed 98-100 %) fra Scharlau og acetonitril af kromatografisk kvalitet fra Sigma-Aldrich.
Gum rosin (kolofonium) prøver (umodificeret og maleinsyre-anhydrid-modificeret fra Soccer, Portugal) og umodificeret kolofonium (Fluka) var opløst i 100 % acetonitril i en koncentration på 1,5 mg/ml.
Kosmetik prøver (1g foundation fra Face, Stockholm) blev spiket med standarder på forskellige koncentrationsniveauer, fra 1 mikrogram/g til 500 mikrogram/g af hver komponent.
2.2.1.3 Syntese af 7-O-DeA
7-O-deA er syntetiseret i to trin som beskrevet i litteraturen (17). En blanding af AbA (5,2 g svarende til 17,19 mmol) og 5 % Pd/C (260 mg, 5 % w/w) blev opvarmet til 245 °C i 1 time under nitrogenflow i en to-halset rundbundet kolbe. Efter afkøling blev det faste stof opløst i ethylacetat (4 x 50 ml eller indtil alt var opløst) og filtreret igennem celite. Den organiske fase blev ekstraheret med 0,25 M natrium hydroxid (2 x 100 ml) efterfulgt af forsuring af den vandige fase med 1 M saltsyre. Råproduktet var derefter isoleret fra den vandige fase ved udrystning med ethylacetat (3 x 50 ml). Den organiske fase indeholdende råproduktet blev tørret med magnesium sulfat og opkoncentreret under reduceret tryk. Oprensning af råproduktet foregik ved hjælp af flash kromatografi (ethylacetat/heman/trifloureddikesyre i forholdet 14/85/1), hvilket gav et udbytte på 2,5 g (46 %) DeA. Dette var i overensstemmelse med tidligere rapporterede data.
Til en blanding af DeA (2,1 g, 6,99 mmol) og kalium hydroxid (0,4 g) i vand (40 ml) blev der drypvis tilsat en opløsning af kalium permanganat (2,8 g, 17,72 mmol) i vand (60 ml) ved stuetemperatur. Slutblandingen blev omrørt i 2,5 time efterfulgt af mætning med svovl dioxid. Et hvidt bundfald blev dannet og centrifugeret fra, vasket med vand og tørret natten over. Stoffet blev oprenset ved hjælp af flash kromatografi (ethylacetat/hexan/ trifloureddikesyre 14/85/1) og gav et udbytte på 1,15 g (52 %) af 7-O-DeA med en renhed på 95 %. Renheden blev bestemt ved Nuclear Magnetic Resonance (NMR) analyse, som er almindelig anvendt til renhedsbestemmelse i forbindelse med synteser. Resultatet var i overensstemmelse med tidligere rapporterede data.
2.2.1.4 Udstyr
Et Agilent 1100 gradient HPLC system blev anvendt til analyse. Der anvendtes et 20 mikroliter injektionsloop, en DAD og en kolonnetermostat sat til 25 °C. HPLC kolonnen var en Prism RP-12 fra Thermo (4,6 mm i.d., længde 150 mm, partikelstørrelse 3 mikrometer). Den stationære fase i kolonnen indeholder både C12-kæder og urea funktioner. Til SPE blev anvendt Oasis MAX kolonner (6cc, 500mg) fra Waters.
2.2.2 Analysemetode
2.2.2.1 Analyse af kosmetikprøver
1 g spiket prøve blev tilsat 20 ml acetonitril og sat i ultralydsbad i 30 min. 500 mikroliter EPA opløsning (928 mikrogram/ml) blev tilsat som intern standard (IS) før ekstraktionen.
Hvert ekstrakt blev overført til plastrør og centrifugeret i 15 min ved 4000 rpm. Supernatanten blev overført til en kolbe, mens bundfaldet (pellet) blev genopløst i acetonitril, ultralydsbehandlet og centrifugeret igen som beskrevet ovenfor. Supernatant fraktionerne blev samlet og inddampet ved hjælp af rotations inddamper til et volumen på cirka 5 ml. Volumenet blev justeret til 7 ml ved tilsætning af acetonitril. MilliQ vand blev derefter tilsat til et slutvolumen på 10 ml indeholdende acetonitril:vand i forholdet 7:3.
Tilsætning af vand gjorde visse af opløsningerne uklare. I de tilfælde blev opløsningen filtreret gennem sprøjtefilter (0,45 mikrometer polypropylene) før fast-fase ekstraktion (SPE).
2.2.2.2 Fast-fase ekstraktion
SPE kolonnen blev konditioneret med 1) 3 ml methanol og 2) 3 ml MilliQ. Prøven blev påsat og kolonnen blev efterfølgende skyllet med 1) 3 ml 50 mM natriumacetat i MilliQ vand for at eliminere meget polære stoffer eller baser, 2) 4 ml methanol for at eliminere hydrofobe stoffer og til sidst 3) 1 ml myresyre (2 %) i methanol. Prøven blev derefter elueret med 2 ml myresyre (2 %) i methanol. Yderligere 1 ml myresyre (2 %) i methanol blev tilsat i tilfælde af gennembrud. Frisk myresyre opløsning blev fremstillet hver dag.
2.2.2.3 Analyse af kolofonium
Intern standard, 200 mikroliter (186 mikrogram) blev tilsat til 2 ml af 1,5 mg/ml gum rosin opløsninger i acetonitril. Gum rosin prøverne blev filtreret og analyseret direkte på HPLC (uden SPE oprensning).
2.2.2.4 HPLC metode
Methanol:vand i forholdet 8:2 med 0,05 % myresyre blev brugt som mobil fase med et flow på 1 ml/min (dette giver et tryk på cirka 240 bar). Eluering foregik ved konstant koncentration og sammensætning af den mobile fase, dvs. isokratisk eluering. Der blev anvendt 4 forskellige bølgelængder 210, 220, 240 og 250 nm. Forholdene mellem absorptionen ved de forskellige bølgelængder blev brugt til identifikation af komponenterne. Der blev anvendt isokratisk eluering for at undgå stigning af basislinjen, hvilket ville have givet anledning til højere LOD værdier. Mængden af myresyre var kritisk for både retentionstid og støj på basislinjen.
Der blev lavet en kalibreringskurve i koncentrationsintervallet fra 0,8 ng/mikroliter til 500 ng/mikroliter for hver komponent. Til kvantificering blev der dagligt lavet et-punkts kalibrering ved tredobbelt injektion af en 250 ng/mikroliter opløsning af komponenterne. Kvantifikationen blev foretaget ved brug af relative respons faktorer analyt/IS.
Specifikke absorptionsforhold (CV mindre end 10 %) blev brugt til identifikation af de forskellige komponenter:
| AbA: | 240nm/250nm: forhold 1,36 |
| DeA: | 220nm/210nm: forhold 0,55 |
| 7-O-DeA: | 220nm/210nm: forhold 0,53; 240nm/210nm: forhold 0,26; 250nm/210nm: forhold 0,45. |
| EPA (IS): | 220nm/210nm: forhold 0,40 |
Til kvantificering af DeA og EPA blev der anvendt arealtoppe ved bølgelængden 210 nm. For AbA og 7-O-DeA anvendtes 240nm.
2.2.3 Resultater
2.2.3.1 Selektivitet
Den udviklede metode bygger på separation og identifikation af de tre udvalgte komponenter i kolofonium (AbA, DeA og 7-O-DeA) (Figur 3). Detektionen af syrerne er foretaget ved de ovenfor angivne bølgelængder. Der blev opnået er basislinje separation af AbA og DeA toppene. Et af problemerne med LC-separation ved UV-detektion er co-eluering mellem pimarinsyre (en harpikssyre, som også findes i kolofonium) og AbA. Den nye metode anvender en LC-kolonne, hvor den stationære fase er mere selektiv overfor molekyleform, hvilket sikrer en separation af pimarinsyre og AbA (dog ikke med total basislinje separation). Med den valgte mobile fase (methanol:vand 8:2 med 0,05 % myresyre) og et flow på 1,0 ml/min, kan de udvalgte harpikssyrer i kolofonium separeres i løbet af 22 min. Det er vigtigt at kontrollere mængden af myresyrer i den mobile fase præcist. For lav koncentration (0,02 %) påvirker separationen i en dårlig retning og ved højt niveau (0,1 %) stiger støj på basislinjen kraftigt. Der blev anvendt isokratisk eluering, da gradient eluering øgede støjen på basislinjen.
2.2.3.2 Separation af komponenter i kolofonium prøver
Basislinje separation af toppe svarende til flere harpikssyrer blev også opnået, hvilket analyse af umodificeret kolofonium viser (Se Figur 4).
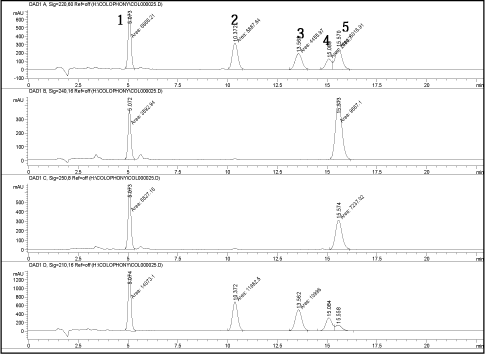
Figur 3. Standard opløsning (500 mikrogram/ml) analyseret med den udviklede metode. 1= 7-O-DeA, 2= DeA, 3= EPA, 4= pimarinsyre, 5= AbA
Krommatogrammer er vist ved 220, 240, 250 og 210 nm
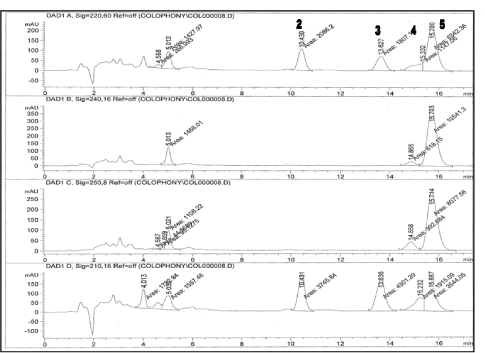
Figur 4: Kolofonium prøve, umodificeret Rosin fra Soccer (1,5 mg/ml) analyseret med den udviklede metode. 2 = DeA, 3 = EPA, 4 = pimarinsyre, 5 = AbA. Den oxiderede harpikssyre 7-O-DeA kunne ikke detekteres. Kromatogrammerne er vist ved 220, 240, 250 and 210 nm
2.2.3.3 Kvantifikation af komponenter i kolofonium prøver
De forskellige analytter er kvantificeret i umodificerede og modificerede kolofonium prøver. Resultaterne er vist i Tabel 1.
Tabel 1. Koncentration af komponenterne i forskellige prøver af umodificeret og modificeret kolofonium (mg/g)
| AbA | DeA | 7-O-DeA | |
| Umodificeret kolofonium af gum rosin type (Soccer) |
255 | 62 | N. D. |
| Umodificeret kolofonium af gum rosin type (Fluka) |
691 | 40 | Not det |
| Malein anhydrid-modificeret gum rosin (Soccer) |
74 | 23 | Not det. |
2.2.3.4 Linearitet
Toparealerne for AbA, DeA og 7-O-DeA varierer lineært med harpikssyre koncentrationen indenfor koncentrationsområdet 10 mikrogram/g til 500 mikrogram/g.
2.2.3.5 Detektionsgrænser
Under anvendelse af den beskrevne metode var instrument detektionsgrænsen (LOD) 4 mikrogram/ml for AbA, 31 mikrogram/ml for DeA og 3 mikrogram for 7-O-DeA.
2.2.3.6 Genfindinger
Genfindingerne blev undersøgt på 3 forskellige koncentrationsniveauer i spiket foundation. De forskellige tilsatte niveauer af de enkelte analytter var 100, 200 og 500 mikrogram/ml. Genfindingen for AbA var 78 % (CV 1 %), DeA var 88 % (CV 5 %), 7-O-DeA var 76 % (CV 2 %) og EPA 97 % (CV 5 %).
2.2.3.7 Præcision
Præcision angiver graden af overensstemmelse mellem uafhængige målinger opnået under fastlagte betingelser. Som oftest udtrykkes præcision som ”mangel på præcision” og beregnes som en spredning mellem måleresultaterrne, hvilket er tilfældet her. Præcision kan bestemmes under både repeterbarheds- og reproducerbarhedsbetingelser. Præcisionen for HPLC metoden var mindre end 10 % (CV) både med hensyn til reproducerbarhed (dag til dag variation, n=6) og repeterbarhed (n=3) beregnet ud fra den relative respons faktor analyt/IS. Præcisionen blev testet med en 250 mikrogram/g standard.
2.3 Fase 3: Validering af den udviklede metode
Metoden udviklet i fase 2 blev valideret for kvantificeringsgrænse, repeterbarhed og genfinding for de udvalgte harpikssyrer og oxidationsprodukt på 2 koncentrations niveauer ved standard addition til kosmetik prøver. En større screening af kolofonium i en række produkter på baggrund af de udvalgte komponenter var ikke mulig på grund af de begrænsede ressourcer i dette projekt. Det anbefales i stedet at indholdet af kolofonium i kosmetikprodukter undersøges i et separat projekt. Enkelte kosmetikprodukter var dog analyseret for indhold af de udvalgte komponenter indenfor dette projekt.
2.3.1.1 Separation af de udvalgte komponenter i kosmetik
Komponenterne blev kvantificeret i forskellige kosmetikprøver i henhold til den udviklede metode. Separationen var fyldestgørende for identifikation og kvantificering i de testede kosmetikprøver. Dette er illustreret med et kromatogram af et voks-strip produkt til hårfjerning (Figur 5).
Kromatogrammet målt ved 210 nm er vist som eksempel.
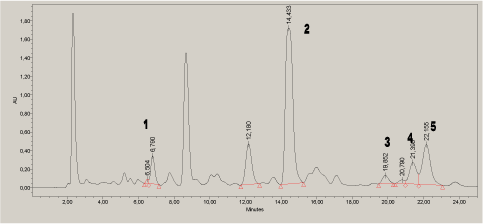
Figur 5. Kromatogram af voks strip ved 210 nm. 1=7-O-DeA, 2=DeA, 3=EPA, 4=pimarinsyre, 5=AbA.
2.3.1.2 Præcision
Repeterbarheden, beregnet som CV, blev bestemt på 2 niveauer baseret på spikede prøver (standard addition). For alle komponenterne var repeterbarheden på lavt niveau (50-90 mikrogram/g for hver komponent) i god overensstemmelse med resultaterne fra fase 2 (se Tabel 2). For det høje niveau (135-250 mikrogram/g) var præcisionen 5-7 % (se Tabel 3).
Tabel 2. Analyser af kosmetikprodukter ved lavt niveau (50 – 90 mikrogram/g) ved standard addition
| komponent | repeterbarhed | % repeterbarhed | % genfinding | LOD | LOQ |
| AbA | 2,4 | 2,4 | 85 | 7,2 | 21,6 |
| DeA | 6,4 | 13 | 84 | 19,2 | 57,6 |
| 7-O-DeA | 4,3 | 11 | 91 | 12,9 | 38,7 |
Tabel 3. Analyser af kosmetikprodukter ved højt niveau (135 – 250 mikrogram/g) ved standard addition
| komponent | repeterbarhed | % repeterbarhed | % genfinding |
| AbA | 13,1 | 6,5 | 94 |
| DeA | 11,2 | 4,8 | 92 |
| 7-O-DeA | 11,8 | 7,5 | 94 |
2.3.1.3 Linearitet og repeterbarhed af HPLC
Lineariteten og repeterbarheden blev testet for den udviklede HPLC metode ved gentagne injektioner (n=10). Metoden er lineær i koncentrationsområdet 1 til 400 mikrogram/g (Korrelationskoeficient, r²: 0,999-1,000). Repeterbarheden af instrumentet var god i området 5-185 mikrogram/ml. AbA: 2-4,5 %, DeA: 0,3-2 %, 7-O-DeA: 0,3-1 % og den interne standard, EPA: 2,5 % ved en koncentration på 125 mikrogram/ml.
2.3.1.4 Detektionsgrænse
Detektionsgrænsen er bestemt som 3 x SD baseret på 6 analyserede prøver spiket på lavt niveau og lå i omkring 7-20 mikrogram/g (se Tabel 2).
2.3.1.5 Kvantifiseringsgrænse
Bestemmelse af metodens kvantifikationsgrænsen var baseret på spikede kosmetikprøver ved brug af ligningen: LOQ = blind + 3 x LOD. Dette svarede til LOQ = 3 x LOD, da der ikke var målbare blindværdier.
2.3.1.6 Genfinding
Genfindingerne, baseret på standard addition, svarede i niveau til de værdier der var fundet i fase 2. Se Tabel 2 og 3. CV for genfindingerne lå på 4-6 %.
2.3.1.7 Identifikation af analytter
De specifikke absorptionsforhold for identifikation af de enkelte analytter blev kontrolleret, da forskellige detektorer blev brugt i fase 2 og fase 3. Der er nogle afvigelser, specielt for DeA og EPA. Det er derfor anbefalet, at de specifikke absorptionsforhold kontrolleres ved hjælp af rene standarder før analysen påbegyndes. CV var mindre end 10 %, hvilket er i overensstemmelse med fase 2 (se Tabel 4).
Tabel 4: Specifikke absorptionsforhold for identifikation angivet for de to forskellige detektorer anvendt i fase 2 og 3.
| AbA | DeA | 7-O-DeA | EPA | |||||
| nm ratio | Fase 2 |
Fase 3 | Fase 2 | Fase 3 | Fase 2 |
Fase 3 | Fase 2 |
Fase 3 |
| 220/210 | 0,55 | 0,83 | 0,53 | 0,53 | 0,4 | 0,06 | ||
| 240/210 | 0,26 | 0,24 | ||||||
| 250/210 | 0,45 | 0,44 | ||||||
| 240/250 | 1,36 | 1,54 | ||||||
2.3.1.8 Kvantifikation af produkter
Forskellige almindelige kosmetik produkter blev analyseret for indhold af de udvalgte harpikssyrer. Baseret på de målte data kan indholdet af kolofonium beregnes (tabel 5). Kun én voks strip havde et signifikant indhold svarende til 7 mg/g kolofonium. For andre produkter kunne indholdet AbA og DeA detekteres (0,5 – 4,5 mikrogram/gram), men koncentrationerne var dog under detektionsgrænsen.
Tabel 5: Kvantifikation af produkter (mikrogram/g)
| Komponenter | Foundation | Lip gloss | Voks strip, normal |
Voks strip, ansigt |
| AbA | <7 | <7 | 2302 | <7 |
| DeA | <19 | <19 | 2517 | <19 |
| 7-O-DeA | <13 | <13 | 27,8 | <13 |
Version 1.0 Marts 2009, © Miljøstyrelsen.