|
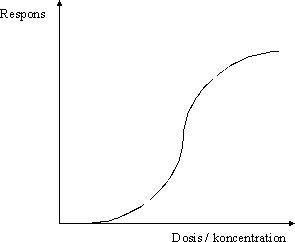
Principper for sundhedsmæssig vurdering af kemiske stoffer med henblik på fastsættelse af kvalitetskriterier for luft, jord og vand 3 Farlighedsvurdering og karakterisering3.1 Dosis-effekt og dosis-respons sammenhænge Formålet med en farlighedsvurdering og farlighedskarakterisering af et givent stof er, på baggrund af en kritisk vurdering af de tilgængelige data vedrørende stoffets toksikologiske effekter og virkningsmekanismer, at vurdere, hvorvidt eksponering for stoffet vil kunne udløse sundhedsskadelige effekter hos mennesker. Ved fastsættelse af kvalitetskriterier tages der altid udgangspunkt i den eksisterende viden, primært viden samlet sammen i kriteriedokumenter, monografier og reviews, hvorfor der i reglen ikke foretages en egentlig toksikologisk bedømmelse af de enkelte studier. Dog søges originallitteraturen altid fremskaffet for de(t) studie(r), der ligger til grund for udpegning af de(n) kritiske effekt(er). Hvor den eksisterende viden er mangelfuld, kompenseres der herfor ved anvendelse af en usikkerhedsfaktor, dette er nærmere beskrevet i 4.4.3. 3.1 Dosis-effekt og dosis-respons sammenhængeFarlighedsvurderingen foretages med udgangspunkt i alle de indhentede data vedrørende stoffets sundhedsskadelige effekter i mennesker og dyr, det vil sige akutte effekter, irritative/ætsende og sensibiliserende egenskaber, effekter observeret i længerevarende undersøgelser med gentagen administration af stoffet, mutagene/genotoksiske og kræftfremkaldende egenskaber, samt påvirkning af fertilitet og fosterudvikling. Endvidere lægges der vægt på de toksikokinetiske data, det vil sige stoffets skæbne i organismen (optagelse, fordeling, metabolisme og udskillelse), samt data vedrørende stoffets virkningsmekanisme(r). Eksponering for et givent stof kan medføre forskellige effekter varierende fra lette gener til dødeligt forløbende forgiftninger. Forløbet er ofte relateret til koncentrationen eller dosis af stoffet. Ved lave doser kan stoffet for eksempel give anledning til forbigående gener, mens der ved højere doser kan optræde alvorlige skader på væv og organer, i værste fald med dødelig udgang. Et enkelt studie (epidemiologisk såvel som dyreeksperimentelt) kan således ofte afsløre forskellige typer af effekter afhængigt af de forskellige koncentrationer eller doser, som mennesker eller dyr har været udsat for i det pågældende studie. Som en følge heraf kan forskellige dosis-effektsammenhænge forventes, hvis stoffet forårsager flere forskellige effekter. Som et led i farlighedsvurderingen foretages der således for hvert enkelt studie en vurdering af dosis-effektsammenhænge, det vil sige en karakterisering af sammenhængen mellem dosis (eller koncentration) af stoffet og de observerede effekter. Derudover foretages også en vurdering af dosis-responssammenhænge, det vil sige en karakterisering af sammenhængen mellem dosis (eller koncentration) af stoffet og forekomst/hyppighed (engelsk: incidence) eller graden/alvorligheden (engelsk: severity) af de observerede effekter. Ved vurdering af alvorligheden af de observerede effekter skelnes der mellem effekter som sådan og skadelige (engelsk: adverse) effekter. Generelt betragtes effekter som følge af eksponering for et givent stof som skadelige, når der er tale om væsentlige forandringer i morfologi, fysiologi, funktion, vækst, udvikling, og/eller levetid hos eksponerede individer, og når forekomsten hos denne gruppe er statistisk signifikant højere end i kontrolgruppen. Sondringen mellem effekt og skadelig effekt kan virke akademisk, men er i mange tilfælde af væsentlig betydning, blandt andet i forbindelse med vurdering af nul-effektniveau og laveste effektniveau i et givent studie (se 3.2), med udpegning af den kritiske effekt (se 3.4), og for størrelsen af usikkerhedsfaktor UFIII der anvendes ved fastsættelse af kvalitetskriterier for luft, jord og drikkevand med henblik på at tage højde for kvalitet og relevans af det tilgængelige datagrundlag (se 4.4.3). Sondringen består i en vurdering af, om de observerede effekter kan tolkes som værende af en egentlig skadelig karakter, eller om der eventuelt er tale om normale uspecifikke reaktioner, som blot er en følge af individernes reaktion på ændringer i ydre omstændigheder (homeostatiske mekanismer). Som eksempel herpå kan nævnes, at en formindsket vægtstigning hos forsøgsdyr kan være en reaktion på foderets smag eller konsistens og dermed ikke nødvendigvis en direkte reaktion på teststoffets toksikologiske effekter. Et andet eksempel er, at ændringer i en enzymaktivitet eller i en klinisk-kemisk parameter, hvilket er lig med en effekt, ikke nødvendigvis betragtes som skadelige, med mindre ændringerne i sig selv giver anledning til sundhedsmæssige påvirkninger af individerne eller er indikatorer herpå. Andre eksempler er misfarvning af et organ eller væv, hvor der ikke samtidig er observeret histologiske eller biokemiske effekter, eller induktion af enzymer involveret i metabolismen af et givent stof. Sondringen mellem effekt og skadelig effekt afhænger selvsagt af de data, der er tilgængelige for vurdering, og sondringen kan således være behæftet med en vis usikkerhed. For eksempel kan der i et givent studie ses en effekt (f.eks. morfologiske forandringer i leveren) hos nogle forsøgsdyr, uden at den pågældende effekt er observeret på et niveau, der er statistisk signifikant forskelligt fra det, der ses i kontrolgruppen. I dette tilfælde vil effekten ikke nødvendigvis tolkes som værende en skadelig effekt som følge af eksponeringen for teststoffet, men kan muligvis tilskrives tilfældige biologiske variationer. Men hvis der for eksempel havde været flere dyr i den pågældende dosisgruppe, så ville effekten måske kunne blive observeret som værende statistisk signifikant forskellig fra observationerne i kontrolgruppen, og i dette tilfælde vil effekten tolkes som værende en skadelig effekt. Der kan også være tale om atypiske effekter, som for eksempel usædvanlige fosterskader der optræder med en hyppighed, der er mindre end det, der i studiet er statistisk signifikant. I sådanne tilfælde vil effekten kunne blive vurderet som skadelig, hvis de få observationer, der ses, ikke med rimelighed kan tilskrives tilfældige biologiske variationer. Der ligger således den begrænsning i farlighedsvurderingen, at man ved gennemførelse af et studie så vidt muligt skal sikre sig at kunne være i stand til at observere en given effekt på et statistisk signifikant niveau med det anvendte antal individer ved de valgte koncentrationer eller doser. Dette kan specielt være et problem ved atypiske effekter, hvor der i givet fald skulle anvendes et uforholdsmæssigt højt antal individer per gruppe eller ved lave koncentrationer/doser, hvor den individuelle følsomhed mellem individer i en udvalgt gruppe kan 'overskygge' en eventuel effekt. Ved farlighedsvurderingen skelnes der endvidere mellem systemiske og lokale effekter. Systemiske effekter optræder i organismen efter optagelse af stoffet via luftvejene, mave-tarmkanalen, og/eller huden. Ved systemiske effekter anses det sædvanligvis at være den samlede dosis af stoffet og sjældent stoffets koncentration i de(t) pågældende medie(r), der er relevant ved fastsættelse af kvalitetskriterier i luft, jord og drikkevand. Under lokale effekter henregnes effekter, der optræder lokalt i luftvejene eller i mave-tarmkanalen samt direkte effekter på hud og øjne. I relation til lokale effekter anses sædvanligvis stoffets koncentration og sjældent den samlede dosis for relevant ved fastsættelse af kvalitetskriterier. I velgennemførte forsøg er doserne (koncentrationerne) valgt således, at der for en given (skadelig) effekt er mindst én dosis med effekt og mindst én dosis, hvor der ikke er effekt. Dosis-responssammenhængen (f.eks. antal dyr hvori den pågældende effekt er observeret eller den procentvise stigning i organvægt i forhold til kontrolgruppens organvægt) kan optegnes i et diagram med logaritmen til dosis ud ad x-aksen og respons i procent op ad y-aksen. Kurveforløbet kan være lineært, kurvet eller U-formet. For de fleste kemiske stoffer forløber dosis-responssammenhængen som en S-formet kurve (figur 3.1.1). Ved de laveste doser er der således ikke observeret effekt, men rent statistisk kan det ikke udelukkes, at en svag effekt måske ville kunne være blevet observeret, hvis der havde været flere dyr i gruppen. Da den S-formede dosis-responskurve gælder for de fleste kemiske stoffers effekter, er denne kurvetype valgt ved illustrationerne af dosis-responssammenhænge i de efterfølgende figurer. For nogle stoffer forløber dosis-responssammenhængen imidlertid anderledes. For eksempel ses for essentielle metaller, at hvis individet får for lidt af et givent essentielt metal, så kan der opstå mangelsymptomer. Omvendt kan der opstå skadelige påvirkninger, hvis individet får for meget af det pågældende essentielle metal. Således har dosis-responssammenhængen for essentielle metallers effekter et U-formet forløb, hvor den højre del af kurven, det vil sige den del, der repræsenterer de toksiske effekter, stadig har det typiske S-formede forløb, som generelt ses for kemiske stoffers effekter. Også i relation til begrebet hormesis ses et U-formet eller omvendt U-formet kurveforløb (se 3.3). Figur 3.1.1 S-formet dosis-respons kurve
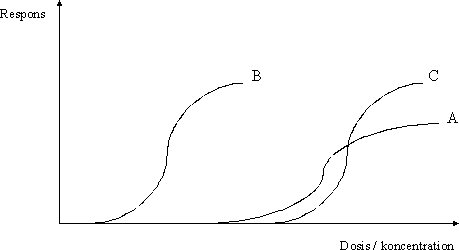
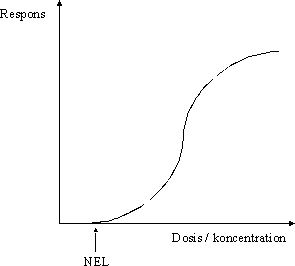
Som følge af at et enkelt studie i princippet kan afsløre forskellige typer af effekter for et givent stof, og at forskellige dosis-responssammenhænge kan forventes for de forskellige effekter, kan der i princippet således optegnes en dosis-responskurve for hver af stoffets forskellige effekter, se figur 3.1.2. For nogle typer af effekter skal der ske en væsentlig øgning af dosis, før der ses et respons, og for disse typer af effekter vil den S-formede dosis-responskurve være forholdsvis flad, kurve A i figur 3.1.2. For andre typer af effekter skal der kun en meget lille øgning af dosis til at fremkalde et markant respons, og for disse typer af effekter vil den S-formede dosis-responskurve være stejl, kurve B og C i figur 3.1.2. Figur 3.1.2 Dosis-respons kurver
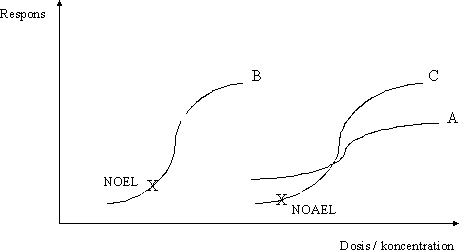
Hvorvidt den S-formede dosis-responskurve for en given effekt er stejl (figur 3.1.2 kurve B og C) eller flad (figur 3.1.2 kurve A) siger ikke umiddelbart noget om den pågældende effekts alvorlighed. Med henblik på vurdering heraf skal der yderligere inddrages en vurdering af ved hvilke dosisniveauer, de enkelte effekter optræder, det vil sige dosis-responskurvens placering i diagrammet i forhold til y-aksen. Jo kortere afstand fra kurven til y-aksen jo lavere koncentrationer (doser) skal der til for at udløse den givne effekt. Et eksempel til illustration af dette forhold er en sammenligning af kurverne B og C i figur 3.1.2. Kurverne B og C har samme stejlhed, og dermed har de respektive effekter B og C samme relative dosis-responssammenhæng. Men da effekt B optræder ved lavere koncentrationer (doser) end effekt C, så kunne effekt B således betragtes som værende mere kritisk end effekt C. Dette vil også ofte, men langt fra altid være tilfældet, idet typen/alvorligheden af effekt også spiller en rolle i vurderingen af den kritiske effekt (se 3.4). Som eksempel på det sidstnævnte kan nævnes, at hvis effekt C er udvikling af tumorer i leveren og effekt B er luftvejsirritation, så vil effekt C selvsagt vurderes som værende en langt alvorligere effekt end effekt B. Det skal dog understreges, at ved fastsættelse af et luftkvalitetskriterium ville det ikke nødvendigvis være effekt C, der, på trods af alvorligheden, ville blive vurderet som værende den kritiske effekt i relation til fastsættelse af et luftkvalitetskriterium og dermed danne grundlaget for herfor. Et andet eksempel er en sammenligning af kurverne A og C i figur 3.1.2. Kurverne A og C har forskellig stejlhed og dermed har de respektive effekter A og C forskellige relative dosis-responssammenhæng. Umiddelbart kunne man fristes til at betragte effekt C som værende mere kritisk end effekt A på grund af en stejlere dosis-responskurve. Dette vil også kunne være tilfældet ved koncentrationer (doser), der er højere end den koncentration (dosis), der svarer til skæringspunktet mellem kurve A og C. Men ved lavere koncentrationer (doser) end den koncentration (dosis), der svarer til skæringspunktet mellem kurve A og C, vil effekt A kunne betragtes som værende mere kritisk end effekt C. Også i dette tilfælde kan der være andre forhold, der gør sig gældende ved en vurdering af de enkelte effekters alvorlighed. 3.2 Fastlæggelse af nul-effektniveau og laveste effektniveauI vurderingen af det enkelte studie indgår også en udpegning af et nul-effektniveau og det laveste effektniveau for en given effekttype samt for stoffet som sådan i det pågældende studie for så vidt, at det overhovedet er muligt på baggrund af data i studiet. For hver enkelt effekt findes teoretisk set et sandt nul-effektniveau (NEL) og laveste effektniveau (LEL), og ideelt set burde det være disse niveauer, der blev fastlagt for en given effekt. For langt de fleste typer af effekter findes der en tærskel, det vil sige en grænse (koncentration/dosis), hvorunder der ikke ses effekt (se 4.2). For nogle typer af effekter antages det, at der ikke findes en tærskel, for eksempel genotoksiske carcinogener, hvor den tilgrundliggende mekanisme for udvikling af tumorer er en beskadigelse af arvematerialet. Denne problemstilling er uddybet i 3.6. Som beskrevet i 3.1 kan dosis-responssammenhængen for mange typer af effekter optegnes som en S-formet kurve i et diagram med dosis (koncentration) ud ad x-aksen og respons op ad y-aksen. Nul-effektniveauet (NEL) for en given effekt er således skæringspunktet mellem den S-formede kurve og x-aksen (figur 3.2.1). Figur 3.2.1 Dosis-respons kurve med identifikation af nul-effektniveau
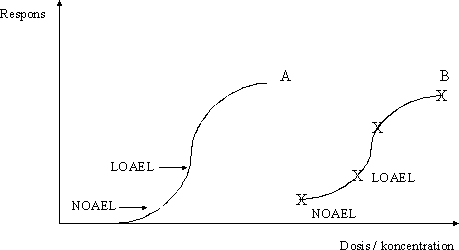
Da det sande nul-effektniveau (NEL) og laveste effektniveau (LEL) er teoretiske værdier, kan det således i praksis ikke lade sig gøre at fastlægge disse niveauer. Det næstbedste ville så være at estimere de niveauer, der ligger tættest på det sande nul-effektniveau (NEL) eller laveste effektniveau (LEL). Dette er imidlertid sjældent muligt i praksis, da en estimering af disse niveauer med en vis grad af sikkerhed i givet fald ville kræve et meget stort antal studier af den pågældende effekt samt et særdeles stort antal dyr og koncentrations-(dosis)-niveauer i det enkelte studie. I praksis fastlægges i et givent studie det observerbare nul-effektniveau (NO(A)EL) og det laveste observerbare effektniveau (LO(A)EL) ud fra de konkrete dosisniveauer, der er anvendt i det pågældende studie. Principperne herfor er beskrevet i det efterfølgende afsnit (3.2.1). Det skal dog bemærkes, at en alternativ metode til fastlæggelse af nul-effektniveau, 'benchmark' metoden (se 3.2.2), i visse tilfælde kan give estimater ('benchmark dose'), der ligger tættere på NEL og LEL. Imidlertid giver det klassiske guideline-forsøgsdesign (se 2.2.1) med 3 doserede grupper og en vehikelkontrolgruppe ikke ideelle data til udarbejdelse af gode dosis-responskurver og dermed estimering af 'benchmark dose'. Hertil er studier med 5-10 dosisgrupper nødvendige. 3.2.1 NO(A)EL og LO(A)ELI konsekvens af sondringen mellem effekt og skadelig effekt (se 3.1) skelnes således også mellem to typer af observerbart nul-effektniveau, NOEL og NOAEL, samt to typer af det laveste observerbare effektniveau, LOEL og LOAEL. Såfremt dosisniveauerne er angivet som koncentrationer anvendes ofte betegnelsen 'koncentration' i stedet for 'niveau', det vil sige NOEC og NOAEC samt LOEC og LOAEC. Termerne defineres som følger: NOEL/C (engelsk: No Observed Effect Level / Concentration) er den højeste dosis / koncentration af et stof, som ikke medfører ændringer i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer i forhold til en sammenlignelig kontrolgruppe. NOAEL/C (engelsk: No Observed Adverse Effect Level / Concentration) er den højeste dosis / koncentration af et stof, som ikke medfører påviselige skadelige ændringer i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer. Der kan ved NOAEL/C ses ændringer i ovennævnte parametre, som ikke vurderes at være af skadelig karakter. LOEL/C (engelsk: Lowest Observed Effect Level / Concentration) er den laveste dosis / koncentration af et stof, som medfører en ændring i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer, og som kan påvises i forhold til en sammenlignelig kontrolgruppe. De ændringer, der ses ved LOEL/C, vurderes ikke at være af skadelig karakter. LOAEL/C (engelsk: Lowest Observed Adverse Effect Level / Concentration) er den laveste dosis / koncentration af et stof, som medfører en skadelig ændring i morfologi, funktionsevne, vækst, udvikling, og/eller levetid hos eksponerede individer og som kan påvises i forhold til en sammenlignelig kontrolgruppe. Det skal understreges at begreberne NO(A)EL og LO(A)EL anvendes med varierende betydning i den videnskabelige litteratur, og at det ikke altid kan afgøres, hvad der menes i de enkelte tilfælde, hvorfor man ofte er nødt til at se nærmere på, om der i det enkelte forsøg er forsøgt en skelnen mellem disse begreber. Med henblik på fastsættelse af kvalitetskriterier for luft, jord og drikkevand tages der oftest udgangspunkt i NOAEL/C eller LOAEL/C, hvorfor disse begreber i det følgende generelt vil være synonymt med observerbart nul-effektniveau henholdsvis det laveste observerbare effektniveau, med mindre der specifikt er tale om NOEL/C eller LOEL/C. Endvidere vil begreberne NOAEL og LOAEL blive anvendt, med mindre der specifikt er tale om koncentrationer. I velgennemførte forsøg er doserne (koncentrationerne) valgt således, at der er mindst én dosis, der resulterer i en observerbar effekt og mindst én dosis, hvor denne effekt ikke observeres. Som beskrevet i 3.1 kan dosis-responssammenhængen for mange typer af effekter optegnes som en S-formet kurve i et diagram med dosis (koncentration) ud ad x-aksen og respons op ad y-aksen. NOAEL og LOAEL for en given effekt vil således ligge et eller andet sted på den S-formede dosis-responskurve, eksempelvis som markeret på kurve A på figur 3.2.2. I praksis er der i det enkelte studie kun få dosisniveauer, og dosis-responskurven vil for eksempel se ud som kurve B på figur 3.2.2. NOAEL for en given effekt vil således være den af de valgte dosisniveauer i studiet, der ikke har medført effekt, og LOAEL i konsekvens heraf det næste dosisniveau i studiet. Dette er illustreret på kurve B på figur 3.2.2 med NOAEL som det nederste punkt på kurven og LOAEL som det næstnederste punkt. Figur 3.2.2 Dosis-respons kurve med identifikation af nul-effektniveauer og laveste effekt niveauer
Som nævnt tidligere (3.1) foretages for det enkelte studie en vurdering af dosis-effekt og dosis-responssammenhænge, ligesom der sondres mellem effekt og skadelig effekt samt mellem systemiske og lokale effekter. Såfremt et enkelt studie afslører forskellige typer af effekter for et givent stof, kan der i princippet optegnes en dosis-responskurve for hver af stoffets forskellige effekter, se figur 3.1.2. Ved fastlæggelse af NOAEL og LOAEL for stoffet som sådan i det pågældende studie skal dernæst foretages en afvejning af de enkelte effekter i forhold til de fastlagte NOAELs og LOAELs. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand vurderes effekter generelt som værende mere kritiske desto lavere doser (koncentrationer), de optræder ved, således at NOAEL for stoffet som sådan i et givent studie ofte, men ikke nødvendigvis altid, vil være identisk med det laveste af de fastlagte NOAELs for enkelteffekter. For de studier, hvor der kan sondres mellem effekter og skadelige effekter, vil NOAEL ikke nødvendigvis være den højeste dosis (koncentration) i studiet, hvor der ikke observeres effekter. Dette forhold er illustreret i figur 3.2.3, hvor kurve B eksempelvis er dosis-responssammenhængen for stigning i aktiviteten af et uspecifikt leverenzym, det vil sige en effekt, og hvor kurve C eksempelvis er dosis-responssammenhængen for irreversible testikelskader, det vil sige en skadelig effekt. I dette tilfælde vil NOAEL for stoffet som sådan i det pågældende studie ligge et eller andet sted på kurve C, mens NOEL vil være at finde på kurve B. Et andet eksempel kunne være, at kurve A er dosis-responssammenhængen for øget (ikke statistisk signifikant) relativ organvægt, og kurve C som ovenfor. I dette tilfælde vil NOAEL for stoffet som sådan i det pågældende studie ligge et eller andet sted på kurve C nedenfor skæringspunktet med kurve A. Figur 3.2.3 Identifikation af nul-effektniveau, sondring mellem effekt og skadelig effekt
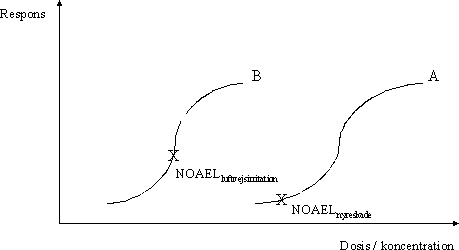
En lang række stoffer kan give anledning til både systemiske og lokale effekter. Eksempelvis medfører indånding af mange organiske opløsningsmidler en påvirkning af centralnervesystemet, hvilket er en systemisk effekt, men kan også give anledning til irritation eller skader i luftvejene, hvilket er lokale effekter. I tilfælde af at det i det enkelte studie ikke kan afklares, hvorvidt de systemiske eller de lokale effekter vurderes som værende mest kritiske, fastlægges NOAEL og LOAEL således for begge typer af effekt. Dette forhold er illustreret i figur 3.2.4, hvor kurve A kunne være dosis-responssammenhængen for en systemisk effekt, for eksempel nyreskade, og kurve B dosis-responssammenhængen for en lokal effekt, for eksempel luftvejsirritation. I praksis er det imidlertid ofte således, at datagrundlaget i det enkelte studie ikke er tilstrækkeligt til at fastlægge både NOAEL og LOAEL for stoffet som sådan i det pågældende studie efter de ovenfor beskrevne principper. I konsekvens heraf vil NOAEL således ofte blive fastlagt som den højeste dosis (koncentration) i studiet, hvor der ikke er observeret effekter, og LOAEL som den laveste dosis (koncentration) i studiet, hvor der er observeret effekter. Figur 3.2.4 Identifikation af nul-effektniveau for systemisk effekt og lokal effekt
3.2.2 Andre metoder til fastlæggelse af nul-effektniveauSom beskrevet ovenfor og illustreret på kurve B i figur 3.2.2 vil NOAEL ofte blive fastlagt som den højeste dosis (koncentration) i studiet, hvor der ikke er observeret effekter, og LOAEL som den laveste dosis (koncentration) i studiet hvor der er observeret effekter. Kurveforløbet for dosis-responssammenhængen og den dertil svarende tærskelværdi kan også beregnes ved hjælp af forskellige matematiske modeller (kurvefitning). Der kan anvendes forskellige statistiske modeller til at beskrive sammenhængen mellem dosis og respons og forskellige fordelinger til at beskrive tilfældige udfald. Fælles for disse modeller og fordelinger er, at de afhænger af de indgående parametre, og kurveforløbene må derfor tilpasses efter de aktuelle måledata. Med henblik på fastlæggelse af et udgangspunkt (engelsk: point of departure) for beregning af den tolerable daglige indtagelse (TDI, se kapitel 4) ville det, ud fra statistiske synspunkter, måske være bedre at tage udgangspunkt i den nedre 95% konfidensgrænse for den dosis (koncentration), som medfører effekt hos 5 eller 10% af dyrene i stedet for at tage udgangspunkt i NOAEL eller LOAEL, som beskrevet i det foregående afsnit. Statistisk set vil denne metode være bedre egnet, hvis der kun indgår et lille antal dyr i studiet, og der indbygges derfor automatisk en slags usikkerhedsfaktor. På den anden side er der nogle andre forudsætninger ved valget af beregningsmetode, som måske kan trække i den anden retning. Den hyppigst anvendte alternative metode til fastlæggelse af et udgangspunkt for beregning af TDI er den såkaldte benchmark-metode. Benchmark-metoden indebærer, at en dosis-responskurve beregnes
ud fra det fulde datasæt for hver effektparameter, og der vedtages en ”kritisk effektstørrelse”. På basis af den beregnede kurve defineres benchmark dose (BMD) som den nedre konfidensgrænse for den
dosis, der fører til den givne kritiske effektstørrelse, det vil sige den incidens (for eksempel 5 eller 10%), der estimeres for en given effekt. Imidlertid giver det klassiske guideline-forsøgsdesign med 3 doserede grupper og en vehikelkontrolgruppe ikke ideelle data til udarbejdelse af gode dosis-responskurver. Hertil er studier med 5-10 dosisgrupper nødvendige. Såfremt det totale antal dyr per forsøg ikke øges, vil et øget antal grupper medføre en formindsket gruppestørrelse, hvorved det ikke længere vil være muligt at fastlægge et NOAEL på sædvanlig vis (se 3.2.1). Det kan således ikke i praksis lade sig gøre at lave et forsøgsdesign, som egner sig til begge fremgangsmåder, eller at bruge de to fremgangsmåder på det samme forsøg. Nødvendig videreudvikling af benchmark metoden omfatter forbedret forsøgsdesign, international enighed om størrelsen af kritisk effekt, samt udvikling af specifikke dosis-respons analysemetoder for forskellige typer af forsøgsdata. Som nævnt fastlægges benchmark dose ved modellering af data fra et givent studie. En hvilken som helst model, som passer til de observerede data, vil give et rimeligt estimat af benchmark dose, og valget af model er tilsyneladende ikke kritisk, idet den estimerede værdi for benchmark dose ligger inden for det observerede dosisinterval. Det skal imidlertid understreges, at benchmark-metoden ikke kan anvendes hvis der i det enkelte studie ikke er et tilstrækkeligt antal dosisniveauer, hvor der observeres effekter. Da dette er tilfældet i de fleste dyreeksperimentelle studier, kan benchmark metoden ikke umiddelbart anvendes som en alternativ metode til fastlæggelse af udgangspunkt for beregning af TDI i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand. Derfor anbefales den traditionelle anvendelse af NOAEL eller LOAEL (se 3.2.1) fortsat som udgangspunkt for beregning af TDI med henblik på fastsættelse af kvalitetskriterier for luft, jord og drikkevand. 3.3 HormesisFænomenet hormesis inden for toksikologien har en lang og kontroversiel historie og går helt tilbage til slutningen af 1800 tallet, hvor det blev beskrevet første gang. Hormesis betegner et fænomen, hvor der ved lave doser ses en stimulation og ved høje doser en inhibition, hvilket giver sig udtryk ved en U-formet eller omvendt U-formet dosis-responssammenhæng. (Calabrese & Baldwin 2001a,b, Calabrese 2001). Der er fremsat en hypotese om, at hormesis repræsenterer en overkompensation mod en påvirkning af homeostasen fremkaldt af for eksempel en mild form for stress (Calabrese 2001). Denne hypotese er ifølge Calabrese (2001) i fuld overensstemmelse med de mange rapporter, der i tidens løb er publiceret, idet det beskedne stimulerende respons observeret efter udsættelse for lave doser af nonessentielle toksiske stoffer (f.eks. cadmium, bly, kviksølv, phenol, bestråling osv.) snarere skyldes en overkompensation mod en påvirkning af homeostasen end en direkte stimulerende effekt af det pågældende stof. Hormesis kendetegnes således ved 1) påvirkning af homeostasen, 2) beskeden overkompensation, 3) reetablering af homeostasen og, 4) processens adaptive natur (Calabrese & Baldwin 2001b). Som nævnt er der publiceret mange undersøgelser, der understøtter, at fænomenet hormesis eksisterer. Men i den toksikologiske litteratur er der kun lidt information om, hvor ofte man kan forvente at observere hormesis. Med henblik på at undersøge, hvor mange studier i den publicerede toksikologiske litteratur der opfyldte nogle specifikke kriterier for evidens af hormesis, udarbejdede Calabrese & Baldwin (2001a) en database under anvendelse af nogle meget strikte 'a priori entry' kriterier (veldefineret NOAEL, 2 doser under NOAEL, og det målte end-point kan udvise både stimulerende og inhiberende respons) samt specifikke evalueringskriterier for evidens af hormesis. Af de i alt 20285 lokaliserede artikler, indeholdt 195 artikler (1%) 668 dosis-responssammenhænge, som opfyldte 'a priori entry' kriterierne. Ved anvendelse af evalueringskriterierne viste det sig, at 245 dosis-responssammenhænge (37% af 668) fra 86 artikler (0,4% af de i alt 20285 lokaliserede artikler) opfyldte kriterierne for evidens af hormesis. I alt 73 forskellige stoffer og blandinger fra en bred vifte af kemiske stofklasser var repræsenteret. Ifølge forfatterne viste resultaterne af analysen således, at når forsøgsdesignet opfylder nogle givne strikte 'a priori entry' kriterier, så er hormesis hyppigt forekommende og bredt repræsenteret afhængig af stof, model og end-point. Det synes således, at hormesis er et ret udbredt fænomen. Med henblik på at vurdere hvorvidt hormesis er et generelt biologisk fænomen uafhængigt af stressfaktorer i miljøet, biologisk end-point, og eksperimentelt modelsystem har Calabrese & Baldwin (2001b) en analyse. I denne analyse indgik 1) den ovenfor beskrevne undersøgelse, 2) en vurdering af 17 meget store studier, hvor hvert studie omfattede data for en række stoffer testet i det samme eksperimentelle modelsystem af den samme undersøgelsesgruppe, 3) en vurdering af farmakologiske data for 24 receptorsystemer for hvilke det bifasiske dosis-responssammenhæng karakteristisk for hormesis er blevet klarlagt, og 4) en vurdering af en database med 1600 dosis-responssammenhænge som er i overensstemmelse med hypotesen for hormesis. Ifølge forfatterne viste resultaterne af denne analyse, at hormetiske effekter repræsenterer evolutionært baserede adaptive responser på miljømæssigt inducerede forstyrrelser i homeostasen. På det nuværende grundlag er det vanskeligt at vurdere, hvilken betydning fænomenet hormesis kan have i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand. Ved fastsættelse af kvalitetskriterierne tages der for et givent stof udgangspunkt i det observerede nul-effektniveau (NO(A)EL) eller det laveste observerede effektniveau (LO(A)EL) for de(n) kritiske effekt(er) (se 3.4). Det vil sige, at kvalitetskriterierne baseres på effekter, som vurderes som værende af en egentlig skadelig karakter og ikke i uspecifikke reaktioner, som blot er en følge af individernes reaktion på ændringer i ydre omstændigheder (homeostatiske mekanismer). På baggrund heraf og sammenholdt med at data indikerer, at hormesis snarere skyldes en overkompensation mod en påvirkning af homeostasen end en direkte stimulerende effekt af det pågældende stof, vurderes det således, at hormetiske effekter umiddelbart er irrelevante i relation fastsættelse af kvalitetskriterier for luft, jord og drikkevand. Det skal endvidere bemærkes, at de milde stresspåvirkninger, som kendetegner hormesis, af nogle vurderes at have en positiv effekt på sundheden og levetiden, idet den øgede forsvarsreaktion i cellen forbedrer livskvaliteten ved at fjerne skader og gøre cellen mere aktiv og effektiv (Rattan 2001). 3.4 Kritisk effektSom det sidste led i farlighedsvurderingen af et givent stof i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand vurderes hvilke(n) effekt(er), der anses for at være mest væsentlig(e) i relation til fastsættelsen af det pågældende kvalitetskriterie, det vil sige en identifikation af de(n) kritiske effekt(er). I de indledende trin i farlighedsvurderingen for et givent stof er der foretaget en nøje vurdering af de enkelte studier, principperne herfor er beskrevet i 3.1. Endvidere er der forsøgt fastlagt et NOAEL og et LOAEL for stoffet som sådan i de enkelte studier, principperne herfor er beskrevet i 3.2. Ved identifikation af de(n) kritiske effekt(er) sammenholdes og vurderes alle de informationer, der er samlet sammen under vurderingerne af alle de enkelte studier. Det vurderes, hvor alvorlige de pågældende effekter er og ved hvilke doser (koncentrationer), de pågældende effekter optræder, ligesom der sondres mellem effekter og skadelige effekter, mellem systemiske og lokale effekter, samt hvorvidt effekter observeret hos forsøgsdyr er relevante for mennesker. For eksempel er relevansen for mennesker af visse forsøgsdyrsspecifikke cancerformer omdiskuteret. Som eksempler kan nævnes mononukleære celleleukæmier, specielle nyretumorer som kun ses hos hanrotter, levertumorer hos gnavere som er en følge af peroxisomproliferation, Leydig-celle tumorer, visse tumorer i thyroidea, samt tumorer i formaven hos gnavere. Den for tiden alment accepterede vurdering af relevansen for mennesker af disse cancerformer er beskrevet i 3.7. Alle disse forhold må naturligvis inddrages i vurderingen af de(n) kritiske effekt(er) samt ved fastlæggelse af NOAEL for de(n) kritiske effekt(er). Principperne for den samlende vurdering af alle stoffets effekter med henblik på identifikation af de(n) kritiske effekt(er) er de samme, som beskrevet for vurderingerne i det enkelte studie (se 3.1). Ligeledes er principperne for fastlæggelse af NOAEL for de(n) kritiske effekt(er) de samme, som beskrevet for vurderingerne i det enkelte studie (se 3.2). I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand vurderes effekter i reglen som værende mere kritiske desto lavere koncentrationer, de optræder ved, således at NOAEL for stoffet som sådan ofte, men ikke nødvendigvis, vil være identisk med det laveste af de fastlagte NOAELs fra de enkelte studier. US-EPA (1998) definerer det kritiske end-point som den effekt, der udviser det laveste LOAEL. 3.5 Sensibilisering for kemiske stofferDet har været diskuteret, hvorvidt sensibilisering er en effekttype, hvor der foreligger en tærskel eller ej. Nedenfor gives en kort opsummering af den foreliggende viden i relation til allergi udløst ved eksponering for kemiske stoffer, som den er fremlagt i et særnummer af tidsskriftet Comments on Toxicicology (Volume 7 1999), herunder en vurdering af tærskelværdi. Allergi defineres generelt som de skadelige effekter, der opstår som følge af stimulation af specifikke immunreaktioner. I relation til såkaldt kemisk allergi udløses disse immunreaktioner i følsomme individer ved eksponering for kemiske stoffer. De mest almindelige reaktioner i relation til kemisk allergi er hudsensibilisering, som resulterer i kontaktdermatitis, og luftvejssensibilisering som er associeret med rhinitis og astma. (Kimber & Dearman 1999a). Det skal nævnes, at ikke kun lavmolekylære kemiske stoffer, men også proteiner kan udløse allergiske reaktioner, men i dette afsnit omtales kun sensibilisering udløst af kemiske stoffer. Sensibilisering ved hudkontakt og luftvejssensibilisering som følge af eksponering for et kemisk stof har en del fælles træk. I begge tilfælde er allergenet et lavmolekylært kemisk stof, som stimulerer en specifik immunreaktion. Kemiske stoffer er haptener og som sådan ikke direkte immunogene. For udløsning af en immunreaktion skal det lavmolekylære kemiske stof først associeres med et makromolekyle (protein), hvorved der dannes et immunogent kompleks, som kan genkendes af immunsystemet. Selve den allergiske reaktion forløber i to faser. I den første fase, induktionsfasen, sker der en allergisk sensibilisering som følge af eksponering for et givent lavmolekylært kemisk stof, således at immunsystemet påvirkes til at respondere mere voldsomt ved en efterfølgende eksponering for det samme kemiske stof. Dette er den anden fase, elicitationsfasen (frembrudsfasen), hvor den sekundære immunreaktion initierer en inflammatorisk reaktion på eksponeringsstedet (hud eller luftveje). Disse inflammatoriske reaktioner fremprovokerer de symptomer, som klinisk genkendes som allergisk kontaktdermatitis eller luftvejssensibilisering. De sekundære reaktioner (elicitationsfasen) hos et i forvejen sensibiliseret individ udløses sædvanligvis ved eksponering for langt lavere doser af et givent stof end de doser, der skal til for at sensibilisere individet (induktionsfasen). Men der er også forskelle, som ikke kun begrundes i de to forskellige eksponeringssteder og dermed resulterende kliniske symptomer, men i relation til de immune og inflammatoriske processer, der er et
resultat af induktionsfasen såvel som elicitationsfasen. Disse forskelle fremgår af de efterfølgende to afsnit, som beskriver kontaktallergi og luftvejssensibilisering. Kontaktallergi er en form for forsinket sensibilisering (engelsk: delayed-type hypersensitivity) og som sådan afhængig af stimulation af specifikke T-lymfocyt reaktioner. Reaktionen kan kort beskrives som følger: Et kemisk allergen passerer den yderste forhornede del af overhuden (stratum corneum), og når frem til epidermis, hvor der sker en direkte interaktion med dendritceller i huden. Dendritcellerne transporteres til lymfeknuder, hvor antigenet inducerer og aktiverer T-lymfocytter, som så deler sig og differentierer. Denne del af reaktionen udgør den cellulære basis for sensibilisering. Når det nu sensibiliserede individ igen eksponeres for det pågældende kemiske stof ved hudkontakt, genkender T-lymfocytterne allergenet og responderer ved at frigøre cytokiner og andre molekylære mediatorer, som initierer den kutane inflammatoriske reaktion, der klinisk genkendes som allergisk kontaktdermatitis. Det er bredt accepteret, at kontaktsensibilisering er karakteriseret ved type 1 immunreaktioner og virkninger af associerede cytokiner. (Kimber & Dearman 1999b). Med hensyn til luftvejssensibilisering som følge af eksponering for kemiske stoffer ved man ikke nær så meget om de tilgrundliggende reaktioner. Ved udløsning af luftvejssensibilisering ved udsættelse for proteiner kan reaktionen kort beskrives som følger: Efter eksponering for et givent protein vil et følsomt individ reagere med dannelse af IgE-antistof, som derefter fordeles systemisk og associeres til mastceller, inklusiv mastceller i luftvejene. Ved en efterfølgende eksponering via inhalation for det samme protein vil allergenet bindes til og sammenlænke IgE, der er associeret med mastcellerne i luftvejene. Dette medfører, at mastcellerne degranulerer og frigør inflammatoriske mediatorer, som udløser en reaktion karakteriseret ved vasodilation og bronchokonstriktion. Hvorvidt kemiske allergener inducerer og eliciterer en luftvejssensibilisering via samme reaktionsmønster som proteiner diskuteres fortsat. Usikkerhederne ligger primært i undersøgelser af sammenhængen mellem kemisk luftvejssensibilisering og dannelse af IgE-antistof. For nogle kemiske allergener (f.eks. visse syreanhydrider og reaktive farvestoffer) er der fundet en god korrelation mellem dannelse af IgE-antistof og de kliniske symptomer for luftvejssensibilisering, mens dette ikke er tilfælde for alle undersøgte kemiske allergener (f.eks. visse diisocyanater). Kimber & Dearman har, på baggrund af den foreliggende viden, det synspunkt, at IgE-antistof spiller en vigtig rolle for kemisk luftvejssensibilisering. (Kimber & Dearman 1999b). Det har været diskuteret, hvorvidt sensibilisering er en effekttype, hvor der foreligger en tærskel eller ej. Basketter et al. (1999) har anført, at da kontaktallergener varierer meget med hensyn til den relative potens, så må det også kunne konkluderes, at de udviser stor variation i relation til en tærskelværdi for både induktion og elicitation. At der findes en tærskelværdi, er vist for nogle få kontaktallergener i humane studier. Evidensen for eksistensen af en tærskel for induktion støttes endvidere af, at selvom folk ofte eksponeres for mange kontaktallergener, så er det normalt kun en lille andel, der bliver sensibiliserede. Ligeledes er der mange studier, der viser, at der også findes en tærskel for elicitationsfasen. Derimod er der kun begrænset viden om, hvorvidt der findes en tærskel for induktion og elicitation af luftvejssensibilisering. Ifølge Basketter et al. (1999) er der al mulig grund til at formode, at der skal være en vis kritisk dosis på et relevant eksponeringssted for at kunne inducere luftvejssensibilisering. Tilsvarende formodes at gøre sig gældende for elicitation af luftvejsreaktionerne i sensibiliserede individer. Udover en kritisk dosis er der også andre faktorer, som har betydning. Blandt andet nævnes det, at eksponeringshyppigheden i relation til dosis er vigtig. Endelig er det også anført, at luftvejssensibilisering sandsynligvis kan induceres via andre eksponeringsveje end inhalation, for eksempel ved hudkontakt, og det understreges, at den kritiske dosis for sensibilisering vil variere afhængigt af eksponeringsvejen. I relation til risikovurdering af kemiske allergener er det anført, at denne vurdering kun kan foretages, hvis potens og tærskelværdi er nøje belyst for det givne kemiske allergen. 3.6 Kvalitativ vurdering af kræftfremkaldende stofferFor langt de fleste typer effekter findes der en tærskel, det vil sige en grænse (koncentration eller dosis), hvorunder der ikke ses effekt(er). Tærskelværdien er således den laveste koncentration eller dosis af stoffet, hvor der optræder en effekt (se 4.2). Sådan forholder det sig også for carcinogener, som ikke er genotoksiske, det vil sige, hvor den tilgrundliggende mekanisme for stoffets indflydelse på udvikling af tumorer ikke er en beskadigelse af arvematerialet. For visse typer af effekter findes der muligvis ikke en tærskel. Sådan forholder det sig antageligt for carcinogener, som er genotoksiske, det vil sige, hvor den tilgrundliggende mekanisme for stoffets effekt på udvikling af tumorer er en beskadigelse af arvematerialet. Dette betyder i teorien, at en hvilken som helst eksponering , hvor lav den end måtte være, vil medføre en risiko for udvikling af tumorer, der er større end nul. Det skal dog understreges, at man internationalt i de senere år har diskuteret og fortsat diskuterer, hvorvidt der foreligger er tærskel for genotoksiske effekter eller ej. For de stoffer, hvor den kritiske effekt har en tærskelværdi, fastlægges TDI med udgangspunkt i et nul-effektniveau (NO(A)EL) eller det laveste effektniveau (LO(A)EL) for den kritiske effekter under anvendelse af en usikkerhedsfaktor (se kapitel 4). For genotoksiske carcinogener fastlægges TDI på baggrund af en kvantitativ risikovurdering (se kapitel 5). Med henblik på fastlæggelse af TDI for kræftfremkaldende stoffer er det således af afgørende betydning, hvorvidt det givne stof vurderes som værende et genotoksisk carcinogen eller ej. Ved den kvalitative vurdering af et stofs kræftfremkaldende effekt tages stilling til to spørgsmål: 1) Er det sandsynligt, at stoffet er kræftfremkaldende hos mennesker? Og 2) har stoffet et genotoksisk potentiale? Afhængigt af evidensen for kræftfremkaldende effekt kan stofferne opdeles i forskellige grupper. Således opererer EU-systemet med 3 forskellige kategorier, US-EPA med 5 forskellige kategorier, og IARC med 4 forskellige kategorier. Placeringen af et stof i en af disse kategorier er ikke baseret på en opdeling efter potens, men er derimod en inddeling efter graden af den dokumentation (omfang, kvalitet og relevans), der findes for den kræftfremkaldende effekt. Disse tre forskellige systemer er således funderet på de samme basale principper. Alligevel er de ikke direkte sammenlignelige, blandt andet som følge af forskelle i antallet af kategorier samt forskelle i kriterierne for indplacering i en given kategori. Også med hensyn mutagene effekter opererer EU-systemet med 3 forskellige kategorier, som ligeledes er en inddeling efter graden af den dokumentation (omfang, kvalitet og relevans), der findes for den mutagene effekt. 3.6.1 EUVed klassificering for kræftfremkaldende effekt i EUs arbejdsgruppe for klassificering og mærkning af kemiske stoffer inddeles stofferne i 3 kategorier under hensyntagen til den nuværende viden (MM 2002): Stoffer, der vides at fremkalde kræft hos mennesket, placeres i kategori 1 (Carc1). Der foreligger tilstrækkelig dokumentation for en årsagssammenhæng mellem menneskets udsættelse for stoffet og udvikling af kræft. Stoffer klassificeres i denne kategori på baggrund af epidemiologiske undersøgelser. Stoffer, der bør anses for at fremkalde kræft hos mennesket, placeres i kategori 2 (Carc2). Der foreligger tilstrækkelig dokumentation til at nære stærk formodning om, at stoffets påvirkning af mennesker kan fremkalde kræft, generelt på grundlag af egnede langtidsforsøg i dyr samt andre relevante oplysninger. Der skal enten foreligge positive resultater fra to dyrearter eller klart positiv effekt fra én dyreart støttet af blandt andet data vedrørende genotoksicitet, metaboliske eller biokemiske undersøgelser, forekomst af godartede tumorer, strukturmæssige ligheder med andre kræftfremkaldende stoffer, eller data fra epidemiologiske undersøgelser, som tyder på en forbindelse. Stoffer, der giver anledning til betænkelighed, da de muligvis kan fremkalde kræft hos mennesket, placeres i kategori 3 (Carc3). Der foreligger ikke tilstrækkelige oplysninger til at foretage en tilfredsstillende vurdering. Der er visse tegn fra relevante dyreforsøg, men disse er utilstrækkelige til at placere dem i kategori 2. Kategori Carc3 omfatter 2 underkategorier: a) Stoffer, som er grundigt undersøgt, men hvor dokumentationen for tumorfremkaldende virkninger er utilstrækkelig til klassificering i kategori 2. Yderligere forsøg forventes ikke at frembringe yderligere oplysninger, der kan være relevante for klassificeringen. b) Stoffer, som ikke er tilstrækkeligt undersøgt. De data, der foreligger, er utilstrækkelige, men giver anledning til bekymring for mennesket. Denne klassificering er foreløbig; yderligere forsøg er nødvendige inden der kan træffes en endelig afgørelse. Ved klassificering for mutagene og genotoksiske effekter i EU's arbejdsgruppe for klassificering og mærkning af kemiske stoffer inddeles stofferne ligeledes i 3 kategorier under hensyntagen til den nuværende viden. Et mutagen er et stof, som fremkalder eller øger antallet af mutationer. En mutation er en permanent ændring i arvemassen eller strukturen i det genetiske materiale i en organisme, som medfører en ændring i organismens fænotypiske egenskaber. Ændringerne kan omfatte et enkelt gen, en gengruppe eller et helt kromosom. (MM 2002): Stoffer, der vides at have mutagene virkninger på mennesket, placeres i kategori 1 (Mut1). Der foreligger tilstrækkelig dokumentation for en årsagssammenhæng mellem stoffets påvirkning af mennesket og arvelige skader på det genetiske materiale. For at kunne placere et stof i kategori Mut1 er positiv dokumentation fra epidemiologiske undersøgelser vedrørende human mutation nødvendig. Eksempler på sådanne stoffer kendes ikke til dato. Stoffer, der bør anses for at have mutagene virkninger på mennesket, placeres i kategori 2 (Mut2). Der foreligger tilstrækkelig dokumentation til at nære stærk formodning om, at stoffets påvirkning af mennesket kan resultere i arvelige skader på det genetiske materiale, generelt på grundlag af: egnede langtidsforsøg i dyr, andre relevante oplysninger. For at kunne placere et stof i kategori 2 er der behov for positive resultater fra undersøgelser, som viser a) mutagene virkninger, b) andre cellulære vekselvirkninger i forbindelse med mutagenicitet i pattedyrs kimceller in vivo, eller c) mutagene virkninger i pattedyrs somatiske celler in vivo kombineret med klare beviser for, at stoffet eller en relevant metabolit når frem til kimcellerne. Stoffer, der giver anledning til betænkelighed, da de muligvis har mutagene virkninger, placeres i kategori 3 (Mut3). Der foreligger dokumentation fra mutagenicitetsundersøgelser, men den er utilstrækkelige til at placere stoffet i kategori 2. For at kunne placere et stof i kategori 3 er der behov for positive resultater fra undersøgelser, som viser a) mutagene virkninger, eller b) andre cellulære vekselvirkninger, som er relevante for mutagenicitet i somatiske celler i pattedyr in vivo. 3.6.2 US-EPAVed klassificering af kræftfremkaldende stoffer i US-EPA inddeles stofferne i 5 grupper (US-EPA 1998): Stoffer, der vides at fremkalde kræft hos mennesker, placeres i gruppe A (human carcinogen). Der foreligger fyldestgørende (sufficient) evidens fra epidemiologiske undersøgelser. Stoffer, der anses for sandsynligt kræftfremkaldende hos mennesker, placeres i gruppe B (probable human carcinogen). Der foreligger fyldestgørende evidens fra undersøgelser af forsøgsdyr eller begrænset (limited) evidens hos mennesker. Gruppe B er opdelt i 2 undergrupper: B1 og B2. Stoffer placeres i gruppe B1, når der er begrænset evidens fra epidemiologiske undersøgelser. Stoffer placeres i gruppe B2, når der er fyldestgørende evidens fra undersøgelser af forsøgsdyr og utilstrækkelige (inadequate) eller ingen data fra epidemiologiske undersøgelser. Stoffer, der anses for muligt kræftfremkaldende hos mennesker, placeres i gruppe C (possible human carcinogen). Der foreligger begrænset evidens fra undersøgelser af forsøgsdyr og ingen evidens hos mennesker. Stoffer, der ikke kan indplaceres i andre grupper, placeres i gruppe D (not classifiable). Der foreligger utilstrækkelige data eller ingen data. Stoffer placeres i gruppe E, når der ikke er evidens for kræftfremkaldende effekt i velgennemførte undersøgelser i mindst 2 forskellige arter eller både i epidemiologiske undersøgelser og undersøgelser af forsøgsdyr. 3.6.3 IARCVed klassificering af kræftfremkaldende stoffer i IARC inddeles stofferne i 4 grupper (IARC 1987): Stoffer, der vides at fremkalde kræft hos mennesker, placeres i gruppe 1 (carcinogenic to humans). Der foreligger fyldestgørende (sufficient) evidens for kræftfremkaldende effekt hos mennesker. Stoffer placeres i gruppe 2, når der ikke er fyldestgørende evidens for kræftfremkaldende effekt hos mennesker og/eller fyldestgørende evidens fra undersøgelser af forsøgsdyr. Gruppe 2 er opdelt i 2 undergrupper: 2A og 2B. Stoffer placeres i gruppe 2A (probably carcinogenic to humans), når der er begrænset (limited) evidens fra epidemiologiske undersøgelser og fyldestgørende evidens fra undersøgelser af forsøgsdyr. Undtagelsesvis kan et stof placeres i gruppe 2A udelukkende på baggrund af begrænset evidens fra epidemiologiske undersøgelser eller udelukkende på baggrund af fyldestgørende evidens fra undersøgelser af forsøgsdyr og andre data, som kan understøtte evidens for kræftfremkaldende effekt. Stoffer placeres i gruppe 2B (possibly carcinogenic to humans), når der er begrænset evidens fra epidemiologiske undersøgelser og ikke fyldestgørende evidens fra undersøgelser af forsøgsdyr. Stoffer placeres også i denne gruppe, når der er utilstrækkeligt (inadequate) evidens fra epidemiologiske undersøgelser eller ingen humane data, men fyldestgørende evidens fra undersøgelser af forsøgsdyr. I nogle tilfælde kan stoffer placeres i denne gruppe, når der er utilstrækkeligt evidens fra epidemiologiske undersøgelser eller ingen humane data, men begrænset evidens fra undersøgelser af forsøgsdyr og andre data, som kan understøtte evidens for kræftfremkaldende effekt. Stoffer, der ikke kan indplaceres i andre grupper, placeres i gruppe 3 (not classifiable). Der foreligger utilstrækkelige data eller ingen data. Stoffer placeres i gruppe 4 (probably not carcinogenic to humans), når der er evidens for, at stoffet ikke har kræftfremkaldende effekt i mennesker og heller ikke i forsøgsdyr. Stoffer kan også placeres i denne gruppe, når der er utilstrækkelig evidens fra epidemiologiske undersøgelser eller ingen humane data, men evidens for at stoffet ikke har kræftfremkaldende effekt i forsøgsdyr samt understøttet i en lang række andre data. 3.6.4 Miljøstyrelsens generelle praksis i relation til kvalitetskriterierI relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand tages der ved vurderingen af kræftfremkaldende effekt ikke kun udgangspunkt i det givne stofs placering i en af EU's, IARC's eller US-EPA's grupper. I vurderingen indgår også nyere data samt andre data som for eksempel stoffets fysisk-kemiske egenskaber, om eksponeringsvejene anvendt i dyreforsøgene er relevante for den generelle befolkning, toksikokinetiske forhold, om stoffet er et direkte reagerende carcinogen, hvilken metabolisering der skal ske for at stoffet bliver til et aktivt carcinogen, om forskelle i metabolisering er speciesafhængige, om stoffet har udvist genotoksiske egenskaber i in vivo tests eller kun i in vitro testsystemer, mekanismen (genotoksisk eller non-genotoksisk) for udvikling af tumorer, om typen af tumorer fundet i en dyreart har relevans for mennesker (se 3.7), og om der er epidemiologiske data til støtte for de dyreeksperimentelle fund. Som det fremgår af ovenstående, indgår mange elementer i vurderingen af, hvorvidt et givent stof skal betragtes som kræftfremkaldende, og hvorledes dette skal indgå i fastsættelsen af kvalitetskriterier. På den baggrund kan det være vanskeligt at beskrive en generel og fast procedure på området, da data kan variere meget fra stof til stof. Som tidligere nævnt er epidemiologiske undersøgelser, der klart viser en sammenhæng mellem eksponering for et givent stof og udvikling af tumorer hos mennesker, af afgørende betydning for vurdering af et stof som kræftfremkaldende hos mennesker. Epidemiologiske undersøgelser, der ikke klart viser en sammenhæng, for eksempel hvor det drejer sig om små risikoforøgelser, kan være til støtte i vurderingen af de dyreeksperimentelle fund og dermed bidrage til, at stoffet vurderes som værende kræftfremkaldende hos mennesker. Det skal understreges, at negative resultater i epidemiologiske undersøgelser ikke beviser et fravær af kræftfremkaldende egenskaber. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand inddrages de vurderinger, der måtte være foretaget af EU, IARC og eller US-EPA. Således vil et givent stof, der en indplaceret i EU's carc1 eller carc2, IARC's gruppe 1 eller gruppe 2A/2B, og/eller US-EPA's gruppe A eller B1/B2, som udgangspunkt blive betragtet som et carcinogen, med mindre der er velunderbyggede informationer og fortolkninger af data, der taler imod denne vurdering. Et eksempel herpå er fortolkning af visse typer af tumorer fundet i en dyreart, som vurderes ikke at have relevans for mennesker (se 3.7). Det skal dog understreges, at et stof som enten ikke er vurderet af EU, IARC og/eller US-EPA eller er indplaceret i en lavere kategori (EU carc3, IARC gruppe 3 og/eller US-EPA gruppe C/D), godt kan betragtes som et carcinogen i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand, såfremt der er velunderbyggede data, der taler herfor. Det kan for eksempel være i tilfælde af, at der er publiceret nye undersøgelser siden en eventuel vurdering er foretaget af EU, IARC og/eller US-EPA, eller der er stor strukturlighed med allerede kendte carcinogener. Endvidere skal der tages stilling til, hvorvidt stoffet vurderes som værende et genotoksisk carcinogen eller ej. I vurderingen heraf indgår først og fremmest de resultater, der er opnået ved testning af stoffets mutagene og genotoksiske potentiale i en række forskellige testsystemer, inklusive in vivo undersøgelser. I relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand tages der som hovedregel udgangspunkt i de vurderinger, der måtte være foretaget af EU, IARC og eller US-EPA. Således vil et givent stof, der i henhold til EU's kriterier er klassificeret for mutagene og/eller genotoksiske egenskaber (mut1, mut2 eller mut3) som udgangspunkt blive betragtet som et muligt humant genotoksisk stof. Det er dog kun et meget lille antal stoffer, der er klassificeres i henhold til EU's kriterier (ingen stoffer er til dato klassificeret mut1), hvorfor vurderingen af et stofs genotoksiske egenskaber må foretages på baggrund af de foreliggende testresultater. Ud fra dette samlede billede vurderes det, om stoffet i relation til fastsættelse af kvalitetskriterier for luft, jord og drikkevand skal betragtes som et genotoksisk eller non-genotoksisk carcinogen. Denne vurdering har som tidligere nævnt afgørende betydning for valg af procedure for estimering af TDI, det vil sige for valget mellem tærskelværdiprincippet, der anvendes ved estimering af TDI for non-genotoksiske carcinogener (se kapitel 4) og den kvantitative risikovurdering, der anvendes ved estimering af TDI for genotoksiske carcinogener (se kapitel 5). Selv om datagrundlaget for et givent stof ikke er tilstrækkeligt med henblik på at afgøre, hvorvidt stoffet kan vurderes som værende et genotoksisk carcinogen, eller datagrundlaget som sådan er utilstrækkeligt til at kunne foretage en kvantitativ risikovurdering for et stof, der vurderes som værende et genotoksisk carcinogen, kan der selvsagt godt være en bekymring i relation til eventuelle kræftfremkaldende egenskaber for det pågældende stof. I disse tilfælde beregnes TDI med udgangspunkt i et NOAEL eller LOAEL (se kapitel 4), og der kompenseres for usikkerheden vedrørende et eventuelt kræftfremkaldende potentiale ved anvendelse af en større usikkerhedsfaktor, end der ellers ville være blevet anvendt (se 4.4.3). 3.7 Effekter hos forsøgsdyr hvor relevansen for mennesker er omdiskuteretGenerelt antages det, at en toksisk effekt hos forsøgsdyr forårsaget af et kemisk stof viser, at stoffet også er toksisk i mennesket. Dette gælder også udvikling af tumorer, selvom type og placering af tumor ikke altid vil være den samme i dyr og menneske. Dog kan visse effekter hos forsøgsdyr have begrænset relevans med hensyn til at forudsige effekten i mennesker. Tumorer, der induceres via en genotoksisk mekanisme vurderes sædvanligvis som værende relevante for mennesket, også når tumorerne opstår i væv, som ikke findes hos mennesket. Non-genotoksiske mekanismer kan omfatte kronisk celleskade, immunosuppression, øget udskillelse af trofiske hormoner samt receptoraktivering. Desuden kan andre mekanismer, som for eksempel CYP450-induktion, ligge til grund for tumorudvikling. Visse specifikke non-genotoksiske mekanismer anses imidlertid ikke som værende relevante for mennesket. I det følgende omtales en del af de tumortyper og mekanismer, hvor relevansen for mennesket sædvanligvis anses for begrænset, eller hvor det ikke umiddelbart kan antages, at dyrefundene er prædiktive for mennesket. Det skal understreges, at der i hvert enkelt tilfælde foretages en nøje vurdering, og at der kan være stor forskel på de enkelte dyrestammers reaktionsmønster. 3.7.1 Leukæmier (mononucleærcelle type) hos Fischer rottenDenne type leukæmi forekommer kun hos rotten og er kun almindelig hos stammen F-344, hvor den naturlige forekomst har været stigende gennem årene. Der er forskel på hyppigheden hos de to køn, og hyppigheden hos hanner er oppe på 50%, mens hyppigheden hos hunner ligger på omkring 30%, med stor variation fra forsøg til forsøg. Det er vist for nogle genotoksiske carcinogener, at de ikke øger forekomsten af mononukleærcelle-leukæmier hos F-344 rotten, mens en række stoffer, som ikke menes at være kræftfremkaldende hos mennesket, øger forekomsten. Som følge af disse forhold anses en øget forekomst af mononukleærcelle-leukæmier hos F-344 rotten for værende af begrænset relevans for mennesket. (Caldwell 1999). 3.7.2 Nyretumorer hos hanrotterVisse nyretumorer hos hanrotter anses som værende ikke relevante for mennesket, idet der er stærk evidens for, at mekanismen for tumordannelsen er specifik for hanrotter. Der er tale om tumorer, der har en sammenhæng med induceret ophobning af proteinet 2-globulin i nyrecellerne. Dette protein dannes i leveren hos hanrotter, mens hunrotter og andre dyrearter, inklusive mennesket, ikke danner 2-globulin. Binding af et kemisk stof til 2-globulin kan føre til, at proteinet ikke nedbrydes som normalt, men ophobes i nyrecellerne. Ophobningen er skadelig for cellerne og medfører nefropati, øget celleproliferation og endelig nyretumorer. Denne mekanisme forekommer ikke hos mennesket. Såfremt det specifikt kan påvises, at denne mekanisme er den egentlige årsag til forekomst af nyretumorer hos dyrene, anses denne effekt at have begrænset relevans for mennesket. IARC har formuleret en række kriterier for, hvornår nyretumorer hos hanrotter kan anses som værende ikke relevant for mennesket (IARC 1999). 3.7.3 Levertumorer hos mus og rotterLevertumorer er et hyppigt fund i gnavere i cancerstudier. Hos mus er den naturlige forekomst af levertumorer meget høj og stærkt påvirkelig af en række faktorer som for eksempel stress og ernæring. Øget forekomst af levertumorer i et musestudie kan derfor meget vel skyldes andre faktorer end teststoffet. Ligeledes er mus meget tilbøjelige til at udvikle levertumorer efter langvarig stimulering af celledelingen eller langvarig enzyminduktion forårsaget af de høje doser af teststof. Denne mekanisme er ikke relevant for mennesket, hvor eksponeringen som regel er meget lavere, end de doser der anvendes ved studier i forsøgsdyr. Et musestudie, hvor den eneste tumorform er levertumorer, anses ofte for at have begrænset relevans for mennesket. (Williams 1997, Carmichael et al. 1997). En lang række kemiske stoffer, der inducerer peroxisomproliferation i leveren, forårsager udvikling af levertumorer hos rotter og mus. Hos mennesket findes receptoren for peroxisomproliferation (PPAR) også, men i lavere niveau end hos gnavere, hvorfor peroxisomproliferation menes at være sjældent forekommende hos mennesket. Tumorer, der er associeret til peroxisomproliferation, anses således for mindre relevante for mennesket. Imidlertid kendes mekanismen for denne form for udvikling af levertumorer ikke, og sammenhængen mellem peroxisomproliferation og udvikling af levertumorer er ikke entydig. For at afvise relevansen af levertumorer, der er associeret til peroxisomproliferation, kræves et stort datagrundlag. (Williams 1997). 3.7.4 Leydig-celle tumorerLeydigceller findes i testis og danner det hanlige kønshormon, testosteron. I cancerstudier med rotter er Leydig-celle tumorer (hyppigst adenom, sjældent carcinom) et hyppigt fund hos ældre dyr. Der er stor forskel på rottestammer, idet Sprague-Dawley har en baggrundsforekomst på 1-5%, mens Fischer-rotten er næsten oppe på 100%. Fischer-rotten anses derfor for at have en atypisk høj tendens til at udvikle Leydig-celle tumorer. Hos mus er hyppigheden generelt lav. Hyppigheden af denne tumortype er meget lav hos mennesket, hvor kun 2% af alle testestumorer stammer fra Leydigceller. For non-genotoksiske stoffer, der fremkalder Leydig-celle tumorer, og hvor mekanismen er undersøgt, er der typisk tale om en induceret øget koncentration af serum-LH (luteiniserende hormon) og dermed øget stimulation af Leydig-cellerne til vækst og deling, eller øget følsomhed for LH hos Leydig-cellerne. Da mennesket besidder det samme hormonsystem, er mekanismen relevant. Det antages imidlertid, at menneskets Leydigceller er langt mindre følsomme for LH-stimulation. Mænd med endokrine lidelser, der medfører konstant øget LH-niveau , får Leydig-celle adenomer med en frekvens på 2-3%. Til sammenligning ses 100% tumorer hos rotter, der får androgenreceptor antagonisten flutamid. En arvelig defekt hos mænd, der medfører konstant LH-receptoraktivering, giver ikke øget forekomst af Leydig-celle tumorer. Epidemiologiske studier af mennesker udsat for stoffer, der fremkalder Leydig-celle tumorer i rotter, har ikke påvist øget forekomst af denne tumor. Fund af Leydig-celle tumorer i et rottestudie kan således have begrænset relevans for mennesket. (Cook et al. 1999). 3.7.5 Thyreoidea tumorerI cancerstudier med rotter er thyreoidea carcinomer hyppige, og en af mekanismerne for udvikling af carcinomer er øget niveau af det thyreoidea-stimulerende hormon (TSH), som udskilles fra hypofysen som led i et feedback system. Hos mennesket er sådanne tumorer derimod ikke almindelige og synes ikke at have relation til øget TSH. Der er flere måder, hvorpå TSH-udskillelsen kan øges. For eksempel kan en øget enzymaktivitet i leveren medføre denne effekt ved, at nedbrydningen af thyreoideahormon øges. Hos mennesket er thyreoidea hormonsystemet det samme som hos rotten, men langt mere robust. Blandt andet er thyreoideahormon hos mennesket langt fastere bundet til protein i plasma. Rottens thyreoidea er endvidere langt mere aktiv og fungerer på et højere stofskifteniveau end hos mennesket. Såfremt et non-genotoksisk stof fremkalder thyreoidea carcinomer hos rotter, og det er vist, at mekanismen er forstyrrelse af thyreoideas hormonbalance og øget TSH, anses relevansen for mennesket for begrænset (IARC 1999). 3.7.6 Tumorer i urinblærenHos rotter og mus kan tilstedeværelsen af blæresten medføre øget celleproliferation og tumordannelse. Dette kan også ske hos mennesket, men for at et kemisk stof i sig selv danner blæresten, skal det
indtages i meget høje doser, hvilket normalt ikke vil være tilfældet hos mennesket. Tumorer hos rotter og mus forårsaget af blæresten er således relevante for mennesket, men disse udvikles ikke, såfremt
stoffet indtages i mængder, der ligger under tærsklen for udvikling af blæresten. 3.7.7 FormavetumorerMavesækken hos rotter og mus er anatomisk forskellig fra menneskets, idet en del af mavesækken hos disse arter er uden kirtler (kaldet formaven). Epithelet i den kirtelløse del af mavesækken ligner det, der beklæder spiserøret. Selv om mennesket ikke besidder et organ lig gnavernes kirtelløse mave, må spiserøret betragtes som analogt med hensyn til epithel. Ofte vil mekanismen for udvikling af formavetumorer være relateret til hyperplasi, som kræver langvarig kontakt. For et stof, der bevirker udvikling af formavetumorer ved direkte kontakt med epithelet, gælder, at da kontakttiden er meget kortere i spiserøret end i mavesækken, vil tumordannelse hos mennesket formentlig ikke ske. I tilfælde, hvor denne specifikke mekanisme kan dokumenteres, vurderes kræftfremkaldende effekt som følge af formavetumorer hos gnavere ikke som relevant for mennesket. (Kroes & Wester 1986). 3.7.8 Øvrige tumortyper og mekanismerUdover de ovenfor anførte eksempler skal det nævnes, at forekomst af mammatumorer hos mus og milttumorer hos F-344 rotter også i en del tilfælde har været vurderet som værende ikke relevante for mennesket. Phaeochromocytom (tumor i binyremarv), ikke ualmindeligt forekommende hos rotter, men sjældent hos mennesket, kan hos rotten være relateret til hypercalcæmi, hvorimod en sådan relation ikke synes at forekomme hos mennesket. Endvidere er det et velkendt fænomen, at kemiske stoffer, der injiceres i forsøgsdyr, kan fremkalde tumorer lokalt på injektionsstedet, for eksempel i muskulatur. Dette vides at forekomme også for stoffer, der ikke er kræftfremkaldende, når de doseres på anden måde, for eksempel oralt. Sådanne lokale tumorer vil ofte blive vurderet som værende ikke relevante for mennesket. 3.7.9 ReferencerBasketter DA, Evans P, Gerberick GF and Kimber I (1999). Chemical allergy: estimation of potency, thresholds and risk assessments. Comm Toxicol 7, 79-89. Calabrese EJ (2001). Overcompensation stimulation: A mechanism for hormetic effects. Crit Rev Toxicol 31, 425-470. Calabrese EJ and Baldwin LA (2001a). The frequency of U-shaped dose responses in the toxicological literature. Toxicol Sci 62, 330-338. Calabrese EJ and Baldwin LA (2001b). Hormesis: A generalizable and unifying hypothesis. Crit Rev Toxicol 31, 353-424. Caldwell DJ (1999). Review of mononuclear cell leukemia in F-344 rat bioassays and its significance to human cancer risk: A case study using alkyl phthalates. Regul Toxicol Pharmacol 30, 45-53. Carmichael NG, Enzmann H, Pate I and Waechter F (1997). The significance of mouse liver tumor formation for carcinogenic risk assessment: results and conclusions from a survey of ten years of testing by the agrochemical industry. Environ Health Perspect 105, 1196-1203. Cook JC, Klinefelter GR, Hardisty JF, Sharpe RM and Foster PMD (1999). Rodent Leydig cell tumorigenesis: A review of the physiology, pathology, mechanisms and relevance to humans. Crit Rev Toxicol 29, 169-261. Goodman JI (1998). The traditional toxicologic paradigm is correct: Dose influences mechanism. Environ Health Perspect 106, 285-288. IARC (1999). IARC Scientific Publications No. 147. Species differences in thyroid, kidney and urinary bladder carcinogenesis. Edited by C.C. Capen, E. Dybing, J.M. Rice and J.D. Wilbourn. Kimber I and Dearman RJ (1999a). Allergic hypersensitivity induced by chemicals: an introduction. Comm Toxicol 7, 5-7. Kimber I and Dearman RJ (1999b). Mechanisms of sensitization to chemical allergens. Comm Toxicol 7, 9-30. Kroes R and Wester W (1986). Forestomach carcinogens: Possible mechanisms of action. Fd Chem Tox 24, 1083-1089. MM (2002). Bekendtgørelse om klassificering, emballering, mærkning, salg og opbevaring af kemiske stoffer og produkter. Bekendtgørelse nr. 329 af 16. maj 2002, Miljøministeriet, Miljøstyrelsen. Rattan S. (2001). Ungdommens kilde. Aktuel Naturvidenskab 3, 25- 27. US-EPA (1998). Ambient water quality criteria derivation for the protection of human health – Technical Support Document. Final draft. United States Environmental Protection Agency, Office of Water 4304, EPA-822-B-98-005. Williams GM (1997). Chemicals with carcinogenic activity in the rodent liver; mechanistic evaluation of human risk. Cancer Lett 117, 175-188.
|