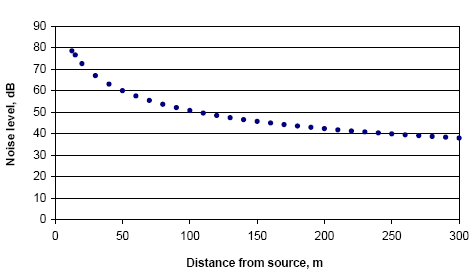
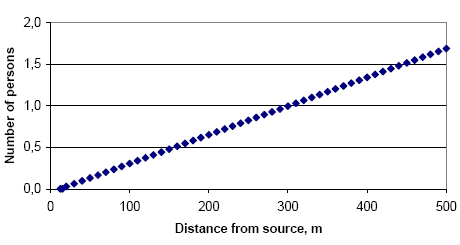
|
Environmental Project no. 996, 2005 Background for spatial differentiation in LCA impact assessment- The EDIP2003 methodologyContents1 Introduction
2 General issues
3 Acidification
4 Terrestrial eutrophication
5 Aquatic eutrophication
6 Photochemical ozone formation
7 Human toxicity
8 Ecotoxicity
9 Integration of external noise nuisance from road and rail transportation in lifecycle assessment
PrefaceThis report was prepared within the Danish LCA methodology and consensus creation project during the period from 1997 to 2003. The report is one out of five technical reports to be published by the Danish Environmental Protection Agency and dealing with key issues in LCA. The reports were prepared as background literature for a number of guidelines on LCA, planned to be published by the Danish Environmental Protection Agency during the autumn of 2004 and spring 2005. The reports present the scientific discussions and documentation for recommendations offered by the guidelines. The reports and guidelines developed within the project are presented in the overview figure below. A primary objective of the guidelines has been to provide advice and recommendations on key issues in LCA at a more detailed level than offered by general literature, like the ISO-standards, the EDIP reports, the Nordic LCA project and SETAC publications. The guidelines must be regarded as a supplement to and not a substitution for this general literature. It is, however, important to note that the guidelines were developed during a consensus process involving in reality all major research institutions and consulting firms engaged in the LCA field in Denmark. The advice given in the guidelines may thus be considered to represent what is generally accepted as best practice today in the field of LCA in Denmark. The development of the guidelines and the technical reports was initiated and supervised by the Danish EPA Ad Hoc Committee on LCA Methodology Issues 1997-2001. The research institutions and consulting firms engaged in the development and consensus process are: COWI, Consulting Engineers and Planners (Project Management) Guidelines and technical reports prepared within the Danish LCA-methodology and consensusproject
In addition to the contribution from all the major Danish research institutions and consulting firms working with life cycle assessment, the work on site-dependent impact assessment in LCA has also had a strong international touch through contributions from prominent European researchers with expertise within the different impact categories covered by the project. The editing of the report by José Potting and Michael Hauschild has thus been based on work performed by the following research teams: Chapter 3 Acidification José Potting (Institute of Product Development (IPU), Technical University of Denmark, now the Center for Energy and Environmental Studies IVEM, University of Groningen, the Netherlands) Wolfgang Schöpp (IIASA, International Institute for Applied Systems Analysis, Laxenburg, Austria) Kornelis Blok (University of Utrecht, Department of Science, Technology and Society, the Netherlands) Michael Hauschild (Institute of Product Development (IPU), Technical University of Denmark) Chapter 4 Terrestrial eutrophication José Potting Wolfgang Schöpp Michael Hauschild Chapter 5 Aquatic eutrophication José Potting Arthur Beusen (RIVM, National Institute of Public Health and the Environment, Bilthoven, the Netherlands) Henriette Øllgaard (The Danish Technological Institute) Ole Christian Hansen (The Danish Technological Institute) Bronno de Haan (RIVM, National Institute of Public Health and the Environment, Bilthoven, the Netherlands ) Michael Hauschild Chapter 6 Photochemical ozone formation Michael Hauschild Annemarie Bastrup-Birk (Danish National Environmental Research Institute) Ole Hertel (Danish National Environmental Research Institute) Wolfgang Schöpp (IIASA, International Institute for Applied Systems Analysis, Laxenburg, Austria) José Potting Chapter 7 Human toxicity José Potting Alfred Trukenmüller (Stuttgart University, Institute of Energy Economics and the Rational Use of Energy, Germany) Frans Møller Christensen (Danish Toxicology Institute) Hans van Jaarsveld (RIVM, National Institute of Public Health and the Environment, Bilthoven, the Netherlands) Stig I. Olsen (Institute of Product Development (IPU), Technical University of Denmark) Michael Hauschild Chapter 8 Ecotoxicity Jens Tørsløv (Danish Hydraulic Institute) Michael Hauschild Dorte Rasmussen (Danish Hydraulic Institute) Chapter 9 Noise nuisance Per H. Nielsen (Institute of Product Development (IPU), Technical University of Denmark) Jens E. Laursen (FORCE Technology) Critical Review Allan Astrup Jensen, FORCE Technology, Henrik Wenzel, IPU and Kim Christiansen, 2.-0 LCA Consultants have assisted in the process by critically reviewing all the guidelines and reports prepared. Authors' prefaceThis Technical Report focuses on site-dependency in impact assessment in LCA. A primary objective of the Technical Report has been to provide the necessary technical background for the guidance provided by the Danish EPA on these matters. At the onset of the work for this particular Technical Report, however, the literature about site-dependency in LCA was limited. This Technical Report therefore represents an exciting and challenging process of method development. The work gained considerable international attention and inspired similar research activity elsewhere which again gave inspiration to our project. Several sets of site-dependent characterisation factors are thus toady available for a number of impact categories. These different sets take their basis in similar types of underlying spatially resolved models, though sometimes are based on slightly different definitions of the impact indicator. Now that the methodology is available, the challenge is to start using it! The results and experience from such practical application will give fresh input to the discussion about the sense and nonsense of site-dependent impact assessment in LCA. It will advance insight in the additional information gained, reductions in spatially determined uncertainties, increase in modelling and parameter uncertainties, and hence the robustness of a site-dependent approach to the different impact categories in LCA. It may also help to settle the – at present slightly theoretical – discussion about the feasibility of site dependency with regard to potential complications in inventory analysis. After all, the proof of the pudding is in the eating of it. José Potting Michael Hauschild October 2003 1 Introduction1.1 Methodology development in progress Authors: José Potting [1] and Michael Hauschild [2] The code of practice of the Society of Environmental Toxicology and Chemistry (Consoli et al. 1993), and the recent international standards and technical reports from ISO (see next section) are widely accepted as general frameworks for Life Cycle Assessment (LCA). These frameworks and technical reports are not detailed methodological references, however, since international agreement is limited to main lines and methodology has not yet been fully developed. A major problem to be solved is the poor accordance between impact as calculated in LCA and the expected occurrence of actual impact. Until recently, Life Cycle Impact Assessment (LCIA) typically focused on substance properties and left out information about the location of the emission and characteristics of – transport to – the receiving environment. Thus LCIA ignored those fate and exposure characteristics which were specified according to the conditions at the relevant locations. Here lies a source of discrepancy between modelled impact and the occurrence of actual impact (Potting and Hauschild (1997a). The need to include information about fate and exposure conditions was foreseen in the development of the EDIP97 methodology, and a site factor was defined for all non-global impact categories to represent the severity of the exposure (Wenzel et al. 1997). While a framework was set up, the development of the methodology was left for later update and EDIP97 provided only some recommendations on how to take in qualitative spatial information in the weighting step. This technical report aims to contribute to a solution of the poor accuracy of the assessed impact in typical LCA resulting from the present disregard of spatial information in LCA. The problem is set more comprehensively in Section 1.3. The subsequent chapters elaborate and present actual methodology to overcome these problems. The reporting in these subsequent chapters takes a rather well informed reader as a starting point, but a general overview of LCA and the impact assessment phase in particular is given in this introduction. Section 1.1 gives some insight in the present state of LCA methodology development as it is still in progress. Section 1.2 provides an introduction in the general framework of LCA. Section 1.3 draws the state-of-the-art of spatial differentiation in LCA when the research for this technical report started. Section 1.4 gives the problem setting and research mandate, and Section 1.5 the outline of this Technical Report. 1.1 Methodology development in progressLCA methodology developed initially mainly in the practice of conducting LCAs. This methodological freedom resulted in LCAs examining the same product, but producing quite different results, sometimes leading to opposite conclusions. Non-explicit choices and assumptions could often be identified as the source of confusion. Absence of a generally accepted methodological framework and the lack of transparency in several LCA reports seriously affected the credibility and further diffusion of LCA. The first initiative to harmonise the scientific conduct of LCA was taken in 1990 by the Society of Environmental Toxicology and Chemistry (SETAC). A series of workshops has resulted in SETACs "Code of practise" with general guidelines for LCA (Consoli et al. 1993). To further take forward methodological development, both the European and North American branch of SETAC installed a number of workgroups in 1993 for a period of 3 years. Most workgroups have documented their results in reports. These reports reflect the consensus and state-of-the-art thinking at that time within the international scientific community about:
Following this three year period, the European branch of SETAC installed new workgroups on several subjects (among which also again one on life cycle impact assessment) for another 3 year period (Udo de Haes et al. 2002). In parallel to the SETAC activities, the International Standard Organisation (ISO) started in 1994 a process of harmonisation and standardisation of the present practice of conducting LCAs. This work was completed in 2000 with a standard on principles and framework (ISO 14040 1997), a standard on goal and scope definition and inventory analysis (ISO 14041 1998) a standard on life cycle impact assessment (ISO 14042 2000) and a standard on life cycle interpretation (ISO 14043 2000). Accompanying these standards are the technical reports illustrating the use of impact assessment (ISO TR 14047), life cycle inventory data format (ISO TR 14048) and goal and scope definition and inventory analysis (ISO TR 14049). There has been quite some relations between SETAC, aiming at scientific development of methodology, and ISO, aiming at harmonisation and standardisation of the present practice. SETAC had the status of a so-called A-liaison organisation within the ISO work on LCA. This means that SETAC has had the right to delegate a representative to ISO meetings that may fully take part in the discussions. The representative had no right to vote, but ISO has sought the full and, if possible, formal backing of SETAC. (Hortensius 1996). There was also quite some overlap in people active in SETAC context as well as in ISO activities. Being from a more recent date than SETACs "Code of practise" (Consoli et al. 1993), the documents ISO 14040, ISO 14041 and ISO14042 also reflect the scientific methodological progress up to around the year 2000, though its main focus is on yet operational methodology. Besides SETAC and ISO, also LCANET has given a major input to the existing consensus regarding method development. LCANET was a concerted action under the DGXII environment and climate research and development program of the European Union with the threefold aim to establish a network of academia and industry and government, to describe the state-of-the art and to identify priority research needs. Expert-meetings were organised around five topics: positioning and application of LCA, goal and scope definition and inventory analysis, life cycle impact assessment and interpretation, work environment, databases and software. For each topic, descriptions of the state-of-the-art and a draft research program were written. These documents were discussed and research priorities were established in a series of workshops. The final results are reported in Udo de Haes and Wrisberg (1997). LCANET had a successor in CHAINET, a concerted action under the same research and development program of the European Union. CHAINET aimed to write a guidebook on the use of different analytical tools and thus had a broader focus thus than LCA only. The guidebook was written interactively with environmental stakeholders and experts of these tools (Wrisberg et al., 2002). The method development and consensus building activities of SETAC have now been continued through the joint Life Cycle Initiative with UNEP with three main pillars of which one is on life cycle impact assessment. The stated aim of the initiative is "to develop and disseminate practical tools for evaluating the opportunities, risks, and trade-offs associated with products and services over their entire life cycle to achieve sustainable development". An element under the initiative is to identify best practice for life cycle assessment within the framework laid out by the ISO standards and to make data and methodology for performing LCA available and applicable worldwide (UNEP 2002). 1.2 General frameworkSETAC's code of practice (Consoli et al. 1993), and the recent international standards and technical reports from ISO (see previous section) are widely accepted as the general framework for LCA. However, these publications do not provide detailed methodological guidance. More comprehensive and detailed guidelines are provided by the Danish EDIP methodology documentation (Wenzel et al. 1997, Hauschild and Wenzel, 1998), the Nordic guidelines (Lindfors et al. 1995), the Dutch LCA guidelines (Heijungs et al. 1992 and lately Guinée, 2002), and the North American publication with guidelines on inventory and principles (Vigon et al. 1993). Large similarities, but also some important differences exist between these "regional standards" for LCA. Those differences arise partly from regional divergences in environmental concerns and control strategies, but express also the methodological immaturity in some areas. SETAC's "Code of practice" (Consoli et al. 1993) distinguishes four methodological phases within LCA: goal and scope definition, life cycle inventory analysis, life cycle impact assessment, and life cycle improvement assessment. In the standard ISO 14040 (1997), life cycle improvement assessment is not longer regarded as a phase on its own, but rather seen as having its influence throughout the whole LCA methodology. Another phase has been added in stead: life cycle interpretations. A brief overview of the general framework is given here. Figure 1.1 presents the ISO framework for LCA (ISO 14040 1997). An important notion of ISO 14040 is the iterative character of LCA. All phases may have to be passed through more than once due to new demands posed by a later phase. Though decisions and actions may follow the interpretation phase, these decisions and action in itself are outside the framework of LCA.
Figure 1.1. The phases of an LCA according to ISO 14040 (1997). The goal definition clarifies the initial reasons, the intended application and the audience of the LCA. The main applications supported by LCA generally ask for a comparative assertion (either comparison of different products that are functionally equivalent, or comparison of the processes within the life cycle of one product). The scope definition specifies the object of the LCA and directs the specific methodology to be followed in the next phases. A particular product can provide different services and a given service can be provided by different products. The object studied in a LCA is actually a product service rather than a product itself. The functional unit, a measure for the service performance of a product, ensures that comparison of products is made on a common basis. The methodological choices about boundaries and procedures for the other phases are according to ISO 14040 specified in scope definition. Inventory analysis identifies and quantifies the resource extractions and consumptions, and the releases to the environment relating to the processes that make up the life cycle of the examined product(s). These extractions, consumptions and releases are also referred to as environmental interventions. The interventions are expressed as quantities per functional unit (and do thus not contain a specification of the temporal and spatial characteristics of these). The life cycle of a product can be divided into stages:
Each stage may consist of a number of processes which each uses one or more inputs from previous processes and gives outputs to one or more next processes. Each input can be followed upstream to its origin and each output downstream to its final end. The total of connected processes is called the product system, process tree or life cycle. Figure 1.2 gives an example of a product system.
Figure 1.2. Life cycle assessment of paper recycling looks at a system which includes forestry and production of virgin fibres and paper additives (the cradle), use of paper, collection, re-pulping and making of new paper, and disposal of worn-out paper fibres (the grave). The paper life cycle draws on other systems like transportation and energy systems which have life cycles of their own, not shown in the figure. Adapted from Hauschild and Barlaz, in prep. The system boundaries determine which processes will be included in the LCA. The definition of the product system and its boundaries takes place in scope definition. Scope definition also decides for which environmental inputs and outputs data should be collected, and about the procedure to allocate these to processes with multiple outputs to next processes. The inventory phase therefore only consists of the actual data collection and data processing. Environmental inputs and outputs have the potential to bring about several kinds of impact on the environment. In impact assessment, the potential contributions from these inputs and outputs to a collection of impact categories are estimated and weighted. As a first step (classification), the environmental inputs and outputs are assigned to the impact categories selected in scope definition. The contribution to an impact category from each input or output is then next modelled (characterisation). In very specific cases and only when meaningful, the modelling results for one impact category or subcategory may be aggregated with those of another one (valuation). Section 1.3 provides some more information about the impact assessment phase. Interpretation is the phase in which the results of the inventory phase and the impact assessment phase are combined in line with the defined goal and scope. Conclusions and recommendations to the decision-maker may be drawn, unless reviewing and revising of previous phases is needed. Both concluding/recommending and reviewing/revising should preferably be based on uncertainty and sensitivity analysis. 1.3 Spatial differentiation and threshold exceedanceThe impact assessment phase initially emerged from the wish to aggregate the large amount of inventory data to a manageable amount of interpretable impact data. For most impact categories, initially rather simple modelling was used to establish characterisation factors. These characterisation factors for emission-related impact categories were limited to equivalency assessment on the basis of intrinsic substance characteristics like the potential to release hydrogen ions (acidification assessment) or no-effect-levels (toxicity assessment). Fate and exposure modelling was not performed, nor was threshold exceedance [3] taken into account since the available data did not allow such evaluation. Already in an early stage, it was recognised that an impact assessment on the basis of substance characteristics limits the environmental relevance of the assessed impact indicators. This environmental relevance was hampered by a lack of fate and exposure modelling, and also impeded by the absence of spatial (and temporal) differentiation in the impact modelling of the non-global categories. (Fava et al. 1993) Schmidt et al. (1996), Giegrich (1996), Potting and Blok (1994 and 1995) and Owens and Rhodes (1995) provide clear examples of the erroneous results this may give rise to in LCIA. Typical characterisation factors for most impact categories did not cover modelling of fate and exposure, nor threshold exceedance and spatial differentiation. Considerable efforts were made though to incorporate generic fate and exposure modelling in the characterisation factors for toxicity assessment (Guinée et al. 1996, Hauschild and Wenzel 1998). The present toxicity factors now usually also cover fate and exposure modelling, and aggregation [4] of the calculated exposure increases from different substances is again based on no-effect-levels. Threshold exceedance, and spatial (and temporal) differentiation in fate and exposure, however, is still not taken into account in impact modelling of toxicity. The issues of spatial differentiation and threshold exceedance are among the most discussed in LCA. Though both are interrelated, they are often addressed as somewhat separate issues. Evaluation of threshold exceedance was initially left out of life cycle impact assessment due to lack of data. Meanwhile, it has for many practitioners turned into a principle in itself that is justified by the reasoning that "less pollution is better". Heijungs et al. (1992) stated: "....LCA is not concerned with the degree to which a no-effect-concentration is actually exceeded, but with the degree to which it is poten.tially filled up...". Udo de Haes et al. (1996) further underpinned this by defining LCA as "...primarily a tool for resource conservation and pollution prevention". For this reason "....all emissions are regarded as relevant on the basis of their intrinsic hazard charac.teristics, whether above or below the no-observed-effect-concentration threshold..." (White et al. 1995). Refraining from spatial differentiation in LCA, i.e. preference for a site-generic impact assessment, was and still is defended with the expected complications in inventory analysis for a more site-specific assessment. The inventory data in LCA are expressed in amounts per functional unit and in principle, nothing is known about the source-strength and variation over time of the examined processes. Due to this lack of differentiation, which is inherent to LCA, it was believed that no environmental concentrations can be predicted (and as a consequence it does neither seem possible to evaluate whether a no-effect-level is surpassed). Spatial differentiation would require for each process in the life cycle more site-specific data (Heijungs et al. 1992). On the other hand, it was commonly recognised that the calculated contributions, except for the global impacts, could be in poor accordance with the expected occurrence of actual impact [5]. The SETAC workgroup on life cycle Impact Assessment advised against evaluation of threshold exceedance. Nevertheless, they identified the elaboration of practical models for inclusion of spatial differentiation into characterisation as one of the main future tasks (Udo de Haes et al. 1996): "There seems to be a great need for further development of the procedure for site-dependent impact assessment. A main challenge then is to prepare relevant maps for the different impact categories, preferably at a world level, with a fair balance between resolving power and feasibility". The methodology presented in this Technical Report is more sophisticated than could have been foreseen in Udo de Haes et al. (1996) and EDIP97, or in Potting and Hauschild (1997b) which outlined a general approach for spatial differentiation in LCA. Presentation of interim-results drew internationally broad attention and triggered similar research activity elsewhere (that the opposite way around gave input to this research). Several sets of characterisation factors are presently available for a number of impact categories that establish the relation between the region of emission and its impact on the receiving environment. These so-called site-dependent factors do usually cover both fate and exposure, and sometimes also the exceedance of thresholds. Potting et al. in Udo de Haes et al. (2002) gives a review. 1.4 Problem-setting and research mandateDue to the lack of spatial and temporal differentiation in inventory analysis, it seems that no environmental concentrations can be predicted and as a consequence it does neither seem possible to evaluate whether a no-effect-level is surpassed. There may therefore be only little accordance between the impact predicted by LCA and the expected occurrence of actual impact. Schmidt et al. (1996), Giegrich (1996), Potting and Blok (1994 and 1995) and Owens and Rhodes (1995) provide clear examples of this. The spotted lack of accordance between the impact calculated by LCA and the expected occurrence of actual impact seriously affects the credibility of LCA. It may cause the wrong products to be taken from the market or the wrong processes within the product's life cycle to be selected for improvement. Enhancement of the impact assessment phase is therefore of vital importance for the credibility of LCA. Only little attempts have been made so far to systematically explore the feasibility of spatial differentiation in LCA. This technical report aims to contribute to a solution of the often poor accuracy of the assessed impact in LCA elaborating and presenting actual methodology to overcome this problem. Acknowledging that current life cycle impact assessment ignores spatially determined differences in exposure of sensitive targets in the environment, and that this often leads to discrepancy between predicted impacts and actually occurring impacts, the research mandate of this study was - to develop LCIA methodology which takes into account differences in exposure - applying this methodology to provide spatially differentiated characterisation factors or site factors to correct existing EDIP97 site-generic characterisation factors for all countries in Europe. 1.5 Outline of this reportThis Technical Report takes a rather well informed reader as a starting point, but a general overview of LCA and the impact assessment phase in particular is given in the previous sections. Chapter 2 elaborates on a number of issues that are of general interest for this report. The next chapters give per chapter details and background information of the methodology developed for a given impact category: acidification in Chapter 3, terrestrial eutrophication in Chapter 4, aquatic eutrophication in Chapter 5, photochemical ozone formation in Chapter 6, human toxicity in Chapter 7, ecotoxicity in Chapter 8, and noise in Chapter 9. This Technical Report contains details and backgrounds, but does not describe in its entirety how to apply the developed methodology. For this, readers are referred to the Guidance Document (Hauschild and Potting 2003) based on this Technical Report. 1.6 ReferencesBarnthouse, L., J. Fava, K. Humphreys, R. Hunt, L. Laibson, S. Noesen, J. Owens, J. Todd, B. Vigon, K. Weitz and J. Young. Life cycle impact assessment: The state-of-the-art. Pensacola (United States of America), Society of Environmental Toxicology and Chemistry – North America, 1997. Christiansen, K., A. de Beaufort-Langeveld, N. van den Berg, R. Haydock, M. ten Houten, S. Kotaji, E. Oerlemans, W-P. Schmidt, A. Weidenhaupt and R. White. Simplifying LCA: Just a cut. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1997. Clift, R., R. Frischknecht, G. Huppes, A-M. Tillman, B. Weidema. Towards a coherent approach to life cycle inventory analysis. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1997. Consoli, F., D. Allen, I. Boustead, J. Fava, W. Franklin, A.A. Jensen, N. de Oude, R. Parrish, D. Postlethwaite, B. Quay, J. Siéguin and B. Vigon (Eds.). Guidelines for life cycle assessment. A code of practice. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe and North America, 1993. Fava, J., F. Consoli, R. Denison, K. Dickson, T. Mohin and B. Vigon (Eds.). A conceptual framework for life cycle impact assessment. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe and North America, 1993. Giegrich, J. Personal communication, Jürgen Giegrich, Institut für Energie- und Umweltforschung, Heidelberg (Germany), 1996. Guinée, J., R. Heijungs, L. Van Oers, D. Van de Meent, T. Vermeire, M. Rikken. LCA impact assessment of toxic releases. Generic modelling of fate, exposure and effect for ecosystems and human beings with data for 100 chemicals. Leiden (the Netherlands), Centre of Environmental Science of Leiden University, 1996. Guinée, J. (ed.): Handbook on life cycle assessment. Operational guide to the ISO standards. Kluwer Academic Publishers, Dordrecht, the Netherlands. ISBN 1-4020-0228-9, 2002. Hauschild, M. and J. Potting. Spatial differentiation in life cycle impact assessment – the EDIP2003 methodology. Guideline from the Danish Environmental Protection Agency, Copenhagen, 2003. Hauschild, M. and Wenzel, H. Environmental assessment of industrial products. Vol. 2: Scientific background. London (United Kingdom), Chapman and Hall, United, 1998. Hauschild M. and Barlaz, M.: Introduction to life cycle assessment. From Christensen, T. and Barlaz, M. (eds.): Solid Waste Technology and Management (in preparation). Heijungs, R., J. Guinée, g. Huppes, R.M. Lankreijer, H.A. Udo de Haes, A. Wegener Sleeswijk, A.M.M. Ansems, P.G. Eggels, R. Van Duin en H.P. de Goede. Environmental life cycle assessment of products. Guide and background (ISBN 90-5191-064-9). Leiden (the Netherlands), Centre of Environmental Science of Leiden Univerisity, 1992 Hortensius, D. Personal communication from Dick Hortensius, employee of the Dutch Normalisation Institute, concerning process and progress in the normalisation of life cycle assessment. Delft (the Netherlands), 1996. ISO 14040. Environmental management - life cycle assessment - principles and framework. International Organisation for Standardisation (ISO), 1997. ISO 14041. Environmental management - life cycle assessment – Goal and scope definition and inventory analysis. International Organisation for Standardisation (ISO), 1998. ISO 14042. Environmental management - life cycle assessment – life cycle impact assessment. International Organisation for Standardisation (ISO), 2000. ISO 14043. Environmental management - life cycle assessment – life cycle interpretation. International Organisation for Standardisation (ISO), 2000. ISO TR 14047. Illustrative examples on how to apply ISO 14042 – life cycle assessment – life cycle impact assessment. Draft technical report. International Organisation for Standardisation (ISO), 2001. ISO TR 14048. Data format for LCA data. Draft technical report. International Organisation for Standardisation (ISO), 2001. ISO TR 14049. Illustrative examples on how to apply ISO 14041 – life cycle assessment – goal and scope definition and inventory analysis. Technical report. International Organisation for Standardisation (ISO), 2000. Lindfors, L-G, K. Christiansen, L. Hoffman, Y. Virtanen, V. Juntilla, O-J. Hanssen, A. Rønning, T. Ekval and G. Finnveden. Nordic Guidelines on life cycle assessment (Nord 1995; 20). Copenhagen (Denmark), Nordic Council of Ministers, 1995. Owens, J.W. and S.P. Rhodes. Discussion paper on the feasibility of developing characterisa.tion models for various impact categories. Submitted to ISO TC207/WG4 on behalf of the USA delegation, May 1995. Potting J. and K. Blok. Spatial aspects of life cycle impact assessment. In: Udo de Haes, H.A., A.A. Jensen, W. Klöpffer and L-G. Lindfors (eds.). Integrating impact assessment into LCA. Proceedings of the LCA symposium held at the fourth SETAC-Europe Congress, 11-14 April 1994 at the Free University of Brussels in Belgium. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1994. Potting, J. and K. Blok. Life cycle assessment of four types of floor covering. Journal of Cleaner Production, Vol. 3 (1995), Issue 4, pp201-213. Potting, J. and M. Hauschild. Predicted environmental impact and expected occurrence of actual impact. Part 1: The linear nature of environmental impact from emissions in life-cycle impact assessment. Int. Journal of Life Cycle Assessment, Vil. 2 (1997a), Issue 3, pp171-177, 1997a. Potting, J. and M. Hauschild. Predicted environmental impact and expected occurrence of actual impact. Part 2: Spatial differentiation in life cycle assessment via de site-dependent characterisation of environmental impact from emissions. Int. Journal of Life Cycle Assessment, Vol. 2 (1997b), Issue 4, pp209-216. Potting, J., W. Klöpffer, J. Seppälä, G. Norris and M. Goedkoop. Climate change, stratospheric ozone depletiom, photooxidant formation, acidification and eutrophication. In: H.A. Udo de Haes et al. Life cycle impact assessment: Striving towards best practice. Brussels (Belgium)/Pensacola (Florida; United states of America), SETAC-Press, 2002. SETAC-EWGCRP. Life cycle assessment and conceptually related programmes. Report from the SETAC-Europe Working Group on Conceptually Related Programmes. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1997. Schmidt, A., B.H. Christensen and A.A. Jensen. Environmentally friendly stoves and ovens (environmental project no. 338; in Danish with English summary). Copenhagen (Denmark), Danish Environmental Protection Agency, 1996. Udo de Haes, H.A. (ed.). Towards a methodology for life cycle impact assessment. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1996. Udo de Haes, H.A. and N. Wrisberg (eds.). Life cycle assessment: state-of-the-art and research priorities. Results of LCANET, a concerted action in the environment and climate programme (DGXII). Bayreuth (Germany), Eco-Informa Press, 1997. Udo de Haes, H.A., G. Finnveden, M. Goedkoop, M. Hauschild, E. Hertwich, P. Hofstetter, W. Klöpffer, W. Krewitt, E. Lindeijer, O. Jolliet, R. Mueller-Wenk, S. Olsen, D. Pennington, J. Potting, B. Steen (eds.): Life Cycle Impact Assessment: Striving towards best practice. ISBN 1-880611-54-6, SETAC Press, Pensacola, Florida, 2002. UNEP (2002): Life Cycle Initiative homepage : http://www.uneptie.org/pc/sustain/lcinitiative/home.htm Vigon, B.W., D. Tolle, B.W. Cornaby, H.C. Latham, C.L. Harrison, T.L. Boguski, R.G. Hunt, and J.D. Sellers. Life cycle assessment: Inventory guidelines and principles (EPA/600/R-92/245). Washington D.C. (United States of America), Office of Research and Development of the Environmental Protection Agency, 1993. Weidema, B.P. (ed.). Environmental assessment of products. A textbook on life cycle assessment (3rd edition). Helsinki (Finland), UETP-EEE, 1997. Wenzel, H., Hauschild M.Z. and Alting, L.: Environmental assessment of products. Vol. 1 - Methodology, tools, techniques and case studies, 544 pp. Chapman & Hall, United Kingdom, ISBN 0 412 80800 5, Kluwer Academic Publishers, Hingham, MA. USA., 1997. White, P., B. De Smet, H.A. Udo de Haes, R. Heijungs. LCA back on track, but is it one track or two? LCA news from SETAC-Europe, Vol. 5 (1995), Issue 3, p2-5. Wrisberg, N., Udo de Haes, H.A., Triebswetter, U., Eder, P. and Clift, R. (eds.): Analytical Tools for Environmental Design and Management in a Systems Perspective. The Combined Use of Analytical Tools. Eco-efficiency in industry and science, vol. 10, Kluwer Academic Publishers, Dordrecht, 2002 2 General issues2.1 Introduction Authors: José Potting [6] and Michael Hauschild [7]The text in this Chapter has been modified from Chapter 7 in Potting (2000). 2.1 IntroductionThis Technical Report explores possibilities and limitations, as well as the relevance of spatial differentiation for characterisation of emission related impact categories in present life cycle impact assessment. Only conceptual thoughts were available at the start of the research. These thoughts have in the course of the research evolved into a sound framework. This chapter discusses a number of issues of general relevance in relation to this framework and the following chapters on individual impact categories. 2.2 The basis for spatial differentiationThe effective implementation of control measures in the industrialised countries has by now led to a situation where risk from a single source to its local environment is prevented in most cases. As elaborated in Potting and Hauschild (1997), many environmental problems nowadays are characterised by the fact that the exposure of any receptor is the result of the - long distance transport of - emissions from very many sources. The background exposure of a single receptor thus usually results from a multiplicity of sources. The other way around, a single source often contributes to the exposure of many receptors because of long distance transport. The consequence of the multiple source, multiple receptor character of present environmental problems is that a single source contributes only very little, in many cases marginally to the exposure of its receptors (except for exposures within the first kilometres from the source). The small or marginal contribution from a single source to exposure of its receptors means that the time behaviour of the emission from that source (i.e. whether it is a flux or a pulse) becomes less important. The temporal variation of the contributions from a single source emission will usually namely to a large extent be cancelled out against the high background exposure from all sources together. Exposure of receptors thus shows a relative invariability in time for the contributions of single sources (this does not imply the opposite reasoning, that the temporal variations in the total exposure of receptors are unimportant). If the exposure of receptors shows a relative invariability in time for the contributions of a single but full source, the same of course inherently holds true for the emission per functional unit. The temporal variation of emissions is thus of minor importance in impact assessment in LCA, except for local impacts or extreme situations (like the very slow emissions of landfill). This is an important learning, because the lack of information about the temporal variation of emissions has long been an issue of intensive debate in LCA. The multiple source, multiple receptor character of present environmental problems also provides the possibility to establish site-dependent characterisation factors. The long distance transport of emissions means that one has to look over several hundreds to thousands kilometres to catch most of the impact from a source. The large impact area of an emission makes the precise location of a source of less importance because the dispersion patterns and impact area of neighbouring sources overlap (i.e. show largely the same gradients). Dispersion patterns and impact area will only start to deviate considerably when sources are located at larger distances from each other. This makes it possible to establish site-dependent characterisation factors that with reasonable to good accuracy estimate the impact from a source located in a given region (as distinct from the characterisation factors for a source in another region). The geographical region of emissions is often already provided by inventory analysis in present LCAs. The lack of temporal and spatial information from inventory analysis has always been put forward as the main reason for LCA not being able to take into account spatial differentiation. As already mentioned before, temporal information is only of limited relevance, while the relevant level of spatial information is usually already available from inventory analysis in the present LCA. Spatial differentiation is thus not seriously limited by a lack of spatial and temporal information from the inventory phase in LCA. The multiple source, multiple receptor character holds for most of the present "non-local" environmental problems that are the result of atmospheric emissions of mainly a limited number of substances with relatively long lifetime (stratospheric ozone depletion, global warming, acidification, tropospheric ozone formation, and terrestrial eutrophication). It is less evident for the problem of toxicity with its large and still growing number of potential toxic substances that have relatively short as well as rather long lifetimes and which are emitted sometimes by a limited number of sources only. It is obvious that in the points presented above about marginal contributions, the temporal aspects and the overlapping impact areas are more "valid" for toxic substances emitted by multiple sources as well as for substances with a long lifetime [8] (than for short-lived substances emitted by few sources only). The multiple source, multiple receptor reasoning may not be valid for all impact categories or for all the substances within an impact category. However, it represents a line of thinking that is considered to roughly describe the state of the present environmental situation. This line of thinking has been one of the main pillars of the methodology development and consensus project. The multiple source, multiple receptor reasoning is most obvious for environmental problems related to air emissions. However, problems related to waterborne emissions (like aquatic eutrophication) tend to show similar characteristics (EEA 1998). The full water system through which a waterborne emission disperses, and the background concentrations from a multiplicity of other sources have to be considered to characterise the full impact from a single source. 2.3 Levels of sophistication and uncertainties in impact modellingPotting and Hauschild (1997) argues that more sophistication should only be added to characterisation for the purpose of increasing the resolving power of the impact modelling. This requires the uncertainties introduced by this additional sophistication to be in balance with the gain of information it provides. The levels of sophistication in impact assessment can roughly be seen in two directions: The extent to which the relevant parameters (or environmental mechanisms) in the causality chain are taken into account in the characterisation factors. That is, whether these factors are based on no, some or full modelling of fate, exposure [9] and effect. The extent to which spatial (and temporal) variation is allowed in each parameter of the modelling underlying the characterisation factors. The characterisation factors developed in this technical report are in general sophisticated in both senses. They cover a large range of the relevant parameters in the causality chain, and they allow a high degree of spatial variation. The term "causality chain" is avoided in ISO terminology (ISO 14042 2000), because complex networks are involved rather than linear chains of environmental mechanisms (Udo de Haes et al. 1999). Though often complex and non-linear, there is nevertheless some sequence in the environmental mechanisms that make an emission to have its final effect. Therefore the term "causality chain" was preferred in this report. The causality chain for global warming could for example be (Udo de Haes et al. 1999): emission of CO2 increased radiative forcing climate change (i.e., temperature rise) rise of sea level flooding of land damage to flora, fauna, and human beings (e.g., year of human life lost). The impact to assess can basically be defined anywhere in the causality chain. There is a tendency in LCA to define the impact (or category indicator in ISO-terminology) as far as possible in the causality chain for the given impact category (Braunschweig et al. 1998, Udo de Haes et al. 1999, Goedkoop et al. 1998). The emissions related to global warming are then no longer aggregated on the basis of their increased radiative forcing (as now typical), for example, but modelled up to the years of human life lost as a consequence of (the complex network of changes caused by) a temperature change that is expected to result from increased radiative forcing. Definition of the impacts further on in the causality chain makes them easier to interpret in the subsequent weighting if this facilitates aggregation across impact categories. Goedkoop et al. (1998) and Hofstetter (1998) therefore express all impact categories in similar damages to humans and ecosystems and resource depletion for example. Such aggregation is largely based on natural sciences and this limits the "uncertainty" (or avoids the lack of credibility) from a value-based aggregation. On the other hand, uncertainties can rapidly become larger if the impact is defined that far in the causality chain where little is known about environmental mechanisms. The site dependent factors in this technical report are defined at different levels in the causality chains. Factors for acidification, terrestrial eutrophication, photochemical ozone and noise are modelled up to and including evaluation of threshold exceedance. The factors for aquatic eutrophication, human and ecotoxicity do not cover an evaluation of threshold exceedance and basically reflect a weighed exposure assessment. The chosen level in each impact category reflect the state-of-the-art modelling for the given impact category (see also next section), and a fair balance is aimed for between gain of information and accuracy in modelling further up in the causality chain. An interesting learning from this technical report is, that the spatial differences become more pronounced, up to a factor thousand between lowest and highest ranking, when indicators are defined further in the causality chain (see also Potting 2000). This technical report shows that spatial differentiation is feasible and provides reasonable to considerable resolving power in LCA. The credibility of LCA results has been and still is discussed intensively among practitioners and users of LCA. A main reason for debate was the poor accordance between the expected occurrence of actual impact and the impact predicted by present life cycle impact assessment (see also Chapter 1). The increased resolving power obtained by spatial differentiation reflects the expected variation in occurrence of actual impact. Spatial differentiation therefore improves the credibility and therewith relevance of the modelling in present life cycle impact assessment. 2.4 The mathematical framework for spatial differentiation The mathematical framework used in Chapter 3, 4 and 5, 6 and 7 differs from the one initially described by Potting and Hauschild (1997a,b) and used in Chapter 8 in that it is more sophisticated. An emission does usually not disperse equally, but will show a spatially differentiated pattern of distribution to its receptors as a result of the processes of environmental fate and transport. The contribution from an emission to exposure increase must preferably be calculated individually for each receptor. Starting from the receptor side, the total exposure of each receptor usually results from the emissions of very many sources. There is also a spatial variation in background exposures among receptors. The background exposure of receptors should preferably also be quantified for each receptor [10]. To assess the impact from a source on all its receptors, ideally, first the impact has to be calculated for each receptor individually, and next these contributions have to be aggregated over all receptors (the procedure is exemplified in Chapter 4 about terrestrial eutrophication):
Formula 2.1 represents a sophisticated approach by considering one source to expose multiple receptors. Such sophisticated approach was until recently regarded by many practitioners impossible in LCA. It would put an infeasible additional data demand on life cycle inventory with regard to the spatial characteristics of the source, and on the impact assessment phase with regard to the background concentrations and sensitivities of receptors. Therefore, initially a simpler approach was proposed by Potting and Hauschild (1997a,b). This approach was based on the assumption that a few standard sources and a few standard target systems or receiving environments can be distinguished. The impact of each source type would sufficiently be described by considering it to have one standard receiving environment (represented by an average receptor instead of by a multiplicity of receptors as in Formula 2.1). Spatial differentiation could be obtained by dividing the world into a limited number of standard source types and standard receiving environments. The impact of a source type on the relevant standard receiving environment could then be characterised by a site factor, possibly distinguished in a fate factor and a factor for the receiving environment (the target factor), which modifies the impact resulting from the conventional site-generic framework:
The fundamental difference between Formula 2.1 and 2.2 is the type of modelling underlying each. Formula 2.2 assumes one source to expose one standard environment existing of one average receptor, whereas Formula 2.1 has a more sophisticated approach by considering one source to expose multiple receptors. It is an important learning of this technical report that the more sophisticated modelling can be used quite well to establish characterisation factors. A number of impact categories is described in models that can be used to establish characterisation factors according to the sophisticated approach. Such sophisticated modelling does not exist for characterisation categories as toxicity or eutrophication via water. However, this expresses the limitations in the relevant field of science rather than limits posed by LCA itself. Spatially differentiated characterisation factors for these characterisation categories can be established according to the more simple approach. Inventory analysis usually provides the needed spatial information, and models for the relevant characterisation categories are often available that facilitates spatial differentiation in LCA. The use of both the sophisticated and simpler approach can be exemplified with the characterisation factors in this technical report. The factors for acidification, terrestrial and aquatic eutrophication and photochemical ozone are established according to Formula 2.1, the factors for noise and ecotoxicity are established according to Formula 2.2, whereas the factors for human exposure increase are a mixture of both frameworks. The hybrid approach for human exposure is chosen to adequately account for exposure increase local to the source and to respect the requirements of feasibility in LCA at the same time (see also Chapter 7). 2.5 Application of site-dependent characterisation factorsApplication of the site-dependent characterisation factors requires data additional to current life cycle impact assessment. This is for the majority of impact categories restricted to the geographical region where an emission takes place. The requirement of additional data is often put forward as an objection against spatial differentiation in impact assessment in LCA. However, the geographical region where an emission takes place is in general already provided by current life cycle inventory analysis or available from the scope definition of the system, since this data is needed to calculate the interventions from transport. More difficult to obtain in LCA is source height and population density near the source as needed for human toxicity (see Chapter 7). Chapter 7 therefore suggests following a default approach unless there are good reasons to deviate. A practical way to avoid needless data gathering is to first perform a site-generic assessment (i.e. without spatial differentiation), and then to retrospectively apply the site-dependent characterisation factors to the processes dominating the considered impact category. In this way, it will probably become clear soon what the dominating processes are for which additional data is required. Such procedure is described in more detail in the guidance document from Hauschild and Potting (2003). These references also provide site-generic characterisation factors (plus their standard deviation). The site-generic factor is based on the spatially differentiated ones. The site-generic factors can be based on the European average, or follow a worst-case approach by taking the highest characterisation factor for the European area. Potting and Hauschild (2003) choose to work with the European average. The level of sophistication in spatial variation is not fixed in LCA. As discussed in By Potting et al. (1999), LCA studies differ in the extent to which data for spatial differentiation are available. Some studies provide a lot of information, whereas other studies cover mainly processes at unknown locations and unknown calendar performance time. Site-generic factors can be used where spatial differentiation is not possible or desired. If studies provide enough data on the other hand, the use of site-dependent characterisation factors can be replaced by a site-specific assessment with the same models as underlying the site-dependent factor. Such site-specific modelling in a life cycle approach is for instance applied in ExternE (Krewit et al. 1998, Rabl et al. 1996). As discussed in Section 2.2, the large impact area of an emission leads to the effect that the dispersion patterns and impact area will only start to deviate considerably when sources are located at larger distances from each other. In a typical LCA, the precise location of a source is therefore of less importance to quantify total impact from a process. However, some applications might need the detailed information from a site-specific impact assessment (see also Section 2.6). A "site-dependent" assessment thus has a level of detail somewhere in between "site-specific" and "site-generic" assessment (the two extremes stretching up a continuum):
All levels of detail may be applied in a particular LCA, though the results at one level should be consistent with the results at another. Each level should preferably take its basis in models similar to the ones underlying the other levels, and the results at each level should preferably point in similar direction as the results at the other levels. This allows a flexible shifting from one to another level of detail, and warrants the credibility of doing so. The site-dependent characterisation factors in this technical report can be used to arrive at site-generic factors, or be replaced by site-specific impact assessment based on the same underlying models. This improves the relevance of the established characterisation factors. 2.6 LCA in relation to RA and EIAIn the discussion about spatial differentiation, that is indirectly relevant for this Technical Report, LCA is typically positioned as distinct from risk assessment (RA) and environmental impact assessment (EIA) due to the apparent misconception that these tools are always local in their perspective. RA and EIA cover a multiplicity of analytical tools that can be quite different in their object and designs. Both EIA and RA can refer to a study of consequences for the environment at varying spatial and temporal scales. Some of the concerns addressed may be local and others regional, possibly all within the same study. Though integrated assessments were incidentally already performed earlier, RA and EIA increasingly tend to involve studies that cover multiple sources. Analytical tools used in EIA and RA can be generic in the way they consider source and receptor characteristics (as for examples EUSES and CalTOX), but typically they operate with a high degree of spatial and temporal differentiation in source and receptor characteristics. The detail in source characteristics is necessary because concentration increases and probabilities of threshold exceedance, particularly close to the source, are highly influenced by the height of release and the temporal variation in the emission. Those characteristics become less dominant at longer distances (see also Section 2.2). The common element in all types of EIA and RA is that they assess risks, often by employing a threshold or quantification of distance from the threshold. As discussed by Potting et al. (1997), LCA theoretically has no limitations to take into account spatial or temporal information and neither to perform threshold evaluation. The other way round, generic EIA and RA also do not take into account spatial and temporal differentiation. EIA and RA can also refrain from threshold evaluation. The differences between LCA, and RA and EIA are thus more given by current practice than based on fundamental incompatibilities. Basically, spatially differentiated RA and EIA approaches, including quantification of threshold exceedance, are used to establish the site-dependent characterisation factors in this technical report. Whereas EIA and RA can be both local and regional in their perspective, LCA is poor in accounting for the contribution of a source to local impact in those situations where concentration increases and the subsequent probability of threshold exceedance are largely determined by that source alone. Concentration increases and probabilities of threshold exceedance are particularly within the first hundreds to thousands meters highly influenced by source characteristics as height of release and temporal variation in the emission. In a typical LCA, this information is only limited available and it will often complicate the analysis to gather additional data. Local impact has often a relatively small share in the full impact of a source. Where the local share is not that small, it is often not just one source in itself that creates ambient concentrations above threshold (see also Section 2.2). The effective implementation of measures to regulate the main sources in many industrialised countries has led to a situation in which risks from those sources on their local environment are usually under control. This will of course not always be the case, and particularly not in developing countries where environmental policy is still limited. However, EIA or RA rather than LCA will be the obvious tool to support decision-making to bring under control the impact from sources on their local environment.
Figure 2.1. The amount of substance(s) emitted from a hypothetical product system and the share received by different environments after emission dispersion. Typical LCA is optimised in another direction than EIA and RA. LCA is an obvious tool to support policy-making directed towards prevention and control of environmental impact resulting from a multiplicity of sources, and where each source impacts on a multiplicity of receptors. A product system consists of a collection of processes (or sources) that are functionally interconnected by the output of one process being the input of another one (see Figure 2.1). A product system functions within a larger economical system of also interconnected processes to which the product system only contributes marginally (in the same way as it only contributes marginally to environmental impact). LCA provides the possibility to prioritise in the sub-system of a product, those processes that make the largest contribution to environmental impact taking into account feedback to other processes and other environmental problems within that product system. It is a tool for source comparison in the context of specific products. The ability to identify the consequences from the changes in one process for other processes, or from the changes in one impact category for other categories is the strength of LCA above EIA and RA. EIA and RA are each optimised in their own directions. They supplement each other and may be used in combination to make solid and informed decision-making. White et al. (1995) provide an interesting overview and discussion of the possibilities and limitations of the different tools in overall environmental management. 2.7 Threshold exceedance in LCAWhere possible, the characterisation factors in this Technical Report take into account the exceedance of thresholds. The discussion whether to perform evaluation of threshold exceedance in LCA is topical for already quite some years now. Unfortunately, the discussion seems to get stuck in a controversy about "less is better" versus "only above threshold" (see also Potting et al. 1999). The impact assessment phase in LCA is relatively young and emerged from the wish to simplify the large amount of data from inventory analysis by aggregation into a manageable amount of impact data. Characterisation factors for most impact categories initially allowed rather simple equivalency assessment on the basis of intrinsic substance characteristics like the potential to release hydrogen ions (acidification assessment) or toxic effect-levels (toxicity assessment) [11]. Assessment of threshold exceedance (i.e. PEC/PNEC 1) was not performed since the available data did not allow such evaluation. Threshold information, usually in the form of a no-effect-level, was used in toxicity assessment only to express the emission of a given substance as a dilution volume of the receiving environment. The basis of equivalency was thus taken in the toxicity potential of each substance. The impact from an emission quantity equal to the no-effect-level was given an impact of one [12], and the impact from any deviating quantity as the ratio of the emission quantity divided by the no-effect-level. Present toxicity factors now also cover fate and exposure modelling, but aggregation of the calculated exposure increases from different substances is still often based on no-effect-levels (PEC/PNEC). Evaluation of threshold exceedance is typically not covered, however, and neither possible with the modelling underlying those toxicity factors. Though evaluation of threshold exceedance was initially not performed due to lack of data, it has meanwhile turned for many practitioners into a principle in itself that is justified by the reasoning that "less pollution is better". An important understanding from the present technical report work is that limiting the threshold discussion into "less pollution is better" versus "only above threshold" is too simple and does not respect the analytical potential of LCA. An example from acidification may clarify this. Let's assume that we have three ecosystems receiving similar quantities of acidifying substance from our functional unit (the contribution of the functional unit is marginal compared to the background load on each ecosystem). Let's further assume that these ecosystems have a priori equal tolerance for acidifying loading (i.e. similar critical loads), but that they are at different levels of background loading:
We want to prioritise for which ecosystem we like to reduce the full acidifying load (background load + marginal increase from our functional unit). The sensitivity of ecosystems, as expressed by their critical loads, does not play any role in yet typical "less is better" acidification factors. However, Heijungs and Huijbregts (1998) propose to establish acidification factors similar as the ones Guinée et al. (1996) established for human toxicity. Such factors express the contribution to the acidifying load of an ecosystem per unit of emission, divided by the critical load for that ecosystem (ΔPAL/CL) [13]. The a priori tolerance of the ecosystems is taken into account, but the background loading does not play a role here. Since all ecosystems have the same critical load, this would result in a similar acidification impact from our functional unit to each of these ecosystems. The practical implication is that the Scottish ecosystem exposed around the critical load is regarded equally important as the German ecosystem exposed far above the critical load (and thus difficult to rescue), and equally important as the Scandinavian ecosystem exposed far below the critical load (and thus hardly in danger). Whether these ecosystems are exposed far below or at or far above their critical loads, they are all characterised as being equally vulnerable in such a "less is better" approach. That might be or might not be a justified choice, but it is at least important to realise that such choice is made by following a "less is better" approach. An "only above threshold" approach as proposed by Pleijel et al. (not published) and followed by Lindfors et al. (1998) typically assess impact only for those ecosystems already exposed above their critical loads (the impact of ecosystems exposed below their critical load is assessed zero). The a priori tolerance of ecosystems and their background loading are taken into account to distinguish ecosystems exposed below from those exposed above their critical load. After the ecosystems are classified, however, the background loading and critical loads do not longer play a role. Whether ecosystems are exposed far above or just above their critical loads, they are all characterised as being equally vulnerable in this approach. Equal priority would be given here to the German and Scottish ecosystem (assumed that the latter is just above its critical load). The characterisation factors of Lindfors et al. (1998) can be adjusted by multiplying the change in exceedance with a severity factor given by the existing background load divided by the critical load for the given ecosystem (Posch 1998). Such sophisticated way to account for background load and a priori tolerance of ecosystems is presaged by Pleijel et al. (1997). The acidification factors in Chapter 4 can be typified as following an "only around threshold approach). This approach takes into account differences between ecosystem in background levels and their a priori tolerance for acidifying loading by basing the characterisation factors on the slope of the curvilinear dose-effect curve. This curve is defined by the critical loads of all ecosystems to which one source contributes. The practical implication is that ecosystems with depositions far above or below their critical loads are not expressed in the characterisation factor. In the above example, this approach would prioritise the Scottish ecosystem (just above its critical load and therefore easy to rescue) rather than the Scandinavian ecosystem or the German ecosystem. In the above example, the value choices underlying the different approaches were illustrated with help of single ecosystems. The emission per functional unit from a given process does usually not contribute to only one ecosystem but to a multiplicity of ecosystems, each with their own critical loads and background loads. Assessing the impact of a process to acidification thus basically involves adding together the dissimilar contributions from one process to a multiplicity of heterogeneous ecosystems (see Formula 2.1 in Section 2.1). Several other possibilities are available to deal with both differences in a priori tolerance for acidifying loading and background loading. The choice for any approach remains in the end a matter of how to value ecosystems with different critical loads as well as different background loads. Some approaches seem more obvious than others. A systematic, quantitative comparison of the actual consequences on the final characterisation factors of each approach would be a helpful input to the discussion about how to deal with thresholds in LCA. The example from acidification applies to all impact categories, and theoretically also to human and eco-toxicity assessment. Although now also EIA and RA have made a start in integrated assessments of human toxicity, however, these models will in practice remain to be less sophisticated than for other impact categories due to the complexity of toxicity assessment. The complexity of toxicity assessment arises from the large number of toxic substances, whereas impact categories as global warming and acidification are determined by a limited number of substances and specific impact mechanisms. 2.8 Site-dependent normalisationNormalisation is an optional step in impact assessment that calculates the assessed impact relative to a selected reference value. This reference value typically refers to impact of the total emission of a reference area (possibly on a per capita basis) (ISO 14042 2000). Different normalisation methods exist. Lindeijer et al. (1996) provides an interesting review in which they discuss some basic issues in normalisation methodology. Thinking about a spatially differentiated normalisation has put another fundamental issue forward. This issue has probably not been discussed before in the literature, but addresses the question whether normalisation should be based on marginal or average impacts. This question is relevant for all normalisation methodologies (and is also not exclusively related to spatial differentiation), but will here be illustrated with the methodology proposed by Wenzel et al. (1997) and Hauschild and Wenzel (1998). Impact categories relate to different spatial scales. Wenzel et al. (1997) and Hauschild and Wenzel (1998) propose to base normalisation on the spatial scale typical for each impact category (that is, the normalisation factor should reflect the level of impact experienced in that region). The global impact level is taken for global impacts as ozone depletion, while for practical reasons the impact level in Denmark is taken for the non-global categories (though the spatial scale of these impact categories can be smaller as well as larger than Denmark). The global impacts will obviously be larger than the regional ones, since more people and larger economic activity are involved. In order to establish a "common scale", the impact in the region defined by the spatial scale of the given impact is divided by the population in that area. This results in the normalisation factors being the impact per inhabitant of Denmark for the non-global categories, and the impact per global citizen for global impacts.
The methodology of Wenzel et al. (1997) and Hauschild and Wenzel (1998) is unique in their use of different reference areas across impact categories. Other methodologies may differ in the chosen reference area, but each methodology usually operates with the same reference area for all impact categories. It is here that normalisation on the basis of marginal or average methodology comes most clearly in sight. The question whether LCA should take a marginal or an average approach was first discussed in the context of inventory analysis. Though being topical for several years, the discussion seems to move to a consensus. Average methodology is seen as connected to LCAs that describe an existing or historical situation (e.g. for environmental reporting or declaration). Marginal methodology is required for LCA's that analyse the consequences from changes in a product system. (Weidema 1998) A marginal normalisation methodology would consider the impact over the full European area that is affected by all emissions of a society, compared to the impact over that area without the emissions from that society (but against the unchanged background of the emissions of other regions). Table 3.2 and 3.3 provide such normalisation factors per country for acidification (see the column "unprotected ecosystem by region"). For acidification, this would for instance mean the impact in the total European area caused by emissions from Denmark and all other regions, compared to the impact in the total European area without the Danish emissions (but with the emissions of all other regions). This increase of impact caused by all Danish emissions divided by the total population in Denmark gives the impact caused per capita [14]: The marginal normalisation factor for the European area could be taken as the average of the marginal normalisation factors for all regions weighed by the emission of each region. Another possibility is to take the average normalisation factor for Europe. A normalisation methodology consistent with Wenzel et al. (1997) and Hauschild and Wenzel (1998) would probably follow an average approach by taking the total impact in an area divided by the population in that same area. The impact area is "defined" by the total area over which an impact more or less clearly extends [15]. For acidification, this would mean for instance the total acidified area in Europe divided by total European population:
It does not make sense to calculate an average normalisation factor for an area smaller than the European area (that is for separate countries). The average normalisation factor for Denmark, for instance, would be the area affected within Denmark divided by Danish population. However, the total impact within Denmark is caused by the emission of all regions and not by the emission from Denmark alone. The meaning of such an average Danish normalisation factor is therefore not very transparant. An average approach as from Wenzel et al. (1997) and Hauschild and Wenzel (1998) puts the emphasis on the impact itself, rather than on the society causing this impact. A possible consequence of such an approach could be to apply several normalisation factors for non-global impact categories. The area where a process takes place gives the relevant normalisation factor. For example, a "European" normalisation factor should be used for impact from acidifying emissions in Europe, whereas the impact from emissions in North America should be normalised with a "North American" factor. The number of required normalisation factors would however become rather large for impact categories that relate to a much smaller than the regional scale, which is unpractical. Furthermore, the set of normalisation factors required for a product system is no longer similar with the set for its alternative. This makes the results of this normalisation step relatively difficult to interpret. Normalisation methodology based on a marginal approach could also choose to operate with several normalisation factors per impact category. However, it is more obvious to work with only one normalisation factor, typically based on the impact from the region where the product is marketed. A relevant question is then whether such normalisation factor should be based on the economic activity of the selected region, or on its consumption. The factors in Table 3.2 and 3.3 relate in this context to the first option, but may be seen, with some caution, as the best estimate of the second option for the time being. Normalisation factors based on consumption should account for the net import and export of impact by the selected region. (Blonk et al. 1997, Lindeijer et al. 1996) As Lindeijer et al. (1996) emphasised, the choice for one or another normalisation method depends amongst others on the following valuation step. It goes beyond the scope of this technical report to further enter into the subject of site-dependent normalisation in the context of valuation, but clearly a better understanding of the aims of valuation, and normalisation in relation to spatial differentiation is needed. The site-dependent normalisation factors for the guidance document follow the method as developed by Wenzel et al. (1998). 2.9 Temporal differentiation in LCATemporal differentiation is an important and intensively discussed issue in LCA. The discussion has focused for a long time predominantly on the lacking time dimension in the inventory data (i.e. whether it is a flux or a pulse). As argued in Section 2.2, the temporal variation of the small (or even marginal) contributions from a single source emission to exposure of their multiple receptors will to a large extent be cancelled out against the very high background exposure from all sources together. The multiple source, multiple receptor perspective of present environmental problems make the lacking time dimension in inventory data to a less dramatic problem (see also Section 2.2). The calendar time to which the different processes in a product system relate is a more important issue when it comes to impact assessment in LCA. The calendar time of a process determines the (estimated) total economic activity with all its emissions being responsible for the total environmenta.l load causing an impact to which background that process adds. The background situation can be rather different between calendar times (and thus between different processes in a product system as discussed below). This could result in rather different factors related to these different calendar times (since the factors do account for the - differing - background situation). The photochemical ozone creation potential from an emission of a volatile organic compound, may be different in 1990 and 2010 due to considerable differences in the background concentration levels of nitrogen oxides posed by the total economy in those years. For instance, the acidification factors from Chapter 3, which are calculated for the reference years 1990 and 2010, show that the difference between different calendar times can be notable. A product system can easily cover a time frame of several decades depending on time-of-use of the product and the time needed for each subsequent process in the product system. For example, a linoleum floor covering will on average first be discarded 15 years after it has been bought (Potting and Blok, 1995). The characterisation factors used to assess the impact from a given process should relate to the calendar time in which that process takes place (a time-dependent characterisation factor). As a matter of fact, also processes themselves can cover a time frame of several years to several decades (and indeed for landfill processes centuries of millennia). A specific type of linoleum will be produced over a certain time interval before the type is taken from the market. This determines the time or calendar interval over which that type is marketed, used, and disposed. Basically, the characterisation factor used to assess the impact from given processes should thus not relate to the calendar time, but to the calendar interval in which a given process takes place. Similar to spatial differentiation, the level of temporal differentiation can be seen as placed on a continuum stretched up by the two extremes "time-specific" and "time-generic". Time-dependent" assessment thus has a level of detail somewhere between those extremes:
An interesting question and subject in need of further research is the level of temporal differentiation needed in LCA. Without going into details, trend analyses show emission projections to be relatively stable over a couple of years (Jol and Kielland 1997). This suggests a time-dependent assessment based on time-intervals of several years to be adequate for LCA. However, the issue was not further elaborated in the work for this Technical Report. 2.10 ReferencesBlonk, H., M. Lafleur, R. Spriensma, S. Stevens, M. Goedkoop, A. Agterberg, B. van Engelenburg, and K. Blok. Three reference levels for normalisation in LCA: Dutch territory 1993/1994, Dutch final consumption 1993/1994, West European territory in the beginning nineties (in Dutch). The Hague (the Netherlands), Ministry of Traffic and Water affairs and Ministry of Housing, Spatial Planning and Environ.ment (VROM), 1997. Braunschweig, A., R. Förster, P. Hofstetter and R. Müller-Wenk. Developments in LCA valuation (ISBN 3-906502-31-7). St. Gallen (Switzerland), Institut für Wirtschaft und Ökologie, 1996. EEA. Europe's environment: The second assessment. Copenhagen (Denmark), European Environmental Agency (EEA), 1998. Goedkoop, M., P. Hofstetter, R. Müller-Wenk, R. Spriensma. The Eco-Indicator 98 explained. Int. Journal of Life Cycle Assessment, Volume 3 (1998), Issue 6, pp352-360. Guinée, J., R. Heijungs, L. Van Oers, D. Van de Meent, T. Vermeire, M. Rikken. LCA impact assessment of toxic releases. Generic modelling of fate, exposure and effect for ecosystems and human beings with data for 100 chemicals. Leiden (the Netherlands), Centre of Environmental Science of Leiden University, 1996. Hauschild, M. and J. Potting. Spatial differentiation in life cycle impact assessment – the EDIP2003 methodology. Guideline from the Danish Environmental Protection Agency, Copenhagen, 2003. Hauschild, M.Z. and Wenzel, H.: Environmental assessment of products. Vol. 2 - Scientific background, 565 pp. Chapman & Hall, United Kingdom, 1998, Kluwer Academic Publishers, Hingham, MA. USA. ISBN 0412 80810 2 Heijungs, R. and M. Huijbregts. Threshold-based life cycle impact assessment and marginal change: incompatible? (CML-SSP Working paper 99.002). Leiden (the Netherlands), Centre of Environmental Science of Leiden University, 1999. Heijungs and Wegener Sleeswijk. The structure of impact assessment: Mutually independent dimensions as a function of modifiers. Int. Journal of Life Cycle Assessment, Vol. 4 (1999), Issue 1, pp2-3. Hofstetter, P. Perspectives in life cycle impact assessment. A structured approach to combine models of the technosphere, ecosphere and valuesphere. Boston (United States of America), Kluwer Academic Publishers, 1998. ISO 14042. Environmental management - life cycle assessment – life cycle impact assessment. International Organisation for Standardisation (ISO), 2000. Jol, A. and G. Kielland. Air pollution in Europe 1997. Copenhagen (Denmark), European Environmental Agency (EEA), 1996. Krewit, W., P. Mayerhofer, A. Friedrich, A. Trukenmüller, and R. Friedrich. Application of the impact pathway analysis in the context of LCA. Int. Journal of Life Cycle Assessment, Vol. 3 (1998), Issue 2, pp86-94. Lindeijer, E., J. Assies, M. Bovy, A. Braunschweig, G. Finnveden, R. Förster, R. Heijungs, P. Hofstetter, A-M Jørgensen, R. Müller-Wenk, B. Stahl,, H.A. Udo de Haes, R. Walz, B.P. Weidema, B. Wilson and N. Wrisberg. Normalisation and valuation. In: Udo de Haes, H.A. (ed.). Towards a methodology for life cycle impact assessment. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1996. Lindfors, LG., M. Alkemark, C. Oscarsson and C. Spännar. A manual for the calculation of ecoprofiles intended for third party certified environmental product performance declarations. Stockholm (Sweden), Swedish Environmental Institute (IVL), 1998. Pleijel, K., J. Altenstedt, H. Pleijel, G. Lövblad, P. Grennfelt, L. Zetterberg, J. Fejes and L-G. Lindfors. A tentative methodology for the calculation of global and regional impact indicators in type-III ecolabels used in Swedish case studies. Stockholm (Sweden), Swedish Environmental Institute (IVL), not published. Potting, J. and K. Blok. Life cycle assessment of four types of floor covering. Journal of Cleaner Production, Vol.3 (1995), Issue 4, pp201-213. Potting, J. and M. Hauschild. Predicted environmental impact and expected occurrence of actual impact. Part 1: The linear nature of environmental impact from emissions in life-cycle impact assessment. Int. Journal of Life Cycle Assessment, Vol. 2 (1997a), Issue 3, pp171-177. Potting, J. and M. Hauschild. Predicted environmental impact and expected occurrence of actual impact. Part 2: Spatial differentiation in life cycle assessment via de site-dependent characterisation of environmental impact from emissions. Int. Journal of Life Cycle Assessment, Vol. 2 (1997b), Issue 4, pp209-216. Potting, J. and M. Hauschild. Levels of sophistication in life cycle impact assessment of acidification. Points of discussion additional to the presentation. In: Bare, J.C., H.A. Udo de Haes and D.W. Pennington (eds.). International workshop on life cycle impact assessment sophistication. Bayreuth (Germany), Eco-Informa Press, in press. Rabl, A. and J.V. Spadaro. Estimates of real damage from air pollution: Site dependency and simple impact indices for LCA. Contribution to the 8th annual meeting of SETAC-Europe, 14-18 April 1998 in Bourdeaux, France. Paris (France), Ecole de Mines de Paris, 1996. Udo de Haes, H.A. (ed.). Towards a methodology for life cycle impact assessment. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1996. Udo de Haes, H.A. and G. Huppes. Positioning of LCA in relation to other environmental decision support tools. In: Udo de Haes, H.A., A.A. Jensen, W. Klöpffer and L-G. Lindfors (eds). Integrating impact assessment into LCA.Proceedings of the LCA symposium held at the fourth SETAC-Europe Congress, 11-14 April 1994 at the Free University of Brussels in Belgium. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1994. Udo de Haes, H.A., O. Jolliet, G. Finnveden, M. Hauschild, W. Krewit and R. Mueller-Wenk (eds.). Best available practice regarding impact categories and category indicators in life cycle impact assessment. Background document for the second working group on life cycle impact assessment of SETAC-Europe. Part 1: Int. Journal of Life Cycle Assessment, Vol. 4 (1999), Issue 2, pp66-74. Part 2: Int. Journal of Life Cycle Assessment, Vol. 4 (1999), Issue 3, pp167-174. Wegener Sleeswijk, A. Regionalisation and fate analysis in LCA. Their relationship and implications (abstract). In: Interfaces in Environmental chemistry and toxicology from the global to the molecular level. Abstract book for the 8th annual meeting of SETAC-Europe, 14-18 April 1998 in Bordeaux (France). Brussels (Belgium), Society of Environmental Toxicology and Chemistry, 1998. Weidema, B. Application typologies for life cycle assessment. Int. Journal of Life Cycle Assessment, Volume 3 (1998), Issue 4, pp237-240. Wenzel, H., Hauschild M.Z. and Alting, L.: Environmental assessment of products. Vol. 1 - Methodology, tools, techniques and case studies, 544 pp. Chapman & Hall, United Kingdom, 1997, Kluwer Academic Publishers, Hingham, MA. USA. ISBN 0 412 80800 5. White, P., B. De Smet, J.W. Owens, P Hindle. Environmental management in an international consumer goods company. Resources, conservation and recycling, Vol. 1995, Issue 14, p171-184. 3 Acidification3.1 Introduction Authors: José Potting [16] Wolfgang Schöpp [17] Kornelis Blok [18] and Michael Hauschild [19] This Chapter is with minor modifications reproduced from an article by the same authors in the Journal for Industrial Ecology. The article is published under the title "Site-dependent life-cycle assessment of acidification" in the Journal for Industrial Ecology Vol. 2 (1998), Issue 2, p63-87. 3.1 IntroductionIn life cycle assessment (LCA) studies, generally no attention is paid to the site where an emission is released. This lack of spatial differentiation affects the relevance of the assessed impact, as was clearly demonstrated by Potting and Blok (1994, 1995). An example regarding acidification may clarify this deficiency. Copper ore contains sulphur that is released as sulphur dioxide (SO2) during the concentration of copper from the ore. The production of 1 kilogram (kg) copper is accompanied by a release of a similar amount of sulphur dioxide, of which 90% on average is captured by emission reducing measures (Potting and Blok 1993). Primary copper production takes place in Albania, Belgium and Finland. The acidifying impact per kilogram of copper is calculated to be the same for all countries if the sensitivities of the areas of deposition are not taken into account. However, sulphur emitted from Albanian copper production deposits on the highly insensitive (calcareous) areas in south Europe, while the Finnish emission deposits on the highly sensitive surrounding regions. The carrying capacity of most of the moderately sensitive West European areas is already exceeded given the lively economic activity in these regions. As a result, sulphur emitted from Belgium copper production has only moderate additional impact. Owens (1997) provides a good review of the constraints imposed on life cycle impact assessment (LCIA) by the lack of spatial and temporal differentiation in life cycle inventory (LCI). Owens is not optimistic about the possibilities of improving LCIA's accuracy in predicting non-global impacts. Other LCIA experts, however, expect that the relevance of LCIA can be enhanced considerably by the introduction into the assessment process of a few site-factors that indicate the susceptibility of the receiving areas to an impact (Potting and Blok 1994, Potting and Hauschild 1997a,b, Udo de Haes 1996, Wenzel et al. 1997). This chapter describes the framework we developed to derive such site-factors for regional impacts, and presents our results from applying this framework in the already existing RAINS model to calculate site-factors for acidification. First, some relevant principles and limitations of LCI and LCIA are outlined. This chapter primarily addresses LCA practitioners, who in general will not be familiar with the RAINS (Regional Air Pollution Information and Simulation) model and its underlying ideas and concepts. Furthermore, the model has recently adapted some important changes in response to rapid scientific developments in the field of the critical load concept. Therefore, a review is provided about the relevant parts of RAINS: emission estimates, dispersion and deposition, and the critical load concept. Results are presented and extensively discussed. Finally some main conclusions are drawn. This chapter is with minor modifications reproduced from an article by the same authors (Potting et al. 1998), which has inspired similar research activities elsewhere. These newer developments are discussed in Chapter 4. 3.2 Typical life cycle impact assessmentLife cycle inventory (LCI) quantifies the emissions per functional unit for each process in the life cycle of a product. The life cycle of an arbitrary product easily covers a multiplicity of processes, and each process may relate to a multiplicity of production sites. This makes collection of actual data for each process a time-consuming (and sometimes even impossible) activity. A regular LCI will therefore often use emission factors to approximate the emission quantities from the actual processes. An emission factor gives the emission quantity for a given substance (in grams) per unit of output (in kg) from a typical process. Spatial and temporal characteristics, and full source strength of the processes thus modelled, are lost in regular inventory analysis (and are often disregarded for the processes underlying the emission factors). To derive the emission quantity per functional unit (f.u.), the amount of process output needed for one functional unit is multiplied with the emission factor:
The result of LCI is a large table that lists the emission quantities per process for each substance. All emission quantities of a given substance are summed up along the life cycle, and are aggregated in the impact assessment phase (LCIA) with the summed emissions of other substances contributing to the same impact. Aggregation is based on equivalency assessment where the emitted quantity of a given substance is multiplied with an equivalency factor that relates this emission to the equivalent emission quantity of a reference substance: Table 3.1. Different sets of Characterisation or Equivalency Factors (Heijungs et al. 1992, Lindfors et al. 1995, Wenzel et al. 1997) and Site Factors (Hauschild and Wenzel 1997) for characterisation of acidification in life cycle assessment.
Several sets of characterisation or equivalency factors for acidification have been proposed (see Table 3.1). All these sets are based on the number of hydrogen ions that can theoretically be released from the substance (expressed in grams SO2-equivalents). The factors from Hauschild and Wenzel (1997) and Lindfors et al. (1995) distinguish roughly between different types of receiving areas but disregard emission dispersion and subsequent deposition. A more sophisticated way to deal with the current lack of spatial differentiation in LCA was proposed by Potting and Blok (1994) and Potting and Hauschild (1997a,b) in a site-dependent approach. After an acidifying substance is emitted, it is dispersed and deposited. Deposition increase together with background deposition may exceed the carrying capacity of the receiving ecosystem and result in impact. Each link in this cause/effect chain can be characterised by a set of descriptors that for acidification are specified by the geographical site where the emission takes place. This geographical site can be characterised by a site-factor that modifies the equivalence factor such that it relates the emission to the impact on its deposition areas: The only additional data needed from inventory analysis to apply Formula 3.4 is the geographical site of emission. This information is in most cases already provided by current LCI, because it is needed, for instance, to calculate the emissions from transport. 3.3 The RAINS modelRelating the site of emissions to the impact on its deposition areas is one of the key elements in the RAINS model. This model (version 7.2) has therefore been used to establish site-factors for acidification. The model was developed by the International Institute for Applied System Analysis (IIASA) in Laxenburg, Austria. RAINS is an integrated assessment model that combines information on regional emission levels with information on long-range atmospheric transport in order to estimate patterns of deposition and concentration for comparison with critical loads and thresholds for acidification, eutrophication and tropospheric ozone formation. A detailed description of the model is provided by Alcamo et al. (1990) and Amann et al. (1995). A short description of the parts of RAINS that are relevant for this chapter is given below. See Chapter 4 for a step by step illustration of the model. 3.4 Emission estimatesTypical distances between an emission source and its locations of deposition are easily several hundreds of kilometres. The geographical scale of analysis, accordingly, has to be large in order to cover most of the impact from an emission source. The RAINS model focuses therefore on the regional dimension (rather than on the local dimension) of the assessed environmental impact. On a regional scale (between 100 to 4,000 km), emissions of sulphur dioxide, nitrogen oxide and ammonia are the principal contributors to acidification. (Alcamo et al. 1990, Barret and Berge 1996) Energy consumption is the main source for emissions of sulphur dioxide and nitrogen oxides. Ammonia emissions stem predominantly from livestock farming and agricultural use of fertiliser. The 7.2 version of the RAINS model incorporates sectoral databases on energy consumption and agricultural activity for 44 regions in Europe. These primary data are based on the national forecasts of the involved countries. Several energy scenarios are provided. Current and future levels of sulphur dioxide, nitrogen oxide and ammonia are estimated by applying emission factors to these primary data. The emission factors are derived mainly from the CORINAIR 1990 emission inventory, but also from national reports and contacts with national experts. The estimated emission levels are modified according to national emission strategies. (Amann et al. 1996) The calculations in this chapter are based on the emission scenarios presented in Table 3.2 (for the year 1990) and Table 3.3 (for the year 2010). IIASA constructed these scenarios for the second interim report to the European Commission, DG-XI, entitled "Cost-effective control of acidification and ground-level ozone" (Amann et al. 1996). A comprehensive clarification of data and assumptions underlying the emission levels in Tables 3.2 and 3.3 can be found in the second interim report. The emission levels for 1990 reflect the actual situation, whereas those for 2010 are a forecast. The 1990 levels of sulphur dioxide, nitrogen oxide and ammonia are in general in good accordance with the results from the CORINAIR 1990 inventory and the EMEP database. The 2010 projections are partly based on officially announced policy targets and national emission ceilings. In addition, they build on detailed forecasts of future economic activities and application of emission control techniques in the various sectors of the economy. 3.5 Exposure assessmentThe 7.2 version of the RAINS model estimates dispersion and deposition of nitrogen and sulphur compounds on grid elements (150 km resolution), resulting from the emissions from 44 regions in Europe. The grid consists of 612 elements covering all 44 European regions, including the European part of the former Soviet Union. Total deposition for one grid element is computed by adding up the contributions from every region and the background contribution for that grid element. The dispersion and deposition estimates are based on a model developed by EMEP (co-operative Program for Monitoring and Evaluation of the long-range transmission of air pollutants in Europe). (Amann et al. 1996, Barret and Berge 1996) The EMEP model is a Lagrangian or trajectory model. In this model, an air parcel is followed on its way through the atmosphere. The EMEP model is a single layer model (see Figure 3.1). It follows air parcels along their (horizontal) travel over the two-dimensional trajectories of atmospheric motion during 96 hours preceding their arrival at a specified grid element. The (horizontal) atmospheric motion is calculated from the wind field at an altitude representing transport within the atmospheric boundary layer. (Alcamo et al. 1990, Amann et al. 1996, Barret and Berge 1996) Main inputs to and outputs from the EMEP parcels are emissions from, and wet and dry depositions on, the underlying grid elements. There is also some exchange with the free troposphere above the atmospheric boundary layer. Lagrangian models do not consider exchanges between air parcels. The size of the air parcels should be large to allow for homogenous atmospheric circumstances. In the EMEP model they measure 150 km by 150 km in the horizontal direction, and have an upper boundary that changes with the mixing height. The EMEP model considers also chemical transformations of sulphur dioxide, nitrogen oxide and ammonia within an air parcel. All chemical processes are described by ordinary first-order differential equations integrated over time. (Alcamo et al. 1990, Amann et al. 1996, Barret and Berge 1996) In Lagrangian models, all atmospheric characteristics, such as concentration and turbulence, are considered to be constant within an air parcel, and for a given time interval. The rate of transformation is determined by the parcel's characteristics at a given moment. At the end of the time interval, the situation within the air parcel changes stepwise with the product of transformation rate and the duration of the time interval. EMEP model calculations are based on input data of actual meteorological conditions that were collected every six hours for the years 1985 through 1995. For each of these years, relationships are established between sources and sinks of pollutants. The results have been averaged over 11 years and rescaled to provide the annual spatial distribution of one unit of emission. The resulting atmospheric transfer matrices are used in the RAINS model to estimate dispersion and deposition on 612 grid elements as a result of the emission in 44 regions. (Amann et al. 1996, Barret and Berge 1996) Figure 3.1. The two dimensional trajectories of atmospheric motion of a air parcel (Alcamo et al. 1990). The dispersion and deposition estimates from the EMEP model have been checked and calibrated more than once with measured annual concentrations and depositions. The EMEP estimates are therefore broadly accepted as the best available. The effect on the uncertainty in the RAINS output data were estimated to be about 10% to 25% for sulphur deposition (Alcamo et al. 1990). The use of `region to grid' atmospheric transfer matrices implicitly assumes that the spatial relative distribution of economic activities and related emissions within a region will not dramatically change in the future. It has been shown that the error introduced by this simplification is within the range of other model uncertainties. (Amann et al. 1996) Table 3.2. Total emissions for the year 1990 are given for each region (column 1) by substance (columns 2, 3 and 4). Columns 5 up to, and including column 13 represent the total area of that (column 5), the total area of ecosystem in that region (column 6), the total area of unprotected ecosystem in that region (column 7) as a percent of total area (column 8), the total area of ecosystem that gets unprotected by the total emission from that region (column 9; normalisation factors), and the acidification factors per substance for that region (columns 10, 11, 12 and 13). The acidification factors for H+ equivalents in column 13 may conditionally be used to approximate acidification factors for other acidifying substances like hydrogen chloride or hydrogen sulphide. Table 3.2. Total emissions for the year 1990 are given for each region (column 1) by substance (columns 2, 3 and 4). Columns 5 up to, and including column 13 represent the total area of that (column 5), the total area of ecosystem in that region (column 6), the total area of unprotected ecosystem in that region (column 7) as a percent of total area (column 8), the total area of ecosystem that gets unprotected by the total emission from that region (column 9; normalisation factors), and the acidification factors per substance for that region (columns 10, 11, 12 and 13). The acidification factors for H+ equivalents in column 13 may conditionally be used to approximate acidification factors for other acidifying substances like hydrogen chloride or hydrogen sulphide. Table 3.3. Total emissions for the year 2010 are given for each region (column 1) by substance (columns 2, 3 and 4). Columns 5 up to, and including column 13 represent the total area of that (column 5), the total area of ecosystem in that region (column 6), the total area of unprotected ecosystem in that region (column 7) as a percent of total area (column 8), the total area of ecosystem that gets unprotected by the total emission from that region (column 9; normalisation factors), and the acidification factors per substance for that region (columns 10, 11, 12 and 13). The acidification factors for H+ equivalents in column 13 may conditionally be used to approximate acidification factors for other acidifying substances like hydrogen chloride or hydrogen sulphide. 3.6 Effect measuresAcid deposition on forest soil may cause an imbalance of nutrients (via leaching of cations), and may mobilise toxic aluminium compounds. These changes can affect tree roots and consequently tree nutrition and water uptake. The resulting decrease in health may lower the ability of trees and other vegetation to cope with stress. (Alcamo et al. 1990) Within certain limits, forest soils are able to carry acid depositions without changing their structure and function. Acid deposition is to some extent neutralised by weathering of base cations, although it is enhanced through the uptake of base cations by vegetation. Deposition of nitrogen does not contribute to soil acidification as long as it functions as fertiliser, or if uptake and denitrification (deposition dependent) are faster than nitrogen deposition. The soil capacity to compensate for acid deposition is therefore described by the critical acid load. The critical acid load links sulphur and nitrogen deposition to the biological effects related to critical aluminium mobilisation, specified at 0.2 equivalents/m³ (Hettelingh et al. 1991, Posch et al. 1995). Figure 3.2 depicts the critical load function for a given ecosystem. There is no exceeding of the critical acid load for any combination of sulphur and nitrogen deposition lying at or below the thick line (representing the critical load function). The critical load function is defined by the maximum critical sulphur deposition (CLmax(S) at a nitrogen deposition of zero), the minimum critical nitrogen deposition (CLmin(N) where nitrogen immobilisation and uptake is equal or larger than the nitrogen deposition), and the slope of the critical load function (that reflects the linearity between nitrogen deposition and denitrification). (Hettelingh et al. 1991, Posch et al. 1995)
Figure 3.2. The relationship between nitrogen and some sulphur depositions and the critical loads for acidifying a hypothetical ecosystems (Posch et al. 1995). Maximum critical sulphur deposition, minimum critical nitrogen deposition, and the slope of the critical load function are characteristics of the considered ecosystem (see Figure 3.2). One EMEP grid element may contain several ecosystems, each with its own critical load function (see Figure 3.3). These critical load functions (weighed for the size of the ecosystems) can be used to construct so called protection isolines for the grid element. Such isolines consist of all combinations of S and N deposition for which a given fraction of ecosystems does not exceed critical loads, and thus in RAINS terminology is assumed to be protected against the adverse effects of acidification (see Figure 3.4) (Posch et al. 1995). The different fractions of protected ecosystem can be plotted in a two-dimensional cross-section against the critical load for actual acidity. A hypothetical example of the resulting cumulative distribution curve is shown in Figure 3.5 (Downing et al. 1993).
Critical load functions for acidification of forest soils have been estimated for the whole of Europe by the Co-ordination Centre for Effects at RIVM in Bilthoven, the Netherlands. In addition, critical load functions have also been established for some heath land (United Kingdom), grassland (United Kingdom and Switzerland), peatland (Switzerland and Estonia) and freshwater (Russia, Finland, Sweden, Norway, Switzerland and the United Kingdom) (Posch et al. 1995). The territory area, the total area of ecosystems, and the area of unprotected ecosystems are given for all 44 RAINS-regions in the reference situation in Table 3.2 (for the year 1990) and Table 3.3 (for the year 2010). More detailed information about critical loads and the construction of cumulative distribution functions can be found in the work of Hettelingh et al. (1991) and Posch et al. (1995). 3.7 Mathematical frameworkThe acidification impact of an emission can be expressed as the area of ecosystem that becomes unprotected as a result of that emission. The mathematical derivation of this area of ecosystem is described here. See Chapter 4 for a step by step illustration. The share of an emission from region (i) that deposits on grid element (j) is given in the RAINS model by the transport coefficient: With the help of the transport coefficient, the total deposition in grid element (j) is calculated from the emission from region (i=1 to n):
The RAINS model compares the summed deposition with the cumulative distribution curve of unprotected ecosystems in grid element (j) in order to determine the area of protected and unprotected ecosystem. The RAINS model considers regions (in most cases identical to countries), rather than the underlying separate economic activities, as sources of impact on grid elements. LCA focuses on separate processes as sources, or actually even smaller than that. LCA deals only with a fraction of the total emission from a process, namely, that emission quantity that is related to one functional unit. This fraction is generally marginal compared to the total emission from that process, and similarly, the total emission from that separate process is in general marginal compared to the total emission from one region. As a consequence, the change of deposition on grid element (j) from that particular process will also be marginal. The relationship between a change in unprotected ecosystems in grid element (j) caused by a change in emission in region (i) may be taken as linear with the transfer coefficient times the first derivative of the cumulative distribution curve for grid element (j = 1 to m), as long as the changes in deposition on grid element (j) remain marginal: The acidification factor (AFs,i in ha/tons) that directly relates a change of emission of substance (s) in region (i) to the change in unprotected ecosystems in its total deposition areas is given by: 3.8 ResultsAcidification factors have been established with the help of the RAINS model by reducing one by one the emission levels of each separate region by 10%, and then relating the result to the reference situation (the initial emission level and area of unprotected ecosystems):
A reduction of 10% has been chosen considering numerical accuracy and the functional form of the cumulative distribution curve for each grid. Calculations have been done for the years 1990 and 2010, and for sulphur dioxide, nitrogen oxide and ammonia. The reference situation and results for the year 1990 are presented in Table 3.2, and for the year 2010 in Table 3.3. 3.9 Variation of acidification factors by regionAs can be seen from Table 3.2, there are large differences among regions in the 1990 acidification factors for sulphur dioxide. The acidification factors for the southern and south-eastern European regions are in general low. This is the combined effect of the insensitivity of the receiving (calcareous) ecosystems for (changes in) acidifying depositions, and the still relatively low emission and related deposition levels in these regions. The acidification factors for the emissions from the Scandinavian and Baltic regions, and in the European part of the former Soviet Union are rather high as a result of deposition on the rather sensitive areas in these regions. The Western and Mid European regions have moderate acidification factors as a result of the large number of ecosystems that is already unprotected because of the rather high emission and related deposition levels in these regions. The 1990 acidification factors for nitrogen oxide show less pronounced differences among regions, and are in all cases lower than the acidification factors for sulphur dioxide. As long as nitrogen functions as a fertiliser, it does not contribute to acidification (note that it may contribute to eutrophication). In addition, nitrogen oxide transport extends on average longer distances than sulphur dioxide transport. This has a "smoothing" effect on the acidification factors. Given that transport distances of ammonia are relatively short, the 1990 acidification factors for this substance show sharper differences than those of nitrogen dioxide. The 2010 acidification factors (Table 3.3) show less sharp differences among regions than for those of 1990 (Table 3.2), but follow roughly the same trends described above. There are also some remarkable changes. This is as such not surprising, given that the reference emission and related deposition situation have changed considerably. As a result, the reference position on the cumulative distribution curve of unprotected ecosystems (see Figure 3.5) may also show important changes for some regions. As shown in Tables 3.2 and 3.3, the acidification factors for sea emissions can be considerable (North Sea and Baltic Sea). It is not the deposition on these sea waters from their own emissions that adds to acidification. Rather, it is the inland wind direction that prevails on sea waters that in the case of both the North and Baltic seas contributes considerably to deposition on the sensitive ecosystems of Scandinavia and the Baltic regions. The emissions from the Mediterranean Sea predominantly deposit on the rather insensitive surrounding regions, while the emissions from the Atlantic Ocean deposit only partly on (sensitive) land. The acidification factors for the Mediterranean and the Atlantic are therefore considerably less than for the North and Baltic Sea. The 1990 and 2010 acidification factors are plotted for each region in Figure 3.6 (sulphur dioxide), Figure 3.7 (nitrogen oxide) and Figure 3.8 (ammonia). The figures give insight into the changes of the acidification factors of regions from 1990 compared to 2010. The factors for Scandinavia are considerably less for 2010 than for 1990, but are still among the highest. The most striking changes occur for the Baltic regions and the former Soviet Union regions (except the Kola/Karelia region). Their factors fall considerably, which places these regions among the large group of regions with moderate to low acidification factors. Also remarkable is the increase of the ammonia factors for the West European countries. The contribution of ammonia to a change in area of unprotected ecosystem becomes apparently more important as sulphur emissions have greatly reduced. 3.10 Robustness of the marginal approachOne of the basic assumptions underlying the presented acidification factors is the marginal contribution that total emissions from separate processes in the life cycle of a product add to the total deposition on receiving grid elements. This assumption follows from a second assumption that separate processes make only a marginal contribution to the total emission from a region. This second assumption is usually true but does not apply for some exceptional cases [20]. Therefore, the robustness of the marginal approach has been investigated by pushing it beyond it limits.
Figure 3.6. Acidification factors for sulphur dioxide in 1990 and 2010.
Figure 3.7. Acidification factors for nitrogen oxide in 1990 and 2010.
Figure 3.8. Acidification factors for ammonia in 1990 and 2010.
Figure 3.9. Unprotected ecosystem by the emissions from each region in 1990 predicted by multiplication with the acidification factors (AF), and predicted by the RAINS model.
Figure 3.10. Unprotected ecosystem by the emissions from each region in 2010 predicted by multiplication with the acidification factors (AF), and predicted by the RAINS model. The acidification factors have been used to estimate for each country the area of unprotected ecosystems from the total emissions from that country by multiplying the country's emission with the appropriate acidification factors. This change in area of unprotected ecosystems has also been determined with the RAINS model by reducting the total emission from each region to zero (see "Total area: unprotected ecosystem by region" in Tables 3.2 and 3.3). The results from both have been plotted in Figures 3.9 and 3.10). The accordance between both ways of prediction appears fairly good (see Figures 3.9 and 3.10). In 1990, the ratio between unprotected area predicted with the RAINS model and by use of the acidification factors is within a factor 1.1 for 23 regions, and within a factor 2 for 16 regions. These results suggest a reasonable to good stability of the estimated acidification factors for moderate changes in the reference situation. However, there are 5 regions with differences larger than a factor of 2: Latvia, Moldova, Kalingrad region, Slovenia and Yugoslavia. In 2010, there are 6 regions showing differences larger than a factor of 2: Belarus, Kola/Karelia region, Remaining Russia, St. Petersburg region, Ukraine and the Atlantic Ocean. The regions with differences larger than a factor of 2 between the RAINS estimates and the estimates with the help of the acidification factors estimates have been examined more closely. For these regions, the ratio between the decrease of unprotected ecosystems area and the emission reduction have been calculated (similar to Formula 3.13) by gradually increasing the emission reduction. The results are presented in Table 3.4. For most regions and most substances, the ratios remain quite stable with increasing emission reduction. The exceptions are restricted to SO2 and apply to Latvia and Slovenia in 1990 and the St. Petersburg region in 2010. As expected, the ratios tend to become unstable for large emission reductions of (in particular) SO2. These results again underline the reasonable to good stability of the estimated acidification factors for moderate changes in the reference situation. 3.11 Acidification factors for substances with very short lifetimesThe RAINS model focuses on the principal contributors to acidification on a regional scale (between 100 to 4,000 km): emissions of sulphur dioxide, nitrogen oxide and ammonia (Alcamo et al. 1990, Barret and Berge 1996). Close to the source (on the local scale), however, other acidifying substances like hydrochloric acid or hydrogen fluoride may also be important (Jaarsveld 1989). These other substances may fully dominate the total acidifying emissions in the life cycle of particular products. The acidification factors from these substances with very short lifetimes have been approximated on the basis of sulphur dioxide and with the help of the RAINS model in order to enable quantification of the acidifying impact from these substances. For each region, the deposition of sulphur (expressed in H+ equivalents) and the related area of unprotected ecosystems have been determined in the reference situation and for the situation in which the emissions from all regions together are reduced by 10%. The acidification factor for the emission of H+ equivalents per region has been calculated from the change in the area of unprotected ecosystems in a region, divided by the change in deposition in that region. The acidification factor for the other acidifying substances may conditionally be calculated from those for H+ equivalents, by multiplying the acidification factors for H+ equivalents with the number of H+ potentially deliverable by, and divided by the molecular mass of that substance. Approximation of the acidification factors of other acidifying substances is only acceptable under the following conditions: a substance fully deposits in the same region as where the source is located, and the deposed substance is fully leached (like sulphur) and not retained in the soil (like phosphor) or taken up by the vegetation (like nitrogen). 3.12 Application of acidification factors in LCAThe application of the acidification factors in LCIA is very simple. An emission in the product's life cycle is multiplied with the acidification factor for that region and substance to derive the estimated acidifying impact of that emission.
The only additional data required, the geographical site or region where an emission takes place, are in general already provided by current LCI. The earlier example on copper production in Albania, Belgium and Finland is used to demonstrate the application of, and result from the acidification factors (see Figure 3.11). Depending on the goal and scope of the study, an average "Western European" acidification factor can be used for emissions with unknown site or region, or sensitivity analysis via a worst case approach (e.g. AF-Kola/Karelia for sulphur) can be followed. Hauschild and Potting (2000), who also give a more elaborated procedure for site-dependent assessment, use the Western European average. The Guidance Document (Hauschild and Potting 2003) based on this Technical Report describes the application procedure for the acidification factors in its entirety.
Figure 3.11. The acidifying impact of 1 kg of primary cathodic copper production in Albania, Belgium, and Finland with regular equivalency assessment (Ip = Ep,s * EqFs; acidification factors from Table 3.2). For all locations similar technologies with similar emission quantities per kilogram of copper are assumed: 100 g SO2 and 10g of NOx (Potting and Blok 1993). Table 3.4. The ratios between the area of unprotected ecosystem and emission reduction for increasing reduction of emission from the given region. The established acidification factors are actually not site-factors but rather replace the product of characterisation or equivalence factor and the site-factors ( EqFs * SFs ) in Formula 3.4. They can be seen as a set of characterisation factors alternative to those in Table 3.1. The characterisation or equivalency factors in Table 3.1 pay no or only limited attention to dispersion and deposition, and the sensitivity of the receiving areas for deposition. However, one may prefer to use one's own set of characterisation or equivalency factors in combination with the acidification factors presented here. This is possible by dividing the acidification factors by the characterisation or equivalency factor for that substance. The resulting site-factors can then be used as modifier of the equivalency factor according to Formula 3.4. An optional step in LCA is normalisation. In normalisation, the assessed impact per functional unit is divided by the impact score of a reference situation (a certain region in a certain period of time) (Udo de Haes et al. 1996). A site-dependent LCIA also requires site-dependent normalisation factors (Wenzel et al. 1997). These factors have been established by comparing the reference situation of unprotected ecosystems with the situation of zero emissions from the given region. The normalisation factors are provided in Table 3.2 and 3.3 under the header "Total area: Unprotected ecosystem by Region" and are not further discussed here. 3.13 DiscussionSome remarks about the stability, feasibility and limitations of application of the established site-factors have already been made in the previous section. Here, a few additional aspects are discussed in more detail. 3.14 Uncertainties in the acidification factorsThe acidification factors are fully based on calculations with the RAINS model. The credibility of, and uncertainties in, these acidification factors are therefore strongly related to the credibility of, and uncertainties in, the model. One of the principal motives for developing the RAINS model was to provide scientific support for the negotiations in Europe under the Geneva Convention on Transboundary Air Pollution. In this role, the RAINS model as well as constituting submodules (like the EMEP atmospheric transfer matrices, and the critical loads compiled by CCE ) have gained broad scientific and political acceptance in Europe. RAINS, like all models, is a simplification of reality and will thus contain uncertainties. The uncertainties in the emission estimates, and dispersion and distribution, were already briefly discussed above. Uncertainties in critical loads have recently been analysed and shown to remain within a factor of 2 for individual ecosystems. However, these uncertainties are cancelled out to a large extent by the number of ecosystems covered (1-36',000) by most individual grid elements and Europe in total (almost 700,000) in the calculated acidification factors (Barkman 1997, Posch et al. 1997). Nevertheless, quantification of combined uncertainties in assessed impact is one of the next steps in the continuing development of the RAINS model and recommended for future update of the presented acidification factors. Although the acidification factors do contain small uncertainties, their introduction in LCIA reduces considerably the errors from the current absence of spatial differentiation. The acidification factors add resolving power of a factor thousand difference between the highest and lowest factors. The gain of information by use of these factors is thus expected to compensate fully for the introduction of new uncertainties. However, it is recommended that a future update of these acidification factors quantifies these uncertainties. The term "deposition" in this chapter refers to annual average deposition. The marginality of total emissions from separate processes justifies another assumption that implicitly underlies the presented framework: for a given moment in time, the contribution from separate processes may be regarded as marginal (though seasonal related emissions might have different deposition patterns). 3.15 Definition of the acidification factorWe defined the acidifying impact of an emission as the area of ecosystem that becomes unprotected as a result of that emission or, put another way, the area of ecosystem that is protected as a result of cutting that emission. The acidifying impact thus refers to a change in risk as a result of a change in emission. As a consequence, not reflected in the acidifying impact are those ecosystems that regardless of the change in emission remain either below or above threshold. A more sophisticated approach would have been to define the acidifying impact in a change of damage rather than in a change of risk. s68 og 68aThe critical load values for a particular ecosystem can only tell whether there is a risk of ecosystem damage Pleijel et al. (not published) advocate another approach to calculate acidification factors (or actually site-factors). They define the acidifying impact of an emission as the contribution it makes to the total deposition on an ecosystem, although they implement this by establishing the share of emission depositing on ecosystems with already exceeded critical load values (with the help of the same submodels underlying the RAINS model). This share times the emission times the relevant characterisation or equivalency factors (according to Table 3.1) gives the acidifying impact. As no calculation has been done, it is not possible to compare their results with the results of the underlying approach. However, it is possible to say something about the relevance of both approaches. Pleijel et al. consider an emission responsible for all depositions above threshold to which it contributes. This could also be interpreted in terms of a linear relation between changes in emissions and resulting damage (although damage may be expected to increase asymptotically with increasing exceeding of critical load values). Our approach considers an emission only responsible for the actual changes in risk it causes, which is useful in a strategy of risk minimisation. The most efficient way to minimise risk integrated over several ecosystems is to give priority to get (and/or keep) depositions below threshold for as many ecosystems as possible. A next sophistication would be to link this marginal approach of risk to damage. 3.16 Usefulness of the acidification factorsLCA has often been criticised for its limited ability to cope with spatial and temporal variation. Owens (1997) discusses four remedies to deal with the poor agreement between the impact as predicted by LCIA and the expected occurrence of actual impact. One of his remedies is to complement LCA with other environmental tools to address the actual relevance of the assessed impact. In this chapter we actually integrate LCA with another environmental tool. The accuracy of prediction with the resulting acidification factors is close to the maximum achievable with presently available environmental tools. Hence deposition and exceeding of critical load values must be analysed over several hundred kilometres in order to cover most of the acidifying impact from an emission. This goes far beyond the abilities of analytical tools as typically used in risk assessment and environmental impact assessment which usually have a range up to 50 km. 3.17 Conclusions and recommendationsAcidification and normalisation factors have been established for 44 regions in Europe to facilitate site-dependent LCIA of acidification. The acidification factors relate the region of emission to the impact on its deposition areas. An emission is multiplied with the acidification factor for the relevant region and substance to derive the estimated acidifying impact of the emission. The only additional required data, the geographical site or region where an emission takes place, is in general already provided by current LCI. The acidification factors show a reasonable to good level of stability of the estimated acidification factors for changes in the reference situation, and the combined uncertainties in the RAINS model are cancelled out to a large extent in the acidification factors because of the large area of ecosystems they cover. On the other hand, the acidification factors add resolving power of a factor thousand difference between highest and lowest factor. The information gain by using these factors in LCA is expected to compensate fully for the accompanying introduction of additional uncertainties. Future update of these acidification factors is recommended to quantify the uncertainties. The acidification factors presented in this chapter are based on the area of ecosystem that becomes unprotected and thus runs the risk of ecosystem damage as a result of that emission. This does not tells us whether such risk actually results in damage or how large the damage will be. The current state-of-the art in the scientific field of critical loads does not yet allow a translation of risk into damage. However, this has to be subject to future update of the established acidification factors. The framework presented here has proven capable of establishing feasible acidification factors for use in LCIA. It is desirable to extend the existing Europe set with factors for the other continents. The RAINS model provides the possibility of doing this for Asia as well. The same framework can also be used to achieve similar factors for other regional environmental impacts. The RAINS version 7.2 provides the possibility to do so for the atmospheric dimension of eutrophication in Europe, while release of a version that is extended to tropospheric ozone formation is on its way. 3.17.1 AcknowledgementResearch for this work was also supported by a Training and Mobility grant from the European Commission, and by the Danish Agency for Industry and Trade under the EUREKA project LCAGAPS. 3.18 ReferencesAlcamo, J., R. Shaw and L. Hordijk (eds.). The RAINS model of acidification. Science and strategies in Europe. Dordrecht (the Netherlands), Kluwer Academic Publishers, 1990. Amann, M., I. Bertok, J. Cofala, F. Gyarfas, C. Heyes, Z. Klimont and W. Schöpp. Cost-effective control of acidification and ground-level ozone. Second interim report to European Commission, DG-X1. Laxenburg (Austria), International Institute for Applied Systems Analysis (IIASA), 1996. Amann, M., M. Baldi, C. Heyes, Z. Klimont and W. Schöpp. Integrated assessment of emission control scenarios, including the impact of troposheric ozone. Water, air and soil pollution, Vol. 85 (1995), Issue 4, pp2595-2600. Barkman, A. Applying the critical loads concept: Constraints induced by data uncertainty. Reports in ecology and environmental engineering (report 1:1997). Lund (Sweden), Dept. of Chemical Engineering II of Lund University, 1997. Barrett, K. and E. Berge (eds.). Transboundary air pollution in Europe. Part 1: Estimated dispersion of acidifying agents and of near surface ozone. EMEP MSC-W status report (research report no. 321996). Oslo (Norway), Norwegian Meteorological Institute, 1996. Downing, R.J., J.P. Hettelingh and P.A.M. de Smet (Eds). Calculation and mapping of critical thresholds in Europe. CCE status report 1993. Bilthoven (the Netherlands), Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and the Environment (RIVM), 1993. EEA. Corinair 90: Summary report no. 3, Copenhagen (Denmark), European Environmental Agency (EEA), 1996. Hauschild, M. and J. Potting. Spatial differentiation in life cycle impact assessment – the EDIP2003 methodology. Guideline from the Danish Environmental Protection Agency, Copenhagen, 2003. Hauschild, M.Z. and H. Wenzel: Environmental assessment of products. Vol. 2 - Scientific background, 565 pp. Chapman & Hall, United Kingdom, 1998, Kluwer Academic Publishers, Hingham, MA. USA. ISBN 0412 80810 2 Heijungs, R. G. Huppes, R.M. Lankreijer, H.A. Udo de Haes, A. Wegener Sleeswijk, A.M.M. Ansems, P.G. Eggels, R. van Duin and H.P. de Goede. Environmental life cycle assessment of products. Guide and backgrounds (ISBN 90-5191-064-9). Leiden (the Netherlands), Centre of Environmental Science of Leiden University, 1992. Hettelingh J-P., R.J. Downing and P.A.M. de Smet. Mapping critical loads for Europe CCE technical report no. 1. Bilthoven (the Netherlands), Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and Environmental Protection (RIVM), 1991. Jaarsveld, J.A. van. Operational atmospheric transport model for priority substances; specifications and guidance for use (report no. 222501002; in Dutch). Bilthoven (the Netherlands), National Institute of Public Health and Environmental Protection (RIVM), 1989. Lindfors, L-G., K. Christiansen, L. Hoffman, Y. Virtanen, V. Juntilla, O-J. Hanssen, A. Rønning, T. Ekvall and G. Finnveden. Nordic guidelines on life cycle assessment (Nord 1995-20). Copenhagen (Denmark), Nordic Council of Ministers, 1995 Owens, W. Life cycle assessment. Constraints on moving from inventory to impact assessment. Journal of Industrial Ecology, Vol. 1 (1997), Issue 1, pp37-49. Pleijel, K., J. Altenstedt, H. Plijel, G. Lövblad, P. Grennfelt, L. Zetterberg, J. Fejes, L-G. Lindfors. A tentative methodology for the calculation of global and regional impact indicators in type-III eco-labels used in Swedish case studies. Stockholm (Sweden), Swedish Environmental Research Institute (IVL), not published. Posch, M, P.A.M. de Smet, J.P. Hettelingh, R.J. Downing (eds.). Calculation and mapping of critical thresholds in Europe. CCE status report 1995. Bilthoven (the Netherlands), Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and the Environment (RIVM), 1995. Posch, M., J.P. Hettelingh, R.J. Downing, and P.A.M. de Smet (Eds). Calculation and mapping of critical thresholds in Europe. CCE status report 1997. Bilthoven (the Netherlands), Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and the Environment (RIVM), 1997. Potting, J. and K. Blok. Environmental interventions from some primary and secondary materials and components in electronic products (report no. 93070; in Dutch). Utrecht (the Netherlands), Dept. of Science, Technology and Society of Utrecht University, 1993. Potting, J. and K. Blok. 1995. Life cycle assessment of four types of floor covering. Journal of Cleaner Production, Vol. 3 (1995), Issue 4, pp201-213. Potting, J. and K. Blok. Spatial aspects of life cycle impact assessment. In: Udo de Haes, H.A., A.A. Jensen, W. Klöpffer and L-G. Lindfors (eds.). Integrating impact assessment into LCA. Proceedings of the LCA symposium held at the fourth SETAC-Europe Congress, 11-14 April 1994 at the Free University of Brussels in Belgium. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1994. Potting, J. and M. Hauschild. Predicted environmental impact and expected occurrence of actual environmental impact. Part 1: The linear nature of environmental impact from emissions in life cycle assessment. Int. Journal of Life Cycle Assessment, Vol. 2 (1997a), Issue 3, pp171-177. Potting, J. and M. Hauschild. Predicted environmental impact and expected occurrence of actual environmental impact. Part 2: Spatial differentiation in life cycle assessment by site-dependent characterisation of environmental impact from emissions. Int. Journal of Life Cycle Assessment, Vol. 2 (1997b), Issue 4, pp209-216. Potting, W. Schöpp, K. Blok and M. Hauschild. Site-dependent life-cycle assessment of acidification". Journal of Industrial Ecology Vol. 2 (1998), Issue 2, pp63-87. Udo de Haes, H.A. (ed.). Towards a methodology for life cycle impact assessment. Belgium, Brussels, Society of Environmental Toxicology and Chemistry – Europe, 1996. Wenzel, H., M.Z. Hauschild and L. Alting: Environmental assessment of products. Vol. 1 - Methodology, tools, techniques and case studies, 544 pp. Chapman & Hall, United Kingdom, 1997, Kluwer Academic Publishers, Hingham, MA. USA. ISBN 0 412 80800 5. 4 Terrestrial eutrophication4.1 Introduction Authors: José Potting [21], Wolfgang Schöpp [22] and Michael Hauschild [23] 4.1 IntroductionThe eutrophying impact usually characterised in life cycle impact assessment refers to eutrophication of aquatic ecosystems. This follows from the modelling of impact that in life cycle assessment typically takes its basis in the composition of aquatic biomass (Wenzel et al. 1997, Heijungs et al. 1992, Lindfors et al. 1995). Until recently, Finnveden et al. (1992) were the only ones explicitly addressing eutrophication of terrestrial ecosystems. They proposed to separate between summing together waterborne and airborne nitrogen emissions [24] because terrestrial ecosystems get eutrophied mainly via nitrogen emissions to air (waterborne nutrients rarely contribute to terrestrial eutrophication) [25]. The contribution to terrestrial eutrophication in the proposal of Finnveden et al. (1992) consists of the airborne nitrogen emission accumulated over the full life cycle of the considered product. Obviously, such an approach does not pay attention to the site where an emission is released, nor to the fate and exposure and sensitivity of the subsequent environments where the released nitrogen is received, and where the loading increase may lead to impact. This lack of spatial differentiation affects the relevance of the assessed impact, as was clearly demonstrated by Potting and Blok (1994, 1995). Potting et al. (1998a,b) developed a framework for constructing factors that relate the region of emission to the impact on its deposition areas (see also Chapter 3). This framework is used to establish characterisation factors for 44 European regions and for the impact categories of acidification, ground-level ozone formation and terrestrial eutrophication. The factors were established with the help of the RAINS model. This chapter reports the results for the terrestrial eutrophication factors. Section 4.2 illustrates RAINS step by step with the several outputs of the model. The procedure for calculating the terrestrial eutrophication factors is exemplified in Section 4.3, and the results are presented and discussed in Section 4.4. Section 4.5 draws some main conclusions. 4.2 The RAINS modelRAINS is an integrated assessment model that combines information on regional emission levels with information on long-range atmospheric transport in order to estimate patterns of deposition and concentration for comparison with critical loads and thresholds for acidification, eutrophication and tropospheric ozone formation. Relating the site of an emission to the impact on its deposition areas is one of the key elements in the RAINS. This model (version 7.2) has therefore been used, similar as for acidification (see Chapter 3), to establish site-factors for terrestrial eutrophication. Alcamo et al. (1990) and Amann et al. (1995) provide a detailed description of the model. Section 3.3 gives a technical summary of the parts of RAINS that are relevant for this chapter. This section illustrates this technical summary step by step with the several outputs of the RAINS model. 4.2.1 Emission estimatesTypical distances between an emission source and its locations of deposition are easily several hundreds of kilometres. The geographical scale of analysis, accordingly, has to be large in order to cover most of the impact from an emission source. The RAINS model focuses therefore on the regional dimension (rather than on the local dimension) of the assessed environmental impact. (Alcamo et al. 1990, Barret and Berge 1996) Though phosphorus is a key nutrient in eutrophication of inland waters (rivers and lakes), it has only minor relevance for terrestrial ecosystems since they are under natural conditions rarely limited in their growth by phosphorus. The soil can provide its own phosphorus by weathering or by mineralisation of indigenous organic matter. There may also be phosphorus input by atmospheric deposition of pollen and dust (for Sweden estimated to be 0.07 kg per hectare per year). Depositions of phosphorus of anthropogenic origin are small [26] compared to phosphorus deposition by pollen or dust. (Chardon 2000, Berdowski and Jonker 1994) Emissions of nitrogen oxides and ammonia are the principal contributors to terrestrial eutrophication. Energy consumption is the main emission source of nitrogen oxides. Ammonia emissions stem predominantly from livestock farming and agricultural use of fertiliser. The 7.2 version of the RAINS model incorporates sectoral databases on energy consumption and agricultural activity for 44 regions in Europe. The calculations in this article are based on the scenarios for nitrogen emissions only. The relevant scenarios are presented in Table 4.1 (for the year 1990) and Table 4.2 (for the year 2010). The emission levels for 1990 reflect the actual situation, whereas those for 2010 are a forecast. Section 3.3 gives more details about the background of the numbers. Table 4.1. Total emissions for the year 1990 are given for each region (column 1) by substance (columns 2 and 3). Columns 4 up to, and including column 9represent the total area of that region (column 4), the total area of ecosystem in that region (column 5), the total area of unprotected ecosystem in that region (column 6) as a percent of total area (column 7), the total area of ecosystem that gets unprotected by the total emission from that region (column 8; normalisation factors), and the terrestrial eutrophication factors per substance for that region (columns 9and 10).
Table 4.2. Total emissions for the year 2010 are given for each region (column 1) by substance (columns 2 and 3). Columns 4 up to, and including column 9represent the total area of that region (column 4), the total area of ecosystem in that region (column 5), the total area of unprotected ecosystem in that region (column 6) as a percent of total area (column 7), the total area of ecosystem that gets unprotected by the total emission from that region (column 8; normalisation factors), and the terrestrial eutrophication factors per substance for that region (columns 9and 10).
Table 4.3. Total nitrogen dioxide emission for Albania, Belgium and Finland in 1990 (Amann et al. 1996) and the breakdown into main economic sectors (abstracted from the RAINS model; version 7.2).
Table 4.3 gives, as an example, the breakdown into the main economic sectors of nitrogen oxide emissions for Albania, Belgium and Finland. Every economic sector can be further broken down into finally the separate sources. The breakdown shows that basically all economic sectors, except transport, have a relative small share in the total nitrogen oxide emission per country. Each of these sectors aggregates the contributions of a large number of underlying individual sources. Obviously, the shares of the individual sources in the total are even smaller if not marginal compared to the sector aggregates. This applies also to the transport sector, which is responsible for more than 50% of all nitrogen oxide emissions. 4.2.2 Exposure assessmentThe RAINS model estimates dispersion and deposition of nitrogen compounds with help of "region to grid" matrices on grid elements (150km resolution) for each of the 44 regions in Europe. The grid consists of 612 elements covering all 44 European regions, including the European part of the former Soviet Union. Figure 4.1 shows the dispersion and deposition resulting from total nitrogen emission in Finland (Section 3.3.2 gives more details about the model) Total deposition within one grid-element is computed by adding together the contributions from each of the 44 regions in Europe (their dispersion and deposition pattern being calculated similarly as done for Finland in Figure 4.1). Figure 4.2 shows the dispersion and deposition from the emissions of all European regions together. The RAINS model offers the possibility for each grid-element to quantify the contributions from each region in Europe. Table 4.4 gives such breakdown for the Finnish grid-element indicated by the arrow in Figure 4.2. The contribution from Finland's own emissions to deposition on this grid-element, as can be seen from Table 4.4, is only a little more than 20% of total deposition (almost 80% thus being imported from abroad). Hence the Finnish contribution to this grid-element stems from total Finnish emissions which sources are distributed over the full Finnish area (although this grid-element, being the economic centre of Finland, may have a relatively high source-density). Table 4.4. The shares of European regions in the total deposition of nitrogen on the southern part of Finland (grid-element x=20, y=25 indicated by the arrow in Figure 4.2) resulting from nitrogen oxide emissions.
Figure 4.1. Dispersion and deposition pattern of the total emission of nitrogen from Finland
Figure 4.2. Total dispersion and deposition from emission of nitrogen from all regions in Europe. The arrow indicates a region in Finland (see text) The region-to-grid matrices in the RAINS model are based on the Lagrangian or trajectory model developed by EMEP (co-operative Program for Monitoring and Evaluation of the long-range transmission of air pollutants in Europe (Barrett and Berge 1996)). Section 3.3.1 gives more details about the background of the model. 4.2.3 Effect measuresNitrogen is an essential nutrient for all species and an increase of nitrogen level is therefore not harmful until a certain level has been reached. Each ecosystem, and as a matter of fact each species being part of that, has an optimum curve for the growth related to increasing nitrogen supplies. The optimum levels differ between the various types of ecosystems as illustrated in Figure 4.3.
If nitrogen levels exceed the optimum for the typical species in a given ecosystem, the growth of those species start to decline whereas growth is stimulated for other species better able to benefit from the increased availability of nitrogen. Downing et al. (1993) suggest critical levels of nitrogen in soil at which changes of vegetation composition are induced. These critical levels are used in RAINS to model critical nitrogen depositions or loads above which these changes of vegetation are expected. Similar to what has been done for acidification, critical loads for eutrophication of terrestrial ecosystems have been estimated for the whole of Europe. Ecosystems covered are mainly forests, but for some countries also heathland and grassland. The critical loads are per grid-element summarised in a cumulative distribution curve for eutrophication (see Figure 4.4). While only few grid-elements contain less than 10 ecosystems, there are several with more than 10,000 ecosystems per grid-element (the highest number being 53,000 in a grid-element shared by the Netherlands, Belgium and Germany). (Posch et al. 1997) The full critical load database in the RAINS 7.2 version was based on almost 700,000 ecosystems (the most recent version relates to over 1.4 million ecosystems; Posch et al. 2002) [27]. Section 3.3.3 gives more details about the background of critical loads for acidification. See Downing et al. (1993) and Posch et al. (1995, 1997, 1999 and 2001) for more details about critical loads for eutrophication. 4.3 Calculation procedureThe total deposition per grid-element (as represented in Figure 4.2) can be compared with the cumulative critical load distribution curve for that grid-element (as in Figure 4.4) to establish the area of ecosystems for which critical loads are exceeded (unprotected ecosystems or UES in RAINS terminology). Proceeding in this manner grid-element by grid-element, the total area of unprotected ecosystem over the whole European domain can be calculated. Figure 4.5 represents the results of this exercise spatially resolved over Europe for the emission situation in 1990.
Figure 4.4. Typical cumulative distribution curve of critical load for eutrophication for a grid element The total area of unprotected ecosystems can be calculated basically for every desired emission situation. This has been used to establish factors that express the area of ecosystems that becomes unprotected against eutrophication as result of small changes in the reference emission situation for 1990. The mathematical derivation of those eutrophication factors is given in Section 4.4, but the calculation procedure will be exemplified here. Figure 4.5 shows the total area of unprotected ecosystem in the whole European domain caused by the European emission level of 1990. This area covered to 76.96 mln ha. We can calculate the area of unprotected ecosystem for exactly the same emission situation as in 1990, except that the total nitrogen oxide emission of 300 kton by Finland is reduced by 10% to 270 kton. The area of unprotected ecosystem for this new situation amounts to 76.62 million ha. The difference between the old reference and the new emission situations is 340,000 ha. This means that as a result of the 10% reduction in emission, the deposition of N will no longer exceed the critical load for 340,000 ha of ecosystem. Figure 4.6 shows where in Europe the reduction in Finnish nitrogen oxide emission leads to a reduction in the area of unprotected ecosystem. The change in total area of unprotected ecosystem in Europe divided by the change in Finnish emissions gives the characterisation factor representing the change in unprotected ecosystem area per unit change of nitrogen dioxide emission in Finland (340,000 ha/30 kton = 11,000 ha/kton = 0.11 m2/g). Figure 4.5. Percentage of ecosystem unprotected Figure 4.6. Percentage of ecosystem eutrophication by emissions in becoming protected by reduction 1990 from all European countries of the total nitrogen dioxide emission from Finland by 10% In the same way, eutrophication factors have been established for nitrogen oxide emissions as well as for ammonia emissions for all European regions. These factors have been calculated for the emission scenarios for 1990 and 2010. The emission scenarios and the calculated factors for terrestrial eutrophication are shown in Table 4.1 and 4.2. The factors for terrestrial eutrophication are thus calculated in exactly the same way as the acidification factors in Chapter 3. Section 3.4 describes in more detail the followed calculation procedure including the mathematical terms. 4.4 ResultsThe established factors for terrestrial eutrophication are given in Table 4.1 (for 1990) and in Table 4.2 (for 2010), and aggregated to provide "site-generic" factors in Table 4.5. There are many similarities but also some notable differences to the acidification factors (see Section 3.4 and 3.5). Similar to the acidification factors, both the 1990 and 2010 factors for terrestrial eutrophication show large differences among regions. The Scandinavian regions and Baltic regions also here have rather high factors due to their high sensitivity, whereas factors for the western and mid-European regions are usually moderate to low. In contrast to the acidification factors, however, the terrestrial eutrophication factors are moderate and even high in several cases for many eastern and southern European regions. Whereas the calcareous soils in southern Europe adequately buffer acidification, it has little influence on the sensitivity of these soils to terrestrial eutrophication. Table 4.5. The mean, standard deviation, minimum and maximum values of the terrestrial eutrophication factors for nitrogen oxide and ammonia, for different selections from the 44 European regions (calculated as simple means and standard deviations and weighted for the national emissions). Total Europe covers all 44 European regions. EU+2 consist of Austria, Belgium, Denmark, Finland, France, Germany (old and new), Greece, Ireland, Italy, Luxembourg, Netherlands, Portugal, Spain, Sweden, Switzerland and United Kingdom. EU consists of the same population minus Norway and Switzerland. East Europe covers Albania, Belarus, Bosnia-Herzegovina, Bulgaria, Croatia, Czech Republic, Estonia, Hungary, Latvia, Lithuania, Macedonia, Moldavia, Poland, Romania, Russia (four regions), Slovakia, Slovenia, Ukraine and Yugoslavia. The nitrogen oxide and ammonia factors for terrestrial eutrophication are for most regions considerably higher than the respective factors for acidification. Since both impacts are expressed in the same unit, this suggests soils to be more sensitive for eutrophication than for acidification from nitrogen deposition. This makes sense, since depositions do not contribute to acidification as long as the nitrogen is assimilated by the ecosystem. However, this assimilation of nitrogen is precisely the ecosystem growth discussed in Section 4.3.3 that can lead to a shift in species composition if the supply of nitrogen exceeds the critical load for eutrophication. Though the nitrogen content of one kg ammonia is 2.7 times higher than for one kg nitrogen oxide, the ratio between the factor for ammonia and for nitrogen oxide is considerably higher for all regions. Due to the less widespread dispersion of ammonia, reductions in emission obviously results in larger deposition reduction locally, which then leads to a stronger increase in the area of ecosystems that becomes protected by the emission reduction. The application of the site-dependent factors will not be discussed here. Instead, the reader is referred to a detailed description and discussion in the Guidance Document (Hauschild and Potting 2003) based on this Technical Report. 4.5 DiscussionThe comments and reflections on the acidification factors in Section 3.4 and 3.5 in Chapter 3 also apply to the terrestrial eutrophication factors discussed here. Chapter 3 is with minor modifications reproduced from the article of Potting et al. (1998). This article and other work by Potting and colleagues on spatial issues in LCA inspired similar research activity elsewhere, and some of the developments since 1998 are discussed here. Krewitt et al. ( 2001) used a different model, the EcoSense model, to calculate the same type of characterisation factors as Potting et al. (1998a,b). However, the effect data for acidification and eutrophication in EcoSense are of tentative quality and the characterisation factors in Krewitt et al. (2001) for these impact categories were later withdrawn (Krewitt 2003). They are therefore not further discussed here. Huijbregts et al. (2000) used the RAINS-model to calculate acidification and terrestrial eutrophication factors, but their definition of the category indicator differs slightly from the one used here and in Chapter 3 in that they do not cover modelling of threshold or critical load exceedance in terms of the area of unprotected ecosystems. Instead, they quantify deposition – that is, increase of exposure – divided by the critical load in grid-elements. Two sets of these so-called Hazard Indices are calculated. The first set covers all ecosystems in the model domain and is independent of the emission levels in the reference situation. The second set covers only those ecosystems that are unprotected or exposed above their critical loads and does depend on the – deposition levels determined by the – emission levels in the reference situation. The reference emission levels and subsequent deposition levels are used to select the ecosystems to be taken into account (in the "Only Above" indices), but they do not play a role as such in the calculation of the hazard indices from Huijbregts et al. (2000). The "Only Above" indices are calculated for 1995 emission levels and the estimated 2000 emission levels The acidification and eutrophication indices or factors of Huijbregts et al. (2000) show considerable, but smaller variation than in the acidification and eutrophication factors in this Technical Report. In a comparison of the 2010 acidification factors for sulphur dioxide, nitrogen dioxide and ammonia with those presented in Chapter 3, Huijbregts et al. (2000) found a poor correlation for the "Above and Below" indices (r2 = 0.01 - 0.33), but a moderate correlation for the "Only Above" ones (r2 = 0.41 - 0.67). Potting (2004) pointed out that the variation in factors seems to become larger as the category indicator is defined closer to the endpoint. This might explain the bad correlation between the factors of Huijbregts et al. and those in this Technical Report. Another reason may be that the estimated emission levels in Potting et al. differ slightly from those in Huijbregts et al., whereas the latter also used a newer set of Critical Load data. It would be useful to compare both indicators on the basis of factors that are calculated with the same input data and the same model version. Goedkoop and Spriensma (1999) in their Eco-indicator 1999 defined their category indicator closer to the endpoint by following a damage approach. They quantified the potentially disappeared fraction of targeted vascular plant species as a function of the deposition of acidifying and eutrophying emissions. The resulting from-midpoint-to-endpoint-factor combines two impact categories. It is based on the Nature Planner from the National Institute of Public Health and the Environment (RIVM) which only covers the Dutch territory. The model of Goedkoop and Spriensma therefore does not allow the calculation of spatially differentiated acidification-eutrophication factors. Since their work, a European database with damage functions has become available (Bakkenes et al. 2002). It would be interesting to connect this database to the RAINS-model. This would facilitate damage factors that are both site-dependent and are based on a full emission-fate-exposure-effect(-damage) modelling. Site-dependent characterisation factors for acidification and terrestrial eutrophication have also been calculated for North America with federal states as the source regions (Bare et al. 2003). These factors represent site-dependent fate factors that express what share of an emission deposits on land. The calculation of the site-generic factors presented in Table 4.5 need some clarification and discussion,. A site-generic terrestrial eutrophication factor becomes necessary if the geographical location of a process is unknown, or known only at an aggregated level (for instance located in West or East Europe). In such a case, one would like to assess the eutrophying impact of the process by means of a factor based on the average of the factors for the regions where the process may be located. However, regions can vary considerably in economic activity. It would therefore be fair to weigh the average for the total regional activities or total regional emissions related to the process. Also this information may be unknown and the best choice is then to use one of the European averages from Table 4.5. For different selections of the 44 European regions, the average, standard deviation, minimum and maximum factor for terrestrial eutrophication has been determined. The site-generic factors in Table 4.5 have been calculated as simple means of the national (regional) factors of Table 4.1 and Table 4.2, and as means weighed by the country or region's total emission. The European averages or site-generic factors in Table 4.5 show, compared to the regional or site-dependent factors on which they are based, a moderate variation in the average factors for clearly different selections of regions. Considering that the standard deviations have the same size as the means, there is not significant difference between the site-generic factors but it is clear that aggregating at this level means that a large spatially determined uncertainty is hidden in the factors. Whether or not to perform a site-dependent impact assessment is still an unsolved point of discussion in life cycle assessment. A considerable group of practitioners would rather avoid the – limited – data gathering required in addition to present typical life cycle assessment. Even though the only additional required data, the geographical site of emission, is in general already provided by current inventory analysis. However, the geographical region of a process may sometimes really be unknown. This is the case in life cycle assessments dealing with processes taking place in the – far – future or for products consisting of materials taken from the spot market (though an average as described above may be appropriate in the latter case). There are also applications where even the effort of limited additional data gathering might exceed the resources of the practitioner [28]. One of the averages from Table 4.5 (e.g. the one for total Europe) can then be chosen as the "site-generic" characterisation factor. Given the large ranges and standard deviations in these European averages or site-generic characterisation factors, a sensitivity analysis with the regional or site-dependent characterisation factors is highly recommended in order to evaluate whether suggested improvement options are indeed significant. The procedure for such a sensitivity analysis is described in detail in the Guidance Document of Hauschild and Potting (2003). 4.6 Conclusions and recommendationsIn the same way as for acidification, characterisation and normalisation factors for terrestrial eutrophication have been established for 44 regions in Europe to facilitate site-dependent LCIA of terrestrial eutrophication. The eutrophication factors relate the region of emission to the impact on its deposition areas. The conclusions about the acidification factors established in Chapter 3 basically also apply for the terrestrial eutrophication factors and are not repeated here. Average factors, standard deviation, minimum and maximum factors for terrestrial eutrophication have been determined for different selections of the 44 European regions. Results show a moderate variation in the average factors for clearly different selections of regions (West and East Europe), but the variation between rather similar selections of regions is small (EU and EU+2). The exact selection of regions becomes thus less important as a rough indication for the unknown area where a process is expected to take place. Whereas even a rough indication of the process' location does result in clearly different averages, the large range and even large standard deviations prevent the differences from being significant. There are applications where even the effort of limited additional data gathering might exceed the resources of the practitioner. One of the averages from Table 4.5 (f.i. the one for total Europe) can than be chosen as a sort of "site-generic" characterisation factor. Given the large ranges and standard deviations in these averages, a sensitivity analysis with the regional characterisation factors is highly recommended in order to evaluate whether suggested improvement options are indeed significant. The procedure for such sensitivity analysis is described in detail in the Guidance Document of Hauschild and Potting (2003). 4.6.1 AcknowledgementResearch for this work was also supported by a Training and Mobility grant from the European Commission, and by the Danish Agency for Industry and Trade under the EUREKA project LCAGAPS. 4.7 ReferencesAlcamo, J., R. Shaw and L. Hordijk (eds.). The RAINS model of acidification. Science and strategies in Europe. Dordrecht (the Netherlands), Kluwer Academic Publishers, 1990. Amann, M., I. Bertok, J. Cofala, F. Gyarfas, C. Heyes, Z. Klimont and W. Schöpp. Cost-effective control of acidification and ground-level ozone. Second interim report to European Commission, DG-X1. Laxenburg (Austria), International Institute for Applied Systems Analysis (IIASA), 1996. Amann, M., M. Baldi, C. Heyes, Z. Klimont and W. Schöpp. Integrated assessment of emission control scenarios, including the impact of troposheric ozone. Water, air and soil pollution, Vol. 85 (1995), Issue 4, pp2595-2600. Bakkenes, M., R. Alkemade, F. Ihle, R. Leemans and J.B. Latour. Assessing effects of forecasting climate change on the diversity and distribution of European higher plants for 2050. Global Change Biology, Vol. 2002, Issue 8, pp390-407. Bare J.C., Norris G.A., Pennington D.W., McKone T. TRACI: The US EPA's tool for the reduction and assessment of chemical and other environmental impacts. Journal of Industrial Ecology, Vol. 6 (2003), Issue 3/4, pp49-78. Barrett, K. and E. Berge (eds.). Transboundary air pollution in Europe. Part 1: Estimated dispersion of acidifying agents and of near surface ozone. EMEP MSC-W status report (research report no. 321996). Oslo (Norway), Norwegian Meteorological Institute, 1996. Berdowski, J.J.M. and W.J. Jonker. Emissions in the Netherlands. Industrial sectors, regions and individual substances (1992 and estimates for 1993; Publication series emission registration no. 21; in Dutch). The Hague (the Netherlands), Ministry of Housing, Spatial Planning and Environment, 1994. Chardon, W.J. The role of the soil in the phosphorous cycling In: Weidema, B.P. and M.J.G. Meusen (eds.) Agricultural data for life cycle assessment. Volume 1 and 2. The Hague (the Netherlands), Agricultural Economics Research Institute (LEI), 2000,p13-24. Downing, R.J., J.P. Hettelingh and P.A.M. de Smet (Eds). Calculation and mapping of critical thresholds in Europe. CCE status report 1993. Bilthoven (the Netherlands), Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and the Environment (RIVM), 1993. Finnveden, G., Y. Andersson-Sköld, M.-O. Samuelsson, L. Zetterberg and L.-G Lindfors. Classification (impact analysis) in connection with life cycle assessment. In: Product life cycle assessment. Principles and methodology (Nord 1992:2). Copenhagen (Denmark), Nordic Council of Ministers, 1992, pp172-231. Finnveden, G. and J. Potting. Eutrophication as an impact category. Int. Journal of Life Cycle Assessment, Vol, 4 (1999), Issue 6, pp311-314. Goedkoop, M. and R. Spriensma. The Eco-indicator 99. A damage oriented method for life cycle impact assessment. Methodology report and annex. Amersfoort (the Netherlands), Pré consultants, 1999. Hauschild, M. and J. Potting. Spatial differentiation in life cycle impact assessment – the EDIP2003 methodology. Guideline from the Danish Environmental Protection Agency, Copenhagen, 2003. Heijungs, R. G. Huppes, R.M. Lankreijer, H.A. Udo de Haes, A. Wegener Sleeswijk, A.M.M. Ansems, P.G. Eggels, R. van Duin and H.P. de Goede. Environmental life cycle assessment of products. Guide and backgrounds (ISBN 90-5191-064-9). Leiden (the Netherlands), Centre of Environmental Science of Leiden University, 1992. Huijbregts, M., W. Schöpp, E. Verkuijlen, R. Heijungs and L. Reijnders. Spatially explicit characterisation of acidifying and eutrophying air pollution in life-cycle assessment. Journal of Industrial Ecology, Vol. 4 (2000), Issue 3, pp125-142. Krewitt, W. A. Trukenmüller, T. M. Bachmann and T. Heck. Country specific damage factors for air pollution; A step towards site-dependent life cycle assessment. Int. Journal of Life Cycle Assessment, Vol. 6 (2001), Issue 4, pp199-210. Krewitt, W. Personal email communication in September 2003 to the SETAC Work Group of Impact Assessment concerning the effect assessment of eutrophication and acidification in EcoSense in relation to the characterisation factors in Krewitt et al. 2001. Lindfors, LG., M. Alkemark, C. Oscarsson and C. Spännar. A manual for the calculation of ecoprofiles intended for third party certified environmental product performance declarations. Stockholm, Swedish Environmental Institute (IVL), 1998. Lindfors, L-G., K. Christiansen, L. Hoffman, Y. Virtanen, V. Juntilla, O-J. Hanssen, A. Rønning, T. Ekvall and G. Finnveden. Nordic guidelines on life cycle assessment (Nord 1995-20). Copenhagen (Denmark), Nordic Council of Ministers, 1995 Posch, M, P.A.M. de Smet, J.P. Hettelingh, R.J. Downing (eds.). Calculation and mapping of critical thresholds in Europe. CCE status report 1995. Bilthoven (the Netherlands), Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and the Environment (RIVM), 1995. Posch, M., J.P. Hettelingh, R.J. Downing, and P.A.M. de Smet (Eds). Calculation and mapping of critical thresholds in Europe. CCE status report 1997. Bilthoven (the Netherlands), Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and the Environment (RIVM), 1997. Posch, M, P.A.M. de Smet, J.P. Hettelingh, R.J. Downing (eds.). Calculation and mapping of critical thresholds in Europe. CCE status report 1999. Bilthoven (the Netherlands), Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and the Environment (RIVM), 1999. Posch, M, P.A.M. de Smet, J.P. Hettelingh, R.J. Downing (eds.). Modelling and mapping of critical thresholds in Europe. CCE status report 2001. Bilthoven (the Netherlands), Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and the Environment (RIVM), 2001. Potting J. Acidification and terrestrial eutrophication. Comparison of different levels of sophistication. Int J. LCA. Accepted, 2004. Potting, J. and K. Blok. 1995. Life cycle assessment of four types of floor covering. Journal of Cleaner Production, Vol. 3 (1995), Issue 4, pp201-213. Potting, J. and K. Blok. Spatial aspects of life cycle impact assessment. In: Udo de Haes, H.A., A.A. Jensen, W. Klöpffer and L-G. Lindfors (eds.). Integrating impact assessment into LCA. Proceedings of the LCA symposium held at the fourth SETAC-Europe Congress, 11-14 April 1994 at the Free University of Brussels in Belgium. Brussels (Belgium), Society of Environmental Toxicology and Chemistry – Europe, 1994. Potting, J., W. Schöpp, K. Blok and M. Hauschild. Comparison of the acidifying impact from emissions with different regional origin in life-cycle assessment. Journal of Hazardous materials, Vol. 1998a, Issue 61, pp155-162. Potting, J., W. Schöpp, K. Blok and M. Hauschild. Site-dependent life impact cycle assessment of acidifciation. Journal of Industrial Ecology, Vol. 2 (1998b), Issue 2, pp.63-87. Wenzel, H., Hauschild M.Z. and Alting, L.: Environmental assessment of products. Vol. 1 - Methodology, tools, techniques and case studies, 544 pp. Chapman & Hall, United Kingdom, ISBN 0 412 80800 5, Kluwer Academic Publishers, Hingham, MA. USA., 1997. 5 Aquatic eutrophication5.1 Introduction Authors: José Potting [29], Arthur H.W. Beusen [30], Henriette Øllgaard [31], 5.1 IntroductionAlthough still of good quality in some regions, European inland waters (fresh waters like rivers and lakes) as well as coastal seas are threatened by a multitude of human activities. The growing number of both users and uses [34] has increased the exploitative pressure on the European waters. This gives more and more conflicts between the various uses and users, and also between man and organisms living in or near those waters. A decline in quantity and quality of the water available for human uses, as well as of the ecological quality of aquatic ecosystems may be the result. A release of pollutants to water may have considerable local impact (in case of a point source), but its main impact seems its contribution to pollution accumulation by other sources upstream and downstream in rivers and finally in coastal waters. Water quality, and eutrophication or nutrient enrichment as part of that, has therewith become a regional topic. Kristensen and Hansen (1994) provide an interesting and comprehensive overview of the state of the environment in European rivers and lakes. EEA (1998) gives a short overview for both marine and inland waters. The focus of this chapter is on life cycle assessment of long-range nutrient enrichment of marine and inland waters. The presently typical way to assess eutrophication in life cycle assessment does not account for spatial variation in emissions, exposure and effect (Section 5.2). This chapter will explore the differences in eutrophying impact from nutrients released at different geographical locations and by different source categories. Section 5.4 describes the CARMEN-model, a model for integrated assessment of eutrophication, which is used to establish spatial resolved exposure factors. The CARMEN-model does not cover an assessment of ecological effect, but Section 5.3 gives a brief review on the issue. Section 5.5 describes how the CARMEN model is used to establish spatial resolved eutrophication factors. The results are presented in Section 5.6 and discussed in Section 5.7. Section 5.8 draws the main conclusions. 5.2 Typical life cycle impact assessmentThe eutrophying impact usually characterised in life cycle impact assessment relates implicitly to eutrophication of aquatic ecosystems. This follows from the modelling of impact that in life cycle assessment typically takes its bases in the composition of aquatic biomass (Wenzel et al. 1997, Heijungs et al. 1992, Lindfors et al. 1995): An increased input of nutrients into an aquatic ecosystem may lead to an increased production of biomass, primarily phytoplankton (see also Section 5.3). The typical composition of phytoplankton, as suggested by Redfield et al. (1993) [35] and commonly used in life cycle impact assessment, consists of carbon, nitrogen and phosphorus in a ratio of 106:16:1. This ratio suggest that 106 mole carbon, 16 mole nitrogen and 1 mole phosphorus is needed for the production of 1 mole of phytoplankton. In life cycle impact assessment of eutrophication, the Redfield ratio is used the other way round to express the contribution of emissions of nitrogen and/or phosphorus to biomass production in terms of the equivalent emission of a reference substance. The reference substance differs between methods, but these methods are further nearly similar. Table 5.1 lists equivalency factors as proposed by Wenzel et al. (1997). Table 5.1. Equivalency factors for nutrient enrichment from Wenzel et al. (1997).
Wenzel et al. (1997) suggest aggregation of the different nitrogen substances separate from those of phosphorus to allow site-considerations in the subsequent weighing of the accumulated substances (see Table 5.1 and Formula 5.1). This separate characterisation of phosphorus and nitrogen does anticipate inland freshwaters typically to be phosphorus limited while marine waters are typically limited by nitrogen. However, Wenzel et al. do also provide factors to add together nitrogen and phosphorus (see Table 5.1 and Formula 5.1): Wenzel et al. (1997) do not distinguish between differences in eutrophying impact from emissions to air, water and soil, and also not distinguish between eutrophication of terrestrial and aquatic ecosystems. Terrestrial ecosystems get eutrophied mainly by airborne emissions [36], [37], however, whereas waterborne nutrients from waste water and originating from agriculture dominate eutrophication of aquatic ecosystems (though stagnant water can be polluted by air depositions as well). The agricultural nutrient data in life cycle inventory usually refer to the amount after plant uptake and binding. This amount can leave the topsoil and may transport to surface water depending on environmental conditions (like soil type or net precipitation). Wenzel et al. (1997) does also not consider variation in environmental conditions. They suppose all types of emissions to fully contribute to eutrophication in general. Similar to Finnveden et al. (1992), we consider eutrophication of terrestrial and aquatic ecosystems as two separate impact categories. This chapter deals with eutrophication of aquatic ecosystems (Chapter 4 addresses terrestrial eutrophication). This chapter focuses on the geographical differences in transport of nutrients through the environment to inland waters and marine waters. The biological aspects of eutrophication will be described first, however. 5.3 EffectThe term "eutrophic" is a biological one and literally means "rich of nutrients", whereas the term "eutrophication" refers to "the process of becoming rich of nutrients" (Dikke van Dalen 1994, Kristensen and Hansen 1994) [38]. This section first elaborates on the nutrients relevant in eutrophication (Section 5.3.1), then describes the potential ecological effects from eutrophication (Section 5.3.2), and finally addresses which nutrients may limit biomass growth (Section 5.3.3). 5.3.1 Relevant nutrientsThe production of biomass in aquatic ecosystems is governed at any time and place by numerous factors simultaneously. However, the overall productivity under otherwise comparable conditions is largely determined by those factors that limit production over a substantial length of time during the main growth period. This long-term limitation is most often – though not always – due to one nutrient being poorly available in the given ecosystem itself and/or not present in sufficient quantities in natural supplies from outside. The concept of "the limiting nutrient" is essential when discussing nutrient enrichment or eutrophication. It states that one nutrient is limiting the growth of primary producers (plants) and thereby indirectly affects the ecosystem and that there is an excess of all other nutrients. An additional amount of the limiting nutrient will lead to increased growth, however, additional amounts of other nutrients will not since they are already in excess. (Finnveden and Potting 1999) Those nutrients most likely to act as limiting factors can be identified by ranking the amount and proportions of all critical elements in aquatic ecosystems against their concentration and proportion in the – unpolluted – water. Hydrogen, oxygen and most salts (like calcium, magnesium, potassium, sodium and chloride) are usually available in abundance and can therefore be eliminated. Carbon dioxide, (bi)carbonates and sulphate are generally available in excess compared to nitrogen and phosphorus in both freshwaters and marine waters. This makes nitrogen and phosphorus the primary candidates for "chronic" nutrient limitation [39]. Both nitrogen and phosphorus exist in many different chemical forms in the aquatic environment. Impact and bio-availability of each form may be different, but they have the ability to interchange. The transformations between the several forms are site-specific and continuous (known as the nitrogen and phosphorus cycle). All forms of nitrogen and phosphorus forms are therefore relevant, and total nitrogen and phosphorus are a good expression for long-term eutrophication. Trace elements may in rare cases be limiting. Primary producers as phytoplankton only need very small quantities of these elements (Hauschild 1998), however, and they are therefore not further addressed here. 5.4 Ecological effectsThe supply of nutrients to water is under natural circumstances in balance with the subsequent growth of biomass such that a stable though variable society of plant and animals is ascertained. Such society is
to some extent able to cope with variations in nutrient supplies. A too large nutrient increase pushes this stable society out of balance, however, and may through a chain of ecological effects provoke a shift
of the biological structure. The effects are generally more apparent in standing or slowly moving waters than in (small) fast flowing rivers (Kristensen and Hansen 1994). The chain of ecological effects is
illustrated in Figure 5.1:
Figure 5.1. Schematic representation of the ecological chain effects as a result of the increase with nutrients or eutrophication of lakes (reproduced from Kristensen and Hansen 1994). See below text for an explanation of the arrows. Standing and shallow clear waters with submerged plants become dominated by phytoplankton after a high nutrient input (→1). Increasing growth of phytoplankton makes the water turbid (→2). The turbidity prevents the light to reach the water-bottom, which makes the submerged plants disappear (→3). The community fish also changes because the predatory fish are unable to see and catch the smaller fish (→4). The fish community becomes dominated by zooplankton-eating species that are more tolerant to the turbidity (→5). Zooplankton eats phytoplankton and the decrease in zooplankton (→6) therefore results in a further increase of phytoplankton (→7). The excess phytoplankton dies and sinks to the bottom (→8). The decay of phytoplankton uses oxygen (→9). The decrease of oxygen leads to further fish kills and disappearance of bottom fauna (→10), and may also evoke a release of phosphorus from the lake bottom (→11). The released phosphorus may be used for a new increase of phytoplankton (→12) after which a new cycle of phytoplankton dying and sinking starts. Topography and the physical/chemical nature of the water influence the impact of nutrients on aquatic biomass growth. It is therefore difficult to define generic nutrient levels above which this growth may become problematic. Kristensen and Hansen (1994) give water quality indications for inland waters and EEA (1998) for marine waters (see Table 5.2). Table 5.2. Water quality categories abstracted from EEA (1998) and Kristensen and Hansen (1994). These levels have to be taken with caution since threshold values are influenced by topography and physical/chemical nature of the water. The nutrient levels refer to the extent of anthropogenic origin rather than to ecological effect.
Based on EEA (1998) Based on Kristensen and Hansen (1994) Drinking Water Directive (80/778/EEC) Kristensen and Hansen (1994) collected and analysed monitoring data about – amongst others – nitrogen and phosphorus concentrations from 800 stations in more than 550 European rivers and 1500 lakes. The annual mean concentrations in the majority of the rivers range from levels of moderate to heavy pollution. Only rivers in northern Europe seem hardly disturbed. Particularly rivers in western Europe show high nitrogen and phosphorus levels. The situation looks somewhat better for European lakes, though the same trends can be observed here. The water quality indications in Table 5.2 relate nutrient levels to the extent of anthropogenic origin rather than to the occurrence of ecological effects caused by biomass growth. Kristensen and Hansen (1994) also describe the biological state or rivers in a number of European countries. Their results are given in Table 5.3 and confirm the trends shown by the monitored data as described in the previous paragraph. EEA (1998) describe the water quality in European seas. They gives annual mean nitrogen and phosphorus concentrations at a number of sample points in west and north European seas and qualitative information about southern seas. Obviously, nutrient concentrations are high and water quality is often poor/bad close to estuaries of rivers (southern part of the North sea, Irish Channel, Waddenzee). The Mediterranean Sea is one of world's most oligotrophic (nutrient-poor) seas. However, eutrophication problems occur in semi-enclosed bays (mainly due to untreated sewage) and uncontrolled expansions of fish farming (Eastern Mediterranean). The northern and west coast of the Adriatic Sea, that receives the nutrient loads of the River Po, is the most endangered in the Mediterranean area. The long residence time of the water in the Black Sea makes this sea highly sensitive to eutrophication. The water quality in this sea and the Sea of Azov is bad, probably due to high inputs from the Danube, Dnieper and Dniester. Table 5.3. Percentage of river reaches in various European countries classified as being of good, fair, poor or bad quality. Of good quality are river reaches with nutrient-poor water, low levels of organic matter, saturated with dissolved oxygen, rich invertebrate fauna, and suitable spawning ground for salmonid fish. River reaches with moderate organic pollution and nutrient content, good oxygen conditions, rich flora and fauna, large fish population are classified as fair. Poor quality river reaches have heavy organic pollution, usually low oxygen concentrations, locally anaerobic sediment, occasional mass occurrence of organism insensitive to oxygen depletion, small or absent fish population, periodic fish kill. Of bad quality are those rivers with excessive organic pollution, prolonged periods of very low oxygen concentration or total deoxygenation, anaerobic sediment, severe toxic input, devoid of fish. (Kristensen and Hansen 1994)
5.4.1 Concept of the limiting nutrientThe concept of "the limiting nutrient" is essential when discussing nutrient enrichment or eutrophication. Generally seawaters are regarded to be "chronic" limited by nitrogen, whereas lakes and larger, slowly flowing rivers are limited by phosphorus. Estuaries can be limited both by nitrogen and phosphorus, whereas small streams and mountainous lakes are usually not limited by nitrogen and phosphorus, but by factors as light and temperature (Miljøstyrelsen 1991, Blau and Seneviratne 1995). The concept of "the limiting nutrient" is a simplification because the limiting nutrient may change over seasons, and also over the years due to earlier loading. (Kristensen and Hansen 1994, Finnveden and Potting 2000) All nutrients can therefore potentially be used for biomass growth, though this maximum will for nitrogen usually not be the case in inland waters and for phosphorus not in marine waters. 5.5 The CARMEN modelCARMEN is an acronym for Cause effect Relation Model to support Environmental Negotiations. It is an integrated assessment model to analyse and evaluate strategies to reduce nutrient loading of inland waters [40] and coastal seas in Europe. The model does actually not contain an assessment of ecological effects, but calculates the change in nutrient loads in ground water, inland waters (river catchments) and coastal seas from changes in nutrient emissions and supplies (i.e. the causes). CARMEN, version 1.0 is used in this study. The causes modelled by CARMEN are atmospheric deposition of nitrogen on soil and coastal seas, phosphorus and nitrogen supply to agricultural soils, and phosphorus and nitrogen in municipal wastewater (see Figure 5.2 and Section 5.4.1). CARMEN models the transport of nutrient by rivers to sea relatively straightforward, but the transport of nutrients from agricultural supply and atmospheric deposition through groundwater drainage and surface runoff, to surface water is modelled spatially resolved over 124320 grid-elements of 10*10 minutes (roughly 100-250km2, depending on the longitude and latitude location of the grid-element). (see Section 5.4.2) Meinardi et al. (1994a,b), Klepper et al. (1995) and Beusen (not published) describe the CARMEN model in detail, but a short overview is given here about estimates of emissions (Section 5.4.1) and transport via groundwater drainage and surface runoff (5.4.2) of nutrients as modelled in CARMEN. 5.5.1 Emission estimatesMain sources for nitrogen in the aquatic environment are – via groundwater drainage and surface runoff – the use of fertiliser and manure in agriculture (diffuse sources), and municipal wastewater and – too a lesser extent – industrial effluent (mainly point sources). Deposition of airborne nitrogen is of some relevance for marine waters, but has usually minor importance for inland waters (except for the indirect contribution via groundwater drainage and surface runoff). Natural nitrogen can be important in regions with low population density. (EEA 1998, Kristensen and Hansen 1994) Figure 5.3 gives an overview of the relative importance of the different sources. Most of the phosphorus loading of inland waters is attributable to discharge from point sources, especially municipal wastewater and – too a lesser extent – industrial effluent. The application of phosphorus in agriculture may also contribute to inland waters through topsoil erosion. Phosphorus will usually not reach the ground water due to strong adsorption to the soil, though it exceptionally occurs after excessive loading on very poor soils (EEA 1998, Klepper et al. 1995, Kristensen and Hansen 1994)
Figure 5.2. Main sources for nitrogen (continuous arrow) and phosphorus (dashed arrow) to soil, groundwater, surface waters and coastal seas addressed in the CARMEN model (Beusen not published). CARMEN models three main sources for nitrogen and phosphorus to surface water (see also Figure 5.2): agriculture, municipal wastewater and atmospheric deposition (only for nitrogen). The several nitrogen and phosphorus supplies have been allocated to each grid-element on the basis of the distribution of land uses in the given grid-element (arable land, grassland, permanent crops, forest, urban area, inland waters, others). 5.5.2 Fate and exposure assessmentThe CARMEN model distinguishes between sources directly and indirectly via inland waters releasing to coastal seas. Oxygen depletion has been a severe problem for several years, but implementation of biological treatment of domestic and industrial wastewater has resulted in that many rivers and lakes are now fairly well oxygenated. However, low oxygen content is a usual phenomenon in the first stretches downstream of point sources for wastewater treatment. Under low oxygen conditions, nitrate is decomposed into nitrogen gas and oxygen used for the biomass decomposition. The nitrogen gas is released to the atmosphere. Removal of nitrogen is modelled for all waters the same in CARMEN by assuming a generic removal of 30% of the nitrogen input (i.e., 70% transports to sea). Figure 5.3. (Figure 9.11 and 9.14 from EEA 1998, p201-202) Phosphorus on its turn may be used for biomass production or temporarily adsorbed in the phosphorus pool in the bottom sediments of lakes and rivers. All phosphorus will remain in the water, however, and in the end transport to marine waters. CARMEN therefore considers no removal of phosphorus (i.e. all of it transports to sea). The strength of CARMEN is a detailed spatial resolved modelling of the transport of nutrients from agricultural supply and atmospheric deposition – through groundwater drainage and surface runoff or topsoil erosion – to surface water. The water flow is the main transport mechanism that brings nutrients – through deep groundwater drainage (nitrogen), runoff (nitrogen) or topsoil erosion (phosphorus) – to surface water. The net precipitation determines the amount of water draining to either groundwater or by surface runoff. The ratio between deep groundwater recharge and surface runoff resulting from net precipitation is determined in CARMEN by: Aquifer type (permeability of subsurface) The flow of deep groundwater to surface water is in addition determined by: Nitrogen transport through deep groundwater and surface runoff to surface water further depends on the amount leaching to the soil water at shallow depth after plant uptake and binding in the topsoil matrix. The share of nitrogen leaching is fixed for arable land by soil type and for agriculturally used grasslands, it is increasing with nitrogen dose for all soils (see also Section 5.7.2.). Phosphorus concentrations in groundwater are usually negligible because of the strong adsorption to the soil. Phosphorus transport to surface water is dominated by erosion of sediment from the topsoil by surface water runoff. (Meinardi et al. 1994/1995) Each of the above factors are modelled by CARMEN spatially resolved over 124320 grid-elements of 10*10 minutes (roughly 100-250km2, depending on the longitude and latitude location of the grid-elements). 5.6 Calculation procedureSite-dependent eutrophication factors have been established with help of the CARMEN model for 32 European countries. The factors relate the amount of nutrient released in a given country to its share to eutrophication of European inland waters [41] and coastal seas (i.e., respectively the share of nutrient indirect to seas through inland waters and the share of nutrient indirectly and directly to seas). The factors quantify the maximum biomass growth these nutrients may contribute to in the receiving water. The factors do not anticipate the deterioration of the water quality as a result of this biomass growth. The eutrophication factors per country were calculated by changing the total amount of either nitrogen or phosphorus from a given source category in one country (other emissions for all countries and other source categories remaining the same). Agricultural supplies of manure or fertiliser to soil, and wastewater releases have been considered as sources for both inland waters and seas. The calculations for coastal seas in addition also addressed atmospheric deposition as a nitrogen source. For each source category, the change in eutrophying loads is calculated spatially resolved over 101 river catchments and 32 coastal seas (see Annex 5.1 and 5.2). Next, the loading increases by this change of one country are accumulated over all river catchments and seas to obtain the factors expressing the share eutrophying respectively inland waters and seas (in kg per kg released). CARMEN models the contributions from nitrogen depositions to eutrophication of inland waters and seas from the actual deposition pattern in Europe. It does not contain the relationships between country emissions and depositions. Huijbregts and Seppälä (2000) recently published data about the nitrogen deposited on European seas as a ratio of the emission in their country of release. These data have been combined with calculations with the CARMEN model to arrive at the loading of separate seas. The data from Huibregts and Seppälä do not consider deposition outside the EMEP model domain, and have therefore been corrected with data from Barrett and Berge (1996) about the percentages exported outside the EMEP model domain. The CARMEN model does not include an assessment whether nutrient loading actually results into biomass growth and what effect this has on the ecological quality of the water. The calculated factors represent the potentially maximum contribution to biomass growth. They are exposure factors rather than expressing ecological effects. 5.7 ResultsThe results of the calculations are abstracted in Table 5.4. Columns 2 to 7 in Table 5.4 give the share of nutrient from a given source category that contributes to eutrophication of inland waters. The last eight columns do the same for marine waters. Table 5.4. The share of nitrogen and phosphorus eutrophying inland waters (Column 2 to 7) and marine waters (Column 8 to 15) for different source categories in different countries (Fert.=fertiliser, Man.=manure, WW=wastewater, NH3=airborne NH3 , NOx=airborne NOx). Hence that the factors for fertiliser and manure relate to nutrient after plant uptake (see Section 5.7.2 for how to arrive at the proper inventory data). There are a number of countries where eutrophication of marine waters by agriculture and wastewater is much smaller than their eutrophication of inland waters (underlined). These numbers relate to countries releasing on seas outside the model domain (they may be replaces with factors for inland waters as best estimate and by a factor 0.7 for nitrogen and a factor 1.0 for phosphor in wastewater). The out-crossed numbers are concluded to be wrong. For Denmark, the factor for marine waters may be lowered to 0.7 for nitrogen and to 1.0 in phosphor in wastewater. The other numbers being striketrough should be replaced by the site-generic factors. Both underlined and out-crossed numbers are excluded from the mean etc.
Hence the factors relating to application of fertiliser and manure refer to the amount of nutrient after plant uptake. This amount is available to leave the topsoil. It is a peculiarity of life cycle assessment to consider the topsoil as part of the economic system or technosphere and the data from inventory analysis should reflect the outputs from the technosphere to the ecosphere. One should thus be careful to apply the factors in Table 5.4 to the proper inventory data (see also Section 5.7.2). The factors in Table 5.4 show eutrophication from fertiliser and manure basically to be similar. There is therefore no reason to employ different factors for these source categories in life cycle assessment. There is neither a difference in the mean factors for agricultural eutrophication of inland waters and marine waters. The eutrophication factors from nitrogen [42] show notable differences for a number of individual countries (i.e. the Netherlands, the Scandinavian countries, the United Kingdom). The main cause is that along the relative long coastline, small water systems are discharging directly to marine waters. Table 5.4 shows the Swiss and Austrian factors for inland waters to deviate from the one for marine waters, while both countries do not have a direct connection to sea. The reason for this deviation is not clear. There are also a number of countries where agricultural eutrophication of seas seems to be much smaller than their eutrophication of inland waters (i.e. Russia, Spain, Portugal, Iceland and Ireland). However, some rivers in these countries release on marine waters that are outside the model domain of CARMEN. The sea factors may here be corrected by replacing them for the inland water factors (which are expected to be a fair estimate). Similar as for nitrogen from agriculture, differences exist between the factors for eutrophication of coastal seas and inland waters by wastewater. These factors express basically the share of nutrient (wastewater) discharged indirectly via inland waters or indirectly plus directly to marine waters. The factors for marine waters and inland waters can be at maximum 0.7 for nitrogen and 1.0 for phosphorus. Hence the Icelandic factors for inland waters are larger then these maximum values and thus probably false. The same applies for Denmark's factors for seas. Small exceedances (like 0.01) of the maximum values (0.7 or 1.00) are expected to be rounding errors. Within source categories, the differences between countries are relatively small. There is a factor 3 for agricultural nitrogen and a factor 7 for phosphorus between lowest and highest factor. The nitrogen factors for the Netherlands and Denmark (both having intensive agriculture) are remarkable low. This is due to the fact that the groundwater recharge of both countries is relative large compared to surface runoff (ratio larger than one). This means that the main part of the nutrients will flow into the groundwater, and due to a time delay of more than 10 years and a mixing with unpolluted groundwater, only a small part of the nutrients will flow out, to the inland waters. The phosphorus factors for Austria and Switzerland are remarkable high. The slope seems here to have a considerable impact on surface runoff and erosion. 5.8 DiscussionSome remarks about the eutrophication factors have already been made in the previous section and will not be repeated here. However, a few additional aspects are discussed in more detail in the following. 5.8.1 Application of the eutrophication factorsThe application of the eutrophication factors in Table 5.5 is very simple. An emission in the product's life cycle is multiplied with the eutrophication factor for that region and substance to derive the estimated eutrophication of that emission. The only additional data required, the country where an emission takes place [43], are in general already provided by current LCI.
The receiving waters can be inland waters on the one hand, and marine waters on the other hand. Both should be regarded in life cycle assessment as subcategories and thus reported separately (i.e. not to aggregate them). It is recommended to refrain from aggregating phosphorus and nitrogen, since nitrogen may at some point leave the aquatic system by nitrification, whereas phosphorus does basically not leave and remain to be available for potential eutrophication. This leads thus all together to two subcategories (inland waters and marine waters) with each two impact indicators (aggregated phosphorus and aggregated nitrogen). The Guidance Document of Hauschild and Potting (2003) based on this Technical Report gives a detailed description of the application procedure for the aquatic eutrophication factors. 5.8.2 Proper inventory dataIt is common practice in life cycle assessment to consider the topsoil of agricultural soils as part of the technosphere. The data in life cycle inventory for nutrient supply in agriculture therefore usually refer to the amount of nutrient available for leaving the topsoil after plant uptake and binding. (Weidema and Meusen 2000). The factors from Table 5.4 also refer to the amount of nutrient available for leaving the topsoil after plant uptake and binding. The eutrophication factors from Table 5.5 can therefore directly be multiplied with that type of inventory data. There is also a number of practitioners who do not consider the topsoil as part of the technosphere or lack data for implementing that, and stop their inventory at the level of nutrient supply. Inventory data reflecting nutrient supplies should first be corrected for plant uptake and binding before applying the eutrophication factors from Table 5.4. The factors in Table 5.5 are based on the relationships employed by CARMEN and can be used for this correction: Table 5.5. The factors expressing the relation between nutrient application and nutrients available for drainage by groundwater and surface runoff or erosion. The factors in this table can be multiplied with the factors in Table 5.4 to obtain the relation between nutrient application and eutrophication of inland waters and coastal seas.
According to Table 5.5, less then 25% of the nitrogen application and 10% of the phosphorus application leaches after plant uptake to soil water at shallow depth and is then available for transport through deep groundwater and surface runoff or erosion. This means that on average less then 12% of the supplied nitrogen and 5% of the supplied phosphorus will end up to be available for biomass growth in inland waters and finally coastal seas. This is in accordance with values given in the literature. (Weidema and Meeusen 2000) A similar situation is at stake for the phosphorus and nitrogen emissions to water. The waterborne nitrogen and phosphorus emissions in life cycle inventory usually refer to the content in effluent after wastewater treatment (unless emissions are released actually directly to surface water). The factors from Table 5.4 refer to releases directly to surface water (after potential purification thus). The eutrophication factors from Table 5.5 can therefore directly be multiplied with this type of inventory data. In case the inventory data do refer to wastewater before purification, the factors in Table 5.6 can be used in connection with Table 5.4 to correct the inventory data. Table 5.6. The expected concentrations (in mg/l) and removal efficiencies (in g/g) for nitrogen and phosphorus in waste after different types of waste water treatment (Hansen, 2000).
5.8.3 Biological oxygen demandDecomposition of organic material used oxygen that is measured as chemical or biological oxygen demand (COD or BOD). Fish kill and disppearance of bottom fauna may occur after prolonged situations of oxygen depletion. This may be the result of prolonged and high nutrient loading. Some methods presently typical for life cycle assessment of aquatic eutrophication therefore suggest considering organic materials, as part of this impact category (Lindfors et al. 1995, Heijungs et al. 1992). This suggestion is not followed here, since organic material often contains nitrogen or phosphorus and is then reflected already in the inventory table as nitrogen and/or phosphorus emission. To also add organic materials to this impact category, would then lead to double counting. Furthermore, organic material is not essentiel for growth of the biomass. Emissions of organic material, which causes BOD/COD, may increase or strengthen the effects and the environmental impacts of nutrient enrichment, especially with regards to oxygen depletion. However, these environmental impact of BOD/COD substances are comparable to the secondary effects of nutrient enrichment. 5.8.4 Related research activitiesDuring and after the work for this chapter, similar research activities were performed by other groups. Huijbregts and Seppälä (2000) used spatially resolved source-receptor matrices to calculate the share of atmospheric emission from source countries that deposits on marine waters. The rest deposits on land, but can transport through deep groundwater and surface runoff to surface water and marine water. The depositions of Huijbregts and Seppälä have been gratefully used here to calculate site-dependent factors that cover the direct and indirect contribution from the atmospheric emissions in all European countries to eutrophication of surface and marine waters. In addition to Huijbregts and Seppälä (2000), Huijbregts and Seppälä (2001) established characterisation factors for aquatic eutrophication, covering fate and potential effect for the Netherlands, Europe and the world. The fate part of these factors seem – at a first sight – less sophisticated than what is proposed in this chapter, whereas the effect factor is similar to what is proposed here. A closer comparison is recommended, however. Bare et al. (2003) calculated similar factors using a similar type of modelling as in this chapter for the federal states in North America. Due to the advanced state of the reporting of the Danish project, it was unfortunately not possible to make a closer comparison here. 5.9 ConclusionsEutrophication factors have been established for 32 countries in Europe to facilitate site-dependent assessment of eutrophication in life cycle assessment (see Table 5.5). The factors relate the country of emission to the potential maximum contribution to biomass growth in the receiving waters. The potential maximum will usually not be the actual contribution. Generally however, seawaters are regarded to be "chronic" limited by nitrogen, whereas lakes and larger, slowly flowing rivers are limited by phosphorus. Interpreting the results from impact assessment can use this and it is therefore recommended to refrain from aggregating nitrogen and phosphorus. Receiving waters can be inland waters and coastal seas (directly or indirect by river discharge to seas). Inland waters and coastal waters are recommended as separate subcategories (aggregating would lead to double counting). The established eutrophication factors are exposure factors rather than expressing the ecological effect of eutrophication and biomass growth. The present state-of-the art in integrated assessment modelling of aquatic eutrophication does not yet allow such effect assessment. The spatial resolved factors in Annex 5.3 to 5.9 may be used for a tentative interpretation in this direction. 5.10 ReferencesBare J.C., Norris G.A., Pennington D.W., McKone T. TRACI: The US EPA's tool for the reduction and assessment of chemical and other environmental impacts. Journal of Industrial Ecology., Vol. 6 (2003), Issue 3/4, pp49-78. Barrett, K. and E. Berge (eds.). Transboundary air pollution in Europe. Part 1: Estimated dispersion of acidifying agents and of near surface ozone. EMEP MSC-W status report (research report no. 321996). Oslo (Norway), Norwegian Meteorological Institute, 1996. Berdowski, J.J.M. and W.J. Jonker. Emissions in the Netherlands. Industrial sectors, regions and individual substances (1992 and estimates for 1993; Publication series emission registration no. 21; in Dutch). The Hague (the Netherlands), Ministry of Housing, Spatial Planning and Environment, 1994. Beusen, A. User manual of CARMEN1 (Draft). Bilthoven (the Netherlands), National Institute of Public Health and Environmental Protection (RIVM), not published. Blau, S. and S. Seneviratne. Acidifiction and eutrophication in life cycle assessments (LCAs). Zürich (Switzerland), Swiss Federal Institute of Technology Zurich, 1995. EEA Europe's environment: The second assessment. Copenhagen (Denmark), European Environmental Agency,1998. Finnveden, G., Y. Andersson-Sköld, M.-O. Samuelsson, L. Zetterberg and L.-G Lindfors. Classification (impact analysis) in connection with life cycle assessment. In: Product life cycle assessment. Principles and methodology (Nord 1992:2). Copenhagen (Denmark), Nordic Council of Ministers, 1992, pp172-231. Finnveden, G. and J. Potting. Eutrophication as an impact category. Int. Journal of Life Cycle Assessment, Vol, 4 (1999), Issue 6, pp311-314. Hansen, O.C: Ole Christian Hansen, unpublished work, 2000. Heijungs, R., J. Guinée, g. Huppes, R.M. Lankreijer, H.A. Udo de Haes, A. Wegener Sleeswijk, A.M.M. Ansems, P.G. Eggels, R. Van Duin en H.P. de Goede. Environmental life cycle assessment of products. Guide and background (ISBN 90-5191-064-9). Leiden (the Netherlands), Centre of Environmental Science of Leiden Univerisity, 1992 Huijbregts, M. and J. Seppälä. Towards region-specific, European fate factors for airborne nitrogen compounds causing aquatic eutrophication. Int. Journal of Life Cycle Assessment, Vol. 5, Issue 2, pp65-67, 2000. Huijbregts, M. and J. Seppälä. Life cycle impact assessment of pollutants causing aquatic eutrophication. Int. Journal of Life Cycle Assessment, Vol. 6, pp339-344, 2001. Jaarsveld, J.A. van, and D. Onderdelinden. TREND: An analtical long-term deposition model for multi-scale purposes (report no. 228603009). Bilthoven (the Netherlands), National Institute of Public Health and Environmental Protection (RIVM), 1990. Klepper, O., A.H.W. Beusen and C.R. Meinardi. Modelling the flow of nitrogen and phosphor in Europe: from loads to coastal seas (RIVM report 461501004). Bilthoven (the Netherlands), National Institute of Public Health and Environmental Protection (RIVM), 1995. Kristensen, P. and H. O. Hansen. European rivers and lakes. Assessment of their environmental state (EEA environmental monographs 1). Copenhagen (Denmark), European Environmental Agency, 1994. Lindfors, L-G, K. Christiansen, L. Hoffman, Y. Virtanen, V. Juntilla, O-J. Hanssen, A. Rønning, T. Ekval and G. Finnveden. Nordic Guidelines on life cycle assessment (Nord 1995; 20). Copenhagen (Denmark), Nordic Council of Ministers, 1995. Meinardi, C.R., A.H.W. Beusen, M.S.J. Bollen and O. Klepper. Vulnerability to diffuse pollution of European soils and groundwater (RIVM report 461501002). Bilthoven (the Netherlands), National Institute of Public Health and Environmental Protection (RIVM), 1994a. Meinardi, C.R., A.H.W. Beusen, O. Klepper and W.J. Willems. Nitrate contamination of European soils and groundwater (RIVM report 461501003). Bilthoven (the Netherlands), National Institute of Public Health and Environmental Protection (RIVM), 1994b. Miljøstyrelsen. Environmental impacts of nutrient emissions in Denmark. Redegørelse fra Miljøstyrelsesn, no. 1). Danish Environmental Protection Agency, Copenhagen, 1991. Paaby H, Møller F, Skop E, Jensen JJ, Hasler B Bruun H, Asman WAH. Omkostninger ved reduktion af næringsstofbelastningen af havområderne – Metode, model, analyse. Faglig rapport fra DMU, nr. 165. Danmarks Miljøundersøgelser, 1996. Redfield, A.C., B.H. Ketchum and F.A. Richards. The influence of organisms on the composition of sea water. In: Proceedings of the 2nd International Water Pollution Conference in Tokyo. Oxford (United Kingdom), Permagon Press, 1993, pp215-243. Samuelsson, M.-O. Life cycle assessment and eutrophication. A concept for calculation of the potential effects of nitrogen and phosphorus. Stockholm (Sweden), Swedish Environmental Research Institute (IVL), 1993. Weidema, B.P. and M.J.G. Meeusen (eds.) Agricultural data for life cycle assessment. Volume 1 and 2. The Hague (the Netherlands), Agricultural Economics Research Institute (LEI), 2000. Wenzel, H., M.Z. Hauschild and L. Alting: Environmental assessment of products. Vol. 1 - Methodology, tools, techniques and case studies, 544 pp. Chapman & Hall, United Kingdom, 1997, Kluwer Academic Publishers, Hingham, MA. USA. ISBN 0 412 80800 5. Wither, P.J.A. Cycling and sources of phosphorus in agricultural systems and to the wider environment: a UK perspective. In: Weidema, B.P. and M.J.G. Meusen (eds.) Agricultural data for life cycle assessment. Volume 1 and 2. The Hague (the Netherlands), Agricultural Economics Research Institute (LEI), 2000, pp13-24. Annex 5.1: The river catchments modelled in CARMEN
Annex 5.2: The coastal seas modelled in CARMEN
6 Photochemical ozone formation6.1 Introduction Authors: Michael Hauschild [44]Annemarie Bastrup-Birk [45] Ole Hertel45 Wolfgang Schöpp [46] José Potting44 6.1 IntroductionThe concentration of ozone in the troposphere rises by about 1% a year over the Northern hemisphere. An important source of this ozone as well as of other reactive substances is the photochemical oxidation of volatile organic compounds and carbon monoxide in the presence of nitrogen oxides and sun light in the boundary layer of the troposphere. The troposphere is the lower stratum of the atmosphere, reaching from the surface of the earth to the tropopause 8-12 km above us and the boundary layer is the lower part of the troposphere (up to an altitude of around 1 km) where full mixing can be assumed and where the exposure of life to the photo oxidants takes place. Due to their reactive nature, the photo-oxidants are injurious to the health of living organisms, vegetation as well as animals and humans. Apart from the general increase in the tropospheric ozone concentration, smog-episodes can occur on a more local scale in cities given a combination of high emission levels and the right meteorological conditions. During smog-episodes, the concentrations of ozone and other photo-oxidants reach extreme levels causing immediate damage to human health. Due to the local and discontinuous nature of photochemical smog-episodes, the focus in life cycle impact assessment is mainly on the more regional increase of tropospheric ozone. 6.1.1 Precursors and photo-oxidantsThe precursors necessary for photochemical ozone formation are nitrogen oxides which have a kind of catalytic effect and volatile organic compounds which are oxidised during the process. While NOx, nitrogen oxides, represents the sum of nitric oxide NO and nitrogen dioxide NO2, there is no universal definition of what a volatile organic compound, VOC is. In the current context, it is defined as an organic compound with a boiling point below 250C (WHO, 1989). For a VOC to contribute to photochemical ozone formation it must either contain hydrogen (i.e. it can not be fully substituted), or contain double bonds (i.e. be unsaturated) (Hauschild and Wenzel, 1998). Methane, which is a VOC according to the definition, is generally treated separately from the rest of the VOCs due on the one side to its very low photochemical reactivity (as reflected in its extremely long atmospheric residence time), and on the other side its large proportion of the total emission of volatile organic compounds. The abbreviation VOC is therefore generally – and also throughout this chapter - used to characterise the volatile organic compounds apart from methane (in some contexts also designated the NMVOCs). While being an intermediate in the atmospheric oxidation of VOCs, carbon monoxide from man-made emissions also contributes to photochemical ozone formation. The photo-oxidants comprise a variety of unstable, oxidising substances formed when VOCs react with oxygen compounds which are naturally present in the troposphere (the most important being the hydroxyl radical, OH). Among the photo-oxidants, the most prominent are ozone, O3, and peroxyacetyl nitrate, PAN, but intermediate products from the oxidation of VOCs also contribute to the injurious effects. It is thus not the VOCs per se which cause the environmental problems associated with photochemical ozone formation, but the products of their conversion. If a direct toxic effect of VOCs occurs, it is treated separately in life cycle impact assessment as a contribution to the impact categories human toxicity and ecotoxicity. 6.1.2 Photochemical ozone formation schemeSome tropospheric ozone arises from the natural transport of ozone from the stratosphere, where it is formed by the action of energetic UV rays from the sun, but the rest is produced in the troposphere by the photochemical ozone formation. Schematically, the photochemical ozone formation can proceeds through the following four steps: 1. Reaction between VOCs or CO and OH to form peroxy radicals, ROO. 2. The peroxy radicals oxidise NO to NO2. 3. NO2 is split by sunlight with formation of NO and release of oxygen atoms. 4. Oxygen atoms react with molecular oxygen, O2, to form ozone. The reaction schemes are complex and difficult to simulate in a model. The heterogeneous spatial distribution of VOC and NOx sources across Europe, and the many hundreds of chemical species involved, makes the photochemical formation of ozone on a regional scale highly non-linear. Apart from the emitted quantity of VOC and NOx, the following factors influence the ozone formation: Meteorological conditions Solar radiation is a driving force behind the photo oxidation but also wind speeds and directions (determining the dispersion of the air pollutants), temperature (influencing reaction kinetics) and precipitation patterns (influencing wet deposition and hence removal of pollutants from the boundary layer) are conditions that influence the photochemical ozone formation Interaction between pollutants The simultaneous concentration of the other relevant pollutants, NOx and individual VOCs, can strongly influence the overall result. The dependence of ozone formation on the precursor emissions (NOx, VOC) is therefore often far from linear. In regions with relatively low emissions, the NOx/VOC ratio is generally low, and ozone formation is limited by NOx. In these regions, reduced NOx emissions will thus reduce ozone formation while reductions in VOC emissions may have little influence on the ozone formation. In regions with extremely high NOx emissions, reduction in the emissions may actually lead to increased ozone formation. Here, the high concentrations of NO may lead to reduction of ozone (through oxidation of NO to NO2). In addition, NO may reduce the concentration of hydroxyl radical which thus limits the potential for oxidation of VOCs. In this situation, reduced emissions of NOx will therefore increase the availability of hydroxyl radical, increasing the oxidation of VOC and hence the ozone formation, unless VOC emissions are reduced at the same time. (Heyes et al., 1997a). Biogenic emissions of VOCs In Europe, the total natural emissions of biogenic VOCs is thought to be around 30 % of the total VOC emissions (Derwent et al., 1991) and this must be represented in a model for ozone formation. For the biogenic (natural) releases of NMVOC only little information is presently available, and often the empirical expressions derived by Lübkert and Schöpp (1989) to describe releases from forested areas are therefore applied. Properties of the individual VOC As mentioned earlier, the photochemical reactivity of methane is much lower than the reactivity of the VOCs. Among the VOCs there is also variation in the potential for formation of ozone as reflected in the individual photochemical ozone creation potentials (POCP) shown in Annex 6.2. A ranking of the different groups of VOCs according to their ozone formation potential per unit weight results in the following sequence (Derwent and Jenkin, 1990):
6.2 Existing systems for characterisation of ozone formationFigure 6.1 shows the cause-impact chain for photochemical ozone formation. The descriptors provided for each link of the chain are characteristics or parameters which influence the behaviour at that point in the chain.
Figure 6.1 Cause-impact chain for photochemical ozone formation. The existing characterisation factors for photochemical ozone formation are all based on the photochemical ozone creation potential, POCP or possibly the Maximum Incremental Reactivity (MIR) of the substances. Both express the short-term contribution to ozone formation. MIR is primarily designed to reflect the contribution during photo-smog episodes while POCP may be calculated with time horizons from 1 to 9 days. While MIR is expressed directly as moles of ozone produced per mole of VOC emitted, POCP is expressed as an equivalent emission of the reference substance ethylene (C2H4) – a strong ozone formation gas. In EDIP97, photochemical ozone formation is characterised using POCP-values for ozone formation over 4-9 days. Some spatial differentiation is built into the EDIP97 through a distinction according to the NOx concentration in the emission area. There are thus two sets of characterisation factors – one for low NOx regions and one for high NOx regions. This is a rather rough distinction since emissions taking place in high NOx regions may well be transported to low NOx regions and oxidised there (and vice versa) but at that time it was considered by the authors to be the best representation of the spatial variability of photochemical ozone formation that would be feasible in life cycle impact assessment. In addition to POCP values for around 90 individual VOCs, EDIP97 also provides aggregated POCP values for some common VOC-mixtures identified by their source rather than their composition. Furthermore, tools are provided for estimation of missing POCP values. Both the POCP and the MIR address impacts at the level called fate-dispersion and distribution in Figure 6.1. They reflect the substance's intrinsic potential for ozone formation under a set of standard conditions regarding the presence of other gases and the meteorological conditions and they focus primarily on the ozone formation during the first days after the emission as relevant for the prediction of photo smog episodes. The behaviour through the first links of the cause-impact chain from emission to the point where the POCP and MIR are modelled, depends to a large extent on the properties of the substance. Further along the chain, the conditions of the environment where the substance is emitted, dispersed, oxidised and where exposure of sensitive target organisms takes place become decisive. Hitherto, it has been seen as impossible to include these descriptors in the modelling of impact, even though they are known to be important – for photochemical ozone formation perhaps more important than the descriptors in the early parts of the cause-impact chain as illustrated by the spatially differentiated factors developed in Section 6.4. It has not been feasible to perform the required spatial differentiation in life cycle assessment, and therefore a low environmental relevance of the modelled impacts has been accepted. For photochemical ozone formation, an additional problem about the existing characterisation models is their inability to represent the impact from NOx. Both POCP and MIR have been developed for ranking of VOCs according to their ozone formation potential and the presence of NOx is considered a prerequisite. Due to the lack of factors for NOx, its contribution to ozone formation is neglected in current life cycle impact assessment and this is a serious flaw as seen from the normalisation references calculated in Section 6.6 showing that the contribution from NOx could be around 2/3 of the total ozone formation in Europe. It has been the purpose of the work performed under the Danish LCA methodology and consensus-creation project to include as much as possible of the later links of the cause-impact chain including this spatially determined variation. The aim has been to include exposure assessment in the characterisation without losing the feasibility in LCIA. In the endeavour to meet these objectives, an integrated impact assessment model was found which supports quantification of the ozone formation potential of both VOC and NOx in a site-dependent manner allowing the development of characterisation factors which include the later links in the cause-impact chain - both exposure assessment and parts of the target system description. This model is described in Section 6.3 6.3 The Rains modelThe RAINS (Regional Air Pollution Information and Simulation) model is an integrated assessment model that combines information on regional emission levels with information on long-range atmospheric transport in order to estimate patterns of deposition and concentration for comparison with critical loads and thresholds for acidification, eutrophication and tropospheric ozone formation. One of the main objectives with the RAINS model is to establish the relationship between the site of an emission and the impact on its deposition areas. The RAINS model (version 6.2) has therefore been seen as an obvious choice to establish spatially differentiated characterisation factors for photochemical ozone formation in parallel to what has been done for acidification (Chapter 3) and terrestrial eutrophication (Chapter 4). Alcamo et al. (1990) and Amann et al. (1995) provide a detailed description of the general model and Section 3.3 gives a summary of those parts of RAINS which are relevant for its use in our context. 6.3.1 Modelling ozone formationRAINS is intended to support the development of cost-effective European abatement strategies for different types of air pollution. For photochemical ozone, the focus is on the long-term ozone levels at the more regional scale. For this purpose, it is crucial with an integrated assessment that represents the relationship between source and receptor regions. For the modelling of ozone formation, RAINS does not apply a mechanistic model of the highly complex reaction schemes behind the formation of ozone and other photo-oxidants. Such models are used for calculation of POCPs but they will not be operational in an integrated assessment model. Instead, RAINS builds on a computationally efficient `reduced-form' model of ozone formation using statistical methods to summarise the response to emission changes given by a more complex reference model, (Heyes et al., 1997b). In the `reduced-form' model as presented in Heyes et al. (1997a), the long-term mean ozone concentration in any one grid cell j is expressed as a function of the annual non methane VOC emission vi and the annual NOx emission ni from emitter country i and the effective NOx emission enj experienced in grid cell j over the period considered (enj is corrected for exchange with the free troposphere): (6.1) where M is the number of emitter countries considered kj includes the effects of background concentrations of O3 and its precursors and natural VOC emissions aijvi provides the linear country-to-grid contribution from VOC emissions in country i (allowing for meteorological effects) bijni provides the linear country-to-grid contribution from NOx emissions in country i (allowing for meteorological effects) cijni2 serves as a correction term to allow for non-linearities occurring close to countries with very high NOx emissions αjenj2 represents the average non-linearity in the O3/NOx relationship experienced along the trajectories arriving at grid cell j and any non-linear effects local to grid cell j dijenjvi allows for interactions between NOx and VOCs along the trajectories The coefficients aij, bij, cij and αj are estimated by linear regression using ni, vi and enj as variables in the more complex reference model. Emission estimates The resulting factors depend on the emission patterns of European countries. They may therefore vary in time, and in order to reveal temporal variation, the factors are calculated for 1990, 1995 and 2010. The underlying per country emissions of non methane VOCs and NOx are based on the inventories of the UN ECE for 1990 and 1995 while the 2010 emission scenario is estimated based on the current reduction plans as expressed by the individual European countries. Meteorological conditions To reduce the influence of annual variations in meteorological conditions, the characterisation factors for each of the emission years 1990, 1995 and 2010 are derived as the average of five different calculations using the meteorological data for the years 1989, 1990, 1992, 1993 and 1994 respectively. Annex 6.1 illustrates the variation between the meteorological years for the calculation of AOT60 for the emission year 1995 6.3.2 Exposure indices in RAINSThe main target organisms of photo-oxidants are human beings and plants. In RAINS, exposure indices have been developed to quantify the exposure of vegetation as well as humans. Vegetation exposure Research on ozone-related vegetation damage makes it possible to define biologically meaningful, but simple, indices to characterise critical ozone exposure and to identify the critical levels of exposure above which by definition, adverse direct effects on receptors, such as certain plant species, may occur. Critical levels to protect vegetation are best established using long term exposure measures. Based on the scientific work on critical levels carried out under the UN/ECE Convention on Long-range Transboundary Air Pollution Working Group on Effects, a number of guideline values have been recommended by WHO (1997). For vegetation, the recommended threshold is 40 ppb and the cumulative exposure index AOT40 – exposure above the threshold of 40 ppb has been accepted as the best available exposure index for damage to crops and natural vegetation (Kärenlampi and Skarby, 1996). The concept of accumulated excess ozone assumes a threshold below which no or small effects occur. The AOT40 uses hourly concentrations during daylight hours over a three-month period (growing season) for herbal plants and crops and a six-month growing period for trees. The AOT40 is calculated as the sum of the differences between the hourly ozone concentrations in ppb and 40 ppb for each hour when the concentration exceeds 40 ppb, using daylight hours only. It is measured as ppbhours or more commonly as ppmhours. Under the UN/ECE Convention on Long-range Transboundary Air Pollution, the critical level for exposure of herbal plants and agricultural crops (relating to a five percent crop loss) has been set at an AOT40 of 3 ppmhours for the growing season and daylight hours, averaged over a 5-year period. For forest trees, a critical level has been set at 10 ppmhours for daylight hours, accumulated over a six month growing season and also averaged over five years (Heyes et al., 1997b). For the currently prevailing European ozone levels, the critical level for crops and natural vegetation is thus stricter than the critical level for forest trees. Therefore the critical level for vegetation exposure applied in RAINS, and hence also in the calculated characterisation factors for vegetation exposure, is the level for crops and natural vegetation. To reduce the influence from fluctuations in meteorological conditions, the ozone exposure in each grid used for calculation of the indices is determined as an arithmetic average of exposures calculated using the meteorology for five separate years in the period 1989-1994. The AOT40 is expressed in two different vegetation-related exposure indices: a cumulative vegetation exposure index calculated as the excess AOT40 (i.e. the AOT40 in excess of the critical level of 3 ppmhours) multiplied by the area of ecosystems exposed to the excess concentration an average vegetation exposure index reflecting the average excess AOT40 in a country. Both indices are based on rural ozone concentrations (Heyes et al., 1997b) Human exposure Ozone reacts with macro-molecules in the tissue of the human air ways. Depending on respiration frequency and ambient ozone concentrations, ozone will react in different parts of the air ways. At high respiration frequency and low ambient concentrations, ozone will mainly react in the upper air ways, whereas high speed of respired air and high ozone concentrations lead to transport of ozone deep into the lungs. A detailed Danish review and evaluation of health effects related to traffic air pollution is found in Larsen et al. (1997). For exposure of human beings, no thresholds for chronic exposure have been established. Following the revised WHO Air Quality Guidelines for Europe (WHO, 1997), a maximum eight-hour average concentration of 60 ppb is proposed as the long-term environmental objective for the EU ozone strategy. The maximum is calculated from running eight-hour averages of the one-hour mean concentrations. The ultimate goal would be to eliminate all excess of this criterion. The modelling of European abatement strategies for individual days over a multi-months period is a rather ambitious task and is not entirely feasible at the moment. In order to simplify the modelling task, the target of no-exceedance of the WHO criterion (60 ppb as maximum eight hours mean concentrations) has been converted into an AOT index for human health in parallel to the AOT40 index for vegetation damages for use in RAINS. The AOT60 is calculated in the same way but applying the threshold of 60 ppb. As a result, an AOT60 (i.e., the cumulative excess exposure over 60 ppb, for practical reasons over a six-month period) of zero is considered as equivalent to the full achievement of the WHO criterion. Any violation of this WHO guideline will consequently result in an AOT60 of larger than zero. It is emphasised by the group behind the RAINS model that the interpretation of the AOT60 index should be cautious. Given the current knowledge on health effects it is not possible to link any AOT60 value larger than zero with a certain risk to human health (Heyes et al., 1997b). Like the AOT40, the AOT60 is expressed in two different exposure indices:
Inaccuracies may occur for grids with major urban areas, where the rural ozone concentrations used for these analyses are lower than the concentrations occurring in the city plumes. The `average' indicator reflects the average exposure of a person in a country, calculated from gridded data. It is important to stress that these indices may not be used to derive estimates of health damage, for which more detailed information is deemed necessary. In the context of this report, these indices provide relative measures to enable a comparison of different scenarios. As for AOT40, ozone exposure in each grid cell used for calculation of the indices is an arithmetic average of exposures calculated using the meteorology for five separate years during the period 1989-1994. The hourly mean tropospheric concentrations of ozone in Europe vary between 0 and 150 ppb, but generally lie in the range 10 to 40 ppb. The background concentrations in Denmark are around 35 ppb (Granby et al., 1998). In the northern part of Europe the concentrations are highest in the summer months (May-August). During this period the ozone concentrations thus frequently exceed thresholds for damages on vegetation and for human health in large parts of Europe. 6.3.3 Validation of the RAINS modelThe performance of the reduced-form model for ozone formation has been validated in terms of its calculation of AOT40 against the results of the full EMEP model in Heyes et al., 1996. Over a realistic range of emissions, it was found to give a very good performance with results on mean ozone concentrations typically deviating less than 1.3% from the full model. A bias appears when the AOT40 approaches zero. In our use, only the excess of 3 ppmhours are counted, so this seems to be a minor problem. The full model takes into account differences in the composition of the VOC emissions for different sectors. Such differences are not included in the reduced-form model and this is a source of deviation from the results of the full EMEP model Overall the performance of the reduced-form model seems satisfactory for our purpose. The reader is referred to Heyes et al., 1996 for further details on the validation. 6.4 Spatially differentiated characterisation factorsThe use of RAINS for calculation of spatially differentiated characterisation factors is described in detail in Chapter 3 for acidification. The procedure is similar for photochemical ozone formation with one main difference. For acidification, the factor for a country is determined by the change in impact caused by a marginal increment of that country's emissions. For photochemical ozone formation, the national factors have been determined through an analytical solution of the differential quotient of the functional expression for AOT40 or AOT60 respectively. The functional form of the AOT40-model is the same as the form of the mean ozone concentration model in equation (6.1): (6.2)
The variables and parameters are explained under equation (6.1). Site-dependent characterisation factors The resulting site-dependent characterisation factors are shown in Table 6.1 for exposure of vegetation and in Table 6.2 for exposure of humans. Table 6.1. Site-dependent characterisation factors for exposure of vegetation to photochemical ozone expressed as cumulative AOT40 in exceedance of the critical level (m2ppmhours/g). Factors calculated for the emission patterns of 1990 and 1995 and the expected pattern of 2010.
Table 6.2. Site-dependent characterisation factors for exposure of humans to photochemical ozone expressed as cumulative AOT60 (persppmhours/g). Factors calculated for the emission patterns of 1990 and 1995 and the expected pattern of 2010.
Site-generic characterisation factors From the site-dependent characterisation factors of Table 6.1 and 6.2 site-generic factors are calculated as the emission-weighted average over all the countries and regions. The site-generic factors are compatible with the site-dependent factors in the sense that they are calculated using the same model and therefore cover the same part of the causality chain underlying photochemical ozone formation impacts. The only difference is that the spatial variation in the included parameters is no longer represented. The site-generic factors can be used if spatial differentiation is unwanted or if the location of some of the processes in the product system is unknown or lies outside Europe. The standard deviation of the site-generic factors reflects the spatially determined variation underlying the site-generic factors. Table 6.3. Site-generic characterisation factors for exposure of vegetation to photochemical ozone expressed as cumulative AOT40 in exceedance of the critical level (m2ppmhours/g). The factors are calculated as the emission-weighted means of site-dependent factors in Table 6.1 for the emission patterns of 1990 and 1995 and the expected pattern of 2010.
Table 6.4. Site-generic characterisation factors for exposure of humans to photochemical ozone expressed as cumulative AOT60 (persppmhours/g). The factors are calculated as the emission-weighted means of site-dependent factors in Table 6.2 for the emission patterns of 1990 and 1995 and the expected pattern of 2010.
6.4.1 Sub categories of ozone formationMeasures of the AOT40 and AOT60 are by no means proportional, and a closer study of the factors for vegetation exposure and for human exposure in Tables 6.1 and 6.2 reveals marked differences in the ranking and relative impact of different emission countries. It is therefore concluded that the exposure of humans and vegetation must be described by each their index and the impact category is split into two sub categories representing respectively: - critical ozone exposure of vegetation - critical ozone exposure of humans Being defined earlier in the cause-impact chain, the EDIP97 impact category photochemical ozone formation covers all impacts from photo-oxidants. In addition to human health and vegetation impacts, also damage to man-made materials is thus represented by the POCP-based impact potential. None of the two sub categories introduced here explicitly covers material damage. Indeed, it is mentioned in Heyes et al., 1997b, that the UN/ECE has proposed a preliminary threshold of an annual mean ozone concentration of 20 ppb based on an acceptable deterioration rate for sensitive organic materials. Separate calculations for material damage require additional information of the distribution of sensitive materials over the grids of the RAINS model. This far, no model calculations have been performed for material damage but until that may happen we suggest to consider damage to materials represented under the human health exposure. Even though the suggested threshold for materials is different, the geographic distribution of sensitive man-made materials is likely to be well represented by the geographic population distribution as already included in the RAINS model. 6.4.2 Characteristics of the new characterisation factorsThe site-dependent characterisation factors presented in Tables 6.1 and 6.2 and the site-generic factors in Tables 6.3 and 6.4 are calculated using a model that covers a larger part of the cause-impact chain in Figure 6.1. In contrast to the old factors that nearly exclusively represent substance-specific characteristics, the new factors also include the dispersion, the concentration increase and the background concentration in the receptor area, the population density or density of vegetation in the receptor area as well as some quantitative concentration-effect relationship for vegetation. The new factors thus eliminate a large part of the spatially determined uncertainty that is included in the EDIP97 factors. Reflecting the actual emission patterns of European countries, the new factors are dependent on changes in these patterns. Therefore, the factors have been calculated for the emission patterns of 1990, 1995 and the expected pattern of 2010. For vegetation exposure, the temporal variability is negligible for most emitter countries but for human exposure, there are significant differences depending on the basis year. In the guideline, the factors based on 1995 emissions is suggested as the default but for emissions taking place in the future, e.g. from the late use stage or disposal stage of the product, the factors based on 2010 could be applied alternatively as part of the sensitivity analysis. The new factors also include the contribution from NOx. From Tables 6.3 and 6.4 it is clear that on a weight basis, the factor for NOx is on average considerably larger than the factor for VOC, so this is also an important improvement compared to the EDIP97 factors. On the other hand, the new characterisation factors do not allow the differentiation between individual VOCs. This aspect is discussed in the next section. 6.4.3 Differentiation between individual VOCsAs mentioned in Section 6.3.1 the ozone formation model applied in RAINS does not allow distinction between individual VOCs. This eliminates a source of variation in the factors but since the variation between the ozone formation potentials lies within a factor 2-4 for most VOCs, this potential error is small compared to the variation eliminated by including the contribution from NOx. Nonetheless, it is suggested to introduce differentiation between the individual VOCs through an efficiency factor s representing the ozone formation potential of a specific VOC (s) relative to the average ozone formation potential of European VOC emissions (6.3) s126The European average POCP was determined for 1985 emissions at 0,40 in Hauschild and Wenzel (1998). Based on this figure and the POCP values applied for characterisation factors in EDIP97, the efficiency factor is calculated and tabulated in Annex 6.3 for the most common individual VOCs as well as for source-specified VOC-mixtures frequently encountered in life cycle inventories. The RAINS model only considers non-methane VOCs and NOx. In comparison, the POCP values used in EDIP97 and other life cycle impact assessment methodologies are also available for methane and carbon monoxide. To include these two substances which may be of importance for some processes, the following assumptions are made: Including carbon monoxide In addition to being a substance emitted from processes with incomplete combustion, carbon monoxide is also an intermediary product in the photochemical oxidation of some VOCs and its atmospheric lifetime is thus within the interval relevant for non-methane VOCs. It is therefore assumed that the dispersion and deposition pattern of carbon monoxide is well represented by the model applied for VOCs and in the calculation of photochemical ozone impact, carbon monoxide is treated as a VOC. Including methane In contrast, methane is an extremely long-lived organic compound with an estimated residence time in the atmosphere of around 10 years. This means that a globally uniform distribution of methane in the atmosphere must be expected and the contribution of methane to ozone formation is rather low at a regional level. In the calculation of characterisation factors it is suggested to base the characterisation factors for methane on the site-generic factors developed for VOCs and correct for the fact that due to the long lifetime of methane, a large part of the ozone formed will expose ocean areas and hence not contribute to exposure of vegetation or humans. A correction factor of 0,5 is proposed. Table 6.5. Site-generic characterisation factors for methane proposed to represent the exposure of vegetation or humans to photochemical ozone. The factors are based on the VOC factors in Table 6.3 and 6.4
6.5 How to perform site-generic and site-dependent characterisationThe site-generic photochemical ozone impacts from a product are calculated applying the new site-generic characterisation factors of Table 6.3, 6.4 or 6.5 in the following formulas: Where: sg POI(veg) is the site-generic photochemical ozone formation impact on vegetation expressed as area exposed above threshold (in m2ppmhours/f.u.) sg POI(hum) is the site-generic photochemical ozone formation impact on human health expressed as persons exposed above threshold (in persppmhours/f.u.) sg POF(veg)VOC is the site-generic photochemical ozone formation factor from Table 6.3 that relates emission of VOCs or CO to the impact on vegetation in the deposition area (in m2ppmhours/g). sg POF(veg)NOx is the site-generic photochemical ozone formation factor for from Table 6.3 that relates emission of NOx to the impacts on vegetation in the deposition area (in m2ppmhours/g). sg POF(hum)VOC is the site-generic photochemical ozone formation factor from Table 6.4 that relates emission of VOCs or CO to the impacts on human health in the deposition area (in persppmhours/g). sg POF(hum)NOx is the site-generic photochemical ozone formation factor from Table 6.4 that relates emission of NOx to the impacts on human health in the deposition area (in persppmhours/g). sg POF(veg)CH4 is the site-generic photochemical ozone formation factor for from Table 6.5 that relates emission of CH4 to the impacts on vegetation in the deposition area (in m2ppmhours/g). sg POF(hum)CH4 is the site-generic photochemical ozone formation factor from Table 6.5 that relates emission of CH4 to the impacts on human health in the deposition area (in persppmhours/g). is a substance-specific efficiency factor from Annex 6.3 expressing the ozone creation potential of the individual volatile organic compound or CO (s) or methane relative to the ozone creation potential of the European average VOC (dimensionless). E is the emission of NOx, CH4 or individual or source-specified VOC or CO (s) according to index (in g/f.u.) The potential spatially determined variation of the site-generic photochemical ozone impacts, can be estimated from the standard deviation given in Table 6.3, 6.4 or 6.5 for each substance. The site-dependent photochemical ozone impacts from a process in the life cycle of a product are calculated applying the new site-dependent characterisation factors of Table 6.1 and 6.2 in the following formulas: Where: sd POI(veg)p is the site-dependent photochemical ozone formation impact on vegetation expressed as area exposed above threshold by the selected process (p) (in m2ppmhours/f.u.) sd POI(hum)p is the site-dependent photochemical ozone formation impact on human health expressed as persons exposed above threshold by the selected process (p) (in persppmhours/f.u.) sd POF(veg)NOx,i is the site-dependent photochemical ozone formation factor from Table 6.1 that relates emission of NOx from country or region (i), where the selected process (p) is located, to the impacts on vegetation in the deposition area (in m2ppmhours/g). sd POF(veg)VOC,i is the site-dependent photochemical ozone formation factor from Table 6.1 that relates emission of VOCs or CO from country or region (i), where the selected process (p) is located, to the impact on vegetation in the deposition area (in m2ppmhours/g). sg POF(veg)CH4 is the site-generic photochemical ozone formation factor CH4 from Table 6.4 that relates emission of CH4 to the impacts on vegetation in the deposition area (in m2ppmhours/g). sd POF(hum)NOx,i is the site-dependent photochemical ozone formation factor from Table 6.2 that relates emission of NOx from country or region (i), where the selected process (p) is located, to the impacts on human health in the deposition area (in persppmhours/g). sd POF(hum)VOC,p is the site-dependent photochemical ozone formation factor from Table 6.2 that relates emission of VOCs or CO from country or region (i), where the selected process (p) is located, to the impacts on human health in the deposition area (in persppmhours/g). sg POF(hum)CH4 is the site-generic photochemical ozone formation factor from Table 6.5 that relates emission of CH4 to the impacts on human health in the deposition area (in persppmhours/g). η is a substance-specific efficiency factor from Annex 6.3 expressing the ozone creation potential of the individual volatile organic compound or CO (s) or methane relative to the ozone creation potential of the European average VOC (dimensionless). Ep is the emission of NOx, CH4 or individual or source-specified VOC or CO (s), according to index, from process (p) (in g/f.u.) Emissions from a non-European or unknown region may as a first approach be calculated using the site-generic factors from Table 6.3, 6.4 or 6.7. The standard deviations on the site-generic factors in these tables give a range of potential spatial variation for the application of the site-generic factor within Europe. Given the size of the variation in emissions and sensitivities within Europe, the site-dependent factor is expected to lie within this range for most regions also in the rest of the world. Expert judgement may be used in the interpretation to assess whether the factor for emissions from processes in non-European regions should be found in the lower or upper end of the range. 6.6 Normalisation referencesThe normalisation reference for photochemical ozone formation is calculated using the new characterisation factors on the national European emission inventories for the corresponding year. Three sets of references are calculated in Annex 6.4 and the reference based on 1995 factors and emissions is retained as the EDIP default reference for normalisation in life cycle impact assessment. Table 6.6. Normalisation references for the two sub categories of photochemical ozone formation calculated for the reference yeras 1990, 1995 and 2010 in Annex 6.4 and expressed as impact per person per year (the person equivalent).
From Table 6.6 it is seen that the photochemical ozone impact was reduced from 1990 to 1995 and is expected to continue the trend towards 2010. 6.7 Interpretation and recommendationsThe new photochemical ozone formation impact potentials are improved in two aspects compared to the impact potentials calculated using the EDIP97 characterisation factors or other systems based on the POCP or MIR approach; the environmental relevance is increased and a part of the spatial variation in sensitivity of the receiving environment is now taken into account. Environmental relevance The environmental relevance is increased because the exposure of the sensitive parts of the environment (vegetation or human beings) is included in the underlying model which now covers most of the causality chain towards the LCA protection areas: Ecosystem health and human health. This is particularly important when weighting factors based on the environmental relevance are used instead of the EDIP default weighting factors (which are based on political reduction targets that for photochemical ozone formation are also aiming for protection of vegetation and human health). In comparison, the EDIP97 factors only cover the potential for formation of ozone. In addition, the contribution of NOx is now included in the impact potentials. The significance of this for a specific product system depends on the quantities of NOx and VOCs emitted. From the calculation of the normalisation references, it is known that on a European level, NOx contributes around twice as much as VOC to photochemical ozone formation and on average the characterisation factor for NOx is more than three times the characterisation factor of VOCs. Spatial variation The spatial variation in exposure for photochemical ozone formation can be large even at the very local scale. The variation in sensitivity between European regions are now presented on a national scale showing a factor 15-20 of difference between least and the most sensitive emission countries for exposure of vegetation and a factor of around 400 times of difference for exposure of humans (the latter reflecting the variation in population density in the deposition areas). This variation is hidden when the EDIP97 factors or similar are used for characterisation. 6.8 ReferencesAlcamo, J., R. Shaw and L. Hordijk (eds.). The RAINS model of acidification. Science and strategies in Europe. Dordrecht (the Netherlands), Kluwer Academic Publishers, 1990. Amann, M., M. Baldi, C. Heyes, Z. Klimont and W. Schöpp. Integrated assessment of emission control scenarios, including the impact of troposheric ozone. Water, air and soil pollution, 85(4), 2595-2600, 1995. Andersson-Sköld, Y., Grennfelt, P., and Pleijel, K.: Photochemical ozone creation potentials: a study of different concepts. J. Air Waste Manage. Assoc. 42(9), 1152-1158, 1992. Derwent, R.G., and Jenkin, M.E.: Hydrocarbon involvement in photochemical ozone formation in Europe. AERE R 13736, AEA Environment and Energy, Harwell Laboratory, Oxfordshire, UK, 1990. Derwent, R.G., Grennfelt, G., and Hov, Ø.: Photochemical oxidants in the atmosphere. Nordic Council of Ministers, Nord 1991:7, Copenhagen, 1991. Derwent, R.G., Jenkin, M.E., Saunders, S.M., et al.: Photochemical ozone creation potentials for organic compounds in northwest Europe calculated with a master chemical mechanism. ATMOS ENVIRON 32 (14-15): 2429-2441, 1998 Granby, K., Kemp, K., Palmgren, F., Hovmand, M. and Egeløv, A.: Measurements of Photochemical Pollutants. In: Fenger, J. (ed.): Photochemical Air Pollution. Danish Aspects. National Environmental Research Institute. - NERI Technical Report 199: 63-101, 1997. Hauschild, M. and Wenzel, H.: Photochemical ozone formation as criterion the environmental assessment of products. In Hauschild, M.Z. and Wenzel, H.: Environmental assessment of products. Vol. 2 - Scientific background, 565 pp. Chapman & Hall, United Kingdom, ISBN 0412 80810 2, Kluwer Academic Publishers, Hingham, MA. USA., 1998. Hettelingh J.-P., Posch M., de Smet P., Downing R.J.: The use of critical loads for emission reduction agreements in Europe. Water, Air and Soil Pollution 85:2381-2388, 1995. Heyes, C., Schöpp, W., Amann: A simplified model to predict long-term ozone concentrations in Europe. WP-96-12, International Institute for Applied Systems Analysis (IIASA), Laxenburg, Austria, 1996. Heyes, C., Schöpp, W., Amann, M., Bertok, I., Cofala, J., Gyarfas, F., Klimont, Z., Makowski, M. and Shibayev, S.: A model for optimizing strategies for controlling ground-level ozone in Europe. Interim report IR-97-002/January, International Institute for Applied Systems Analysis (IIASA), Laxenburg, Austria, 1997a. Heyes, C., Schöpp, W., Amann, M., Bertok, I., Cofala, J., Gyarfas, F., Klimont, Z., Makowski, M. and Shibayev, S.: Simultaneous optimization of abatement strategies for ground-level ozone and acidification. Interim report IR-97-090/December, International Institute for Applied Systems Analysis (IIASA), Laxenburg, Austria, 1997b. Kärenlampi L. and Skarby L., (eds), 1996. Critical levels for Ozone in Europe: Testing and Finalizing the Concepts. UN-ECE Workshop Report. Dept. of Ecology and Environmental Science, University of Kuopio, Finland, 1996. Larsen, P.B., Larsen, J.C., Fenger, J., and Jensen, S.S.: Health evaluation of air pollution from road traffic (In Danish: Sundhedsmæssig vurdering af luftforurening fra vejtrafik). Miljøprojekt nr. 352, Danish Environmental Protection Agency, Copenhagen, 1997. Lübkert, B. and Schöpp, W.: A model to calculate natural (volatile organic compounds) VOC emissions from forests in Europe, wp-89-82, 65 pp, w89082, IIASA, Laxenburg, Austria, 1989 Posch, M., Hettelingh, J.P., Downing, R.J. and P.A.M. de Smet (eds): Calculation and mapping of critical thresholds in Europe. CCE status report 1997. Co-ordination Centre for Effects (CCE) at the National Institute of Public Health and the Environment (RIVM), Bilthoven (the Netherlands), 1997. Wenzel, H., Hauschild M.Z. and Alting, L.: Environmental assessment of products. Vol. 1 - Methodology, tools, techniques and case studies, 544 pp. Chapman & Hall, United Kingdom, ISBN 0 412 80800 5, Kluwer Academic Publishers, Hingham, MA. USA., 1997. World Health Organization: Indoor air quality: organic pollutants. EURO Reports and Studies 111, WHO Regional Office for Europe, Copenhagen, 1989 WHO: Ozone. In: Air Quality Guidelines for Europe, pp 315-326. WHO Regional Publications, European Series No 23, Copenhagen, 1987 Annex 6.1 Influence from meteorological conditionsCalculation of AOT60 for individual European countries based on 1995 emissions and meteorological data for five different years. Annex 6.2 POCP values for individual VOCsPOCP values from different sources (Derwent and Jenkin, 1990, Derwent et al., 1998, Andersson-Skjöld et al., 1992). EDIP97 applies the low NOx values from Andersson-Skjöld et al., 1992 for emissions from low NOx areas and the values from Derwent and Jenkin, 1990 for high NOx areas. In EDIP97, all values are scaled down by a factor 100 so the reference substance ethylene has a POCP of 1,00.
Annex 6.3 Efficiency factors for individual VOCs and source-specified VOCsThe dimensionless efficiency factor is representing the efficiency of individual VOCs relative to the European average VOC in contributing to ozone formation. It is derived as the quotient between the respective POCP-factors for 4-9 days in high NOx-areas (the EDIP97 characterisation factors for high NOx-areas, Wenzel et al., 1997).
Annex 6.4 Normalisation references for photochemical ozone formationNormalisation references for photochemical ozone formation calculated with the new characterisation factors and the national European emission inventories for each of the years 1990, 1995 and 2010.The reference based on 1995 factors and emissions is retained as default factor for normalisation in life cycle impact assessment. Impacts on vegetation 7 Human toxicity7.1 Introduction Authors: José Potting (editor) [47] Alfred Trukenmüller [48]Frans Møller Christensen [49] 7.1 IntroductionSeveral sets of human toxicity factors are presently in use in life cycle assessment, most notably those of Guinée et al. (1996), as updated by Huijbregts (1999), the factors of Hauschild and Wenzel (1998) and of Hertwich et al. (2001). All these sets of human toxicity factors follow a framework similar to the one as described in the technical guidance documents on risk assessment released by the European Commission (EC 1994, 1996). These documents are written in close parallel with the enactment of European legislation on management of risks from pesticides, and from society's use of chemicals.
Figure 7.1. Steps in the risk management process (modified from Van Leeuwen and Hermens 1995) Figure 7.1 gives a general scheme for the steps in the risk management as described in the technical guidance documents of the European Commission (EC 1994, 1996). The human toxicity factors of Guinée et al. (1996), Hauschild et al. (1998), Huijbregts (1999) and Hertwich et al (2001) take their basis in those steps encircled in Figure 7.1. First, the increase of environmental exposure or concentration is predicted for one unit of emission of a given substance (exposure assessment), and in parallel the no-effect-concentration or safe dose is predicted (effect assessment). Next, the predicted environmental concentration or exposure (PEC) is divided by the predicted no-effect-level (PNEC) or safe dose. In this way, characterisation factors are obtained following a framework (PEC/PNEC) similar to the one for risk characterisation (PEC/PNEC). The exposure assessment underlying the characterisation factors for use in life cycle assessment calculates similar exposure increases for all releases of the same quantity and substance. Disregarded are the circumstances under which these emissions take place. This contradicts what we intuitively would expect. An example for atmospheric emissions may illustrate this: Exposure increases from an emission released at moderate height will close to the source be considerably higher than those from an elevated release, but lower than those from an emission released at ground level or indoors. The traditional characterisation factors also do not take into account spatial differences in for example atmospheric conditions and population densities between areas. Section 7.2 evaluates the need for spatial differentiation in characterisation factors for human toxicity, and continues by exploring the feasibility of a framework [52] based upon Potting et al. (1999) to establish site-dependent factors that assesses the increases of human exposure from air emissions. The framework takes into account variation in release height, atmospheric conditions, and population densities in the receiving areas. The framework is used to calculate site-dependent factors that quantify the increase of accumulated human exposure from an emission at a given location. Those factors can be used as an exposure factor in combination with the existing characterisation factors for human toxicity from Wenzel et al. (1997). The Guidance Document of Hauschild and Potting (2003) to this Technical Report describes in its entirety how to apply the developed methodology. In contrast to a risk characterisation as described in the technical guidance documents of the European Commission (EC 1994, 1996), the characterisation factors used in life cycle assessment do usually not account for background exposures. They are based on exposure increases rather than actual exposures (sum of the background exposure and the exposure increase). As a consequence, exceedance of the no-effect-levels is not taken into account in these characterisation factors. The predicted exposure increases (PEC) are only weighted with the relevant no-effect-concentration (NEC) for the given substances (PEC/NEC). This weighting is needed to aggregate exposures to different substances. A lively debate is going on whether or not life cycle assessment should perform an evaluation of threshold exceedance. This issue is closer examined in Section 7.3 in which information is also provided for a selection of substances that may facilitate a qualitative threshold evaluation. 7.2 Exposure assessment [53]7.2.1 IntroductionHuman toxicity is a complex impact category. This complexity arises, amongst others, from the numerous possible routes through which an emission can lead to human exposure. Figure 7.2 gives a simplified overview. There are three main routes of human exposure to environmental pollutants: Inhalation of air (part of the exposures via the environment in Figure 7.2), Ingestion of food, water and sometimes even soil (all part of the exposures via the environment in Figure 7.2), and Penetration of the skin after contact with polluted surfaces, air, and sometimes also soil or water (part of consumer exposures in Figure 7.2).
Figure 7.2. Overview of the main routes of human exposure to toxic substances (Van Leeuwen and Hermens 1996). The exposure of humans to environmental pollutants usually takes place via more than one route at the same time (multi-route exposure), but one exposure route is often dominating over the other exposure routes. A typical life cycle assessment focuses on inhalation and ingestion, though skin exposure may be of major relevance within the product system some products (like cloths or cosmetics). Hauschild and Wenzel (1998) provide characterisation factors for over 100 substances to characterise the toxic impact from emissions to air, water and soil. An emission can have a direct effect through exposure to the medium to which it is initially released. However, emissions have often also indirect effects through re-distribution of the substance to another medium than the one to which the substance was initially emitted. The factors of Hauschild and Wenzel (1998) distinguish therefore further between inhalation of air, and ingestion of water and soil. Table 7.1 gives an overview of the resulting nine different characterisation factors, while Annex 7.1, 7.2 and 7.3 list the factors per substance. Hauschild and Wenzel (1998) can be consulted for an extensive description of the backgrounds for these human toxicity factors. Table 7.1: Overview of the characterisation factors provided by Hauschild and Wenzel (1998).
The factors of Hauschild and Wenzel (1998) are based on similar exposure increases for all releases of the same quantity and substance. As clarified in the introduction, this contradicts with what we intuitively expect. Section 7.2.2 evaluates the need for spatial differentiation in the several sub-categories. Section 7.2.3 up to and including Section 7.2.7 explore the feasibility of a framework based upon Potting et al. (1999) to establish site-dependent factors that assess the increases of human exposure from air emissions. 7.2.2 The need for spatial differentiationThe degree to which a source contributes to exposure depends to a large degree on the properties of the substances, the characteristics of the source and the characteristics of the receptor (see also Potting and Hauschild 1997). The characterisation factors of Hauschild and Wenzel (1998) account fully for substance information, but address only to a limited extent information about source and receptor. In addition, Hauschild and Wenzel (1998) do not cover variation in characteristics of sources and receptors. Guinée et al. (1996) allow for some spatial differentiation by distinguishing between exposures resulting from emissions to agricultural soil, industrial soil and to other soils. Huijbregts (1999) continues with this differentiation into soil types, and adds a differentiation into emissions to fresh water and seawater. Differentiation between emissions to different water and soil types may be a useful supplement to the characterisation factors of Hauschild and Wenzel (1998). However, the characterisation factors of both Guinée et al. (1996) and Huijbregts (1999) integrate all types of exposure resulting from emission to the given medium into one characterisation factor. This makes them incompatible to use in combination with the dis-aggregated factors from Hauschild and Wenzel (1998). In the discussions about priority setting for the research underlying Section 7.2, we estimated direct exposures through ingestion of both fresh water and seawater to be small compared to indirect exposures after redistribution (most of the industrialised countries after all purify their water before supplying it as drinking water). Further, we estimated indirect exposure through ingestion of food from agricultural soils by far dominant compared to indirect exposure through ingestion of food from other soils. Differentiation between different water and soil types was therefore not prioritised and is not further addressed in this chapter. Better foundation for refraining from further differentiation in soil and water types is recommended for future research. [54] Distinction between geographic locations of emission is another type of differentiation discussed in priority setting for the research underlying this chapter. We considered this differentiation of major importance for exposures from atmospheric emissions (see Section 7.2.3 up to Section 7.2.7), but of less relevant for exposures from emissions to water and soil. The present state-of-the-art does moreover not allow spatial resolved modelling of direct exposures from emissions to water and soil, and indirect exposures form emissions to all media. Deliberating the importance of the possible and/or relevant spatial differentiation in the several exposure routes, we decided to focus the research underlying Section 7.2 on spatial differentiation in direct human exposure from emissions to air. The results of this research are reported in Section 7.2.3 up to 7.2.7. However, better foundation for refraining from spatial differentiation of exposures from emissions to soil and water is recommended for future research. 7.2.3 Human exposure from air emissions [55]Among the different exposure routes, inhalatory exposures have as a unique feature that they are ubiquitous and can thus not be avoided once a substance is present in air (Williams sine dato). Inhalatory exposures also result in most cases directly from emissions to air rather than being the result of re-distribution between the different environmental media. This chapter focuses only on inhalatory exposures from atmospheric releases. Dispersion and dilution in air are quick processes and the concentration increases at ground level are close to the source very much influenced by wind speeds and source characteristics like height and dynamics of the release (see Figure 7.3). The effective- release height is decisive for the concentration increase at ground level. A moderately high release (25m) has its peak concentration increase within 0.5km from the source, while the peak concentration from a high release (>150m) is a hundred times lower and occurs within 5km. The concentration increase from an emission at ground level (<1m) has its peak within a few metres from the source and is here a thousand times higher than for a similar emission released at a height of 25 metres. Close to the source, the concentration is governed by dilution and the concentration increases are here for all substances almost equal when released at similar release height. The release height becomes less important at increasing distance, however, and removal processes start to take over as is illustrated in Figure 7.4 for the short-lived substance hydrogen chloride and the long-lived substance benzene. Removal processes are largely substance dependent. They are the combined result of deposition and chemical transformations. The emissions of substances with different removal characteristics begin to show their own concentration pattern, and concentration patterns for the emissions of the same substance released at different heights start to converge. At large distances, the height of release becomes negligible and removal processes finally take fully over (see Figure 7.5). Figure 7.3. Concentration increase at ground level versus distance local to the source (from 0 to 5km) from an emission of one gram per second in the Netherlands. Concentration increases have been calculated with the OPS model (Van Jaarsveld 1990, Van Jaarsveld en de Leeuw 1993). The numbers on the y-axis have to be multiplied with the factor for the given release height in the legends to obtain the proper order of magnitude. Figure 7.4. Concentration increase at ground level versus distance semi-local to the source (from 5 to 50km) from an emission of one gram per second in the Netherlands. Concentration increases have been calculated with the OPS model (Van Jaarsveld 1990, Van Jaarsveld en de Leeuw 1993). Figure 7.5. Concentration increase at ground level versus distance local to the source (from 50km to several hundred to thousand kilometres) from an emission of one gram per second in the Netherlands. Concentration increases have been calculated with the OPS model (Van Jaarsveld 1990, Van Jaarsveld en de Leeuw 1993). Atmospheric conditions also influence concentration increases. Wind velocity is of major importance close to the source, whereas precipitation (important for wet deposition) and hours of sunshine (important for photo-chemical transformation) become more important at larger distances. Annual mean atmospheric conditions differ from region to region. Notably wind speeds and precipitation tend to be high in maritime and low in continental climates. The population densities in the area with increased concentrations are the last factor determining the increase of human exposure. Population densities vary considerably over Europe (see Figure 7.6). Western Europe is far more densely populated than North-East and Northern Europe. Neighbouring regions differ less dramatically. Population densities near to the source are of special interest since concentration increases are highest here. Built-up areas show large differences in population densities. (Tobler et al. 1995, Stanners and Bourdeau 1995, EEA1998)
Figure 7.6. Estimate of population densities for 1994 from Tobler et al. (1995). Locations of the Northern, Central, Southern European and maritime sites are indicated with capital letters. Potting et al. (1999) presented a framework to establish site-dependent factors to be used in life cycle assessment that assess the accumulated human exposure increase from outdoor emissions by taking into account all factors described above. This framework could not be applied in its initial form and has therefore been adapted slightly for the research reported here. It consist of 5 steps:
Consecutively, for each source type:
Section 7.2 focuses fully on assessment of exposure increase (Step 1 to 3), but refrains from evaluating whether the exposure situation is above or below a threshold value (step 4). The site-dependent factors that are established in step 5, therefore quantify accumulated exposure increase rather than human toxicity. The site-dependent exposure factors are used in combination with the old characterisation factors in Annex 7.1-7.3 7.2.4 Identification of source types, and classification of processesNear to a source, the release height is decisive for the concentration increases at ground level. The release height for transport will typically be near to ground level (<1m). The information about the release height of industrial processes is usually not available in life-cycle assessment and can vary considerably between sources. The types of industrial processes are on the other hand typically known in life cycle assessment since this is one of its basic informations. An interesting question is therefore whether it is possible to identify source types (or classes of release height) to which industrial processes can be allocated. The Dutch Emission Registration (DER) maintains a rather unique database that contains detailed information about the water and air emissions from over 700 major industrial sources in the Netherlands. Amongst others, the data cover release height, emission type (substance, quantity, flow, temperature), process type and industrial sector. The information is provided mainly by industry itself on a voluntary basis, and to a limited extent also by authoritative bodies like those for water management. All relevant data are recorded for the individual emission points and the individual equipment within each company. (Berdowski et al. 1995) Data from 1994 have been used to analyse whether a relation exists between the height of release points and their connected processes. The median and mean height of release have been determined for records belonging to each industrial sector and for all industrial sectors together. The Statistical Analysis System (SAS Software Release 6.11) was used to analyse the data. The results are presented in Table 7.2. Though these results are expected to give a good indication of release heights, they should only be taken as indicative due to data problems [56] and because they represent data from Dutch industry only. Particularly for countries outside Europe, North America and Japan, the release heights may be considerably lower. Table 7.2: The median, mean and range of release height for each industrial sector in a database containing detailed information about the air emissions from over 700 major industrial sources in the Netherlands based on analyses of data from the Dutch Emission Registration. The overall mean height is 31m with a standard deviation of nearly the same size. However, the median counts only 20 metres due to the majority of individual sectors that have considerably lower means. The mean is pulled up by a few sectors with very high emission points: "Cokes and crude oil refinery/processes", "centralised electricity production & district heating", and "waste processing" (mainly incineration). Excluding these sectors gives a mean [57] height of release for the remaining observations of 25m. The median, mean and range of release height for each industrial sector have been calculated including as well as excluding the own production of energy (steam and/or electricity). The influence of own energy production on the median and mean is for most sectors minor. For a few sectors, the high release heights disappear from the observations without dramatically changing the median and mean when own energy production is excluded. Only the mean height of release for the wood & furniture industry falls considerably (from 20m to 11m). However, we have to take into account the very small number of observations for this sector. The agreement between the mean and the median is for most sectors reasonably good. Also the standard deviations for most sectors are relatively small when the number of observations is reasonable. Most sectors have a relatively low mean height of release: The "extraction of gas, minerals, otherwise" (15m), "textile industry" (17m), "leather preparation" (19m), "wood & furniture industry" (20m), "paper industry " (22m), "printing houses" (15m), "plastic and rubber processing" (15m), metal processing (13m) and the group of "remaining industrial sectors" (17m). The release heights of the percentiles in the higher end are usually also relatively low. Some sectors contain also a few observations of relative high releases, but these observations are in most cases related to own energy production. The "leather preparation" contains only three observations from which two were very low (6 and 7m) and one higher (45m). The "food and tobacco industry" (25m), "chemical industry" (30m) and "glass, pottery, stone and cement industry" (31m) have moderate mean heights of release that are not much influenced by excluding own energy production. The production of sugar, fish flour & glue etc., grass and pulp drying and coffee roasting somewhat pull up the mean height for the "food and tobacco industry". The maximum release height of 100m relates to sugar production. The "chemical industry" contains production of many different chemicals. Most sub-sectors have moderate release heights, but those for the production of inorganic basic chemical and petrochemical products are somewhat higher, while the medians are higher and maximums even very high. The high to very high sources are in most cases related to processes involving some kind of combustion process. The mean release height is relatively high for the "crude oil refinery and processing" (91m), but surprisingly enough rather modest for "electricity production and heating" and "waste processing (mainly incineration)". The mean for "electricity production and heating" increases considerably, however, when the release heights are weighted with the annual production of electricity (133m). Something similar is the case for the "crude oil refinery and processing" and "waste processing". Since these two sectors cover output of several different products, however, it was impossible to calculate a weighted mean. The release heights of "waste processing" are high for processes related to waste incineration (93m). The above results show that the majority of Dutch industry has relatively moderate release heights. This is further underlined by the fact that these results relate to the 700 main industrial sources while the less important ones are not covered. There are only few industries with high sources. The results refer to height of release rather than to "effective release height". The effective release height also includes the plume rise as a result of its heat content. It is actually the effective release height that is relevant for dispersion and dilution calculations. Based on this, three release heights have been chosen for which accumulated exposure increases will be calculated: 1m for traffic, 25m representing the majority of industry, and 150m being a reasonable estimate for high sources. All results here refer to Dutch industry that may not appropriately reflect industry in other countries due to differences in legislation and production capacities and technologies. But the three classes of release heights are expected to be relevant for Europe. 7.2.5 Accumulated human exposure increase local to the source (from 0 to 10km)An atmospheric emission leaves its source in a concentrated form that is sometimes visible as a plume. The plume usually mixes with the surrounding air before it results in concentration increases at ground level. Wind speeds largely determine how fast the plume dilutes, whereas the release height also influences how fast the plume reaches ground level. This is typically modelled with a Gaussian plume approach (Harssema 1995). Atmospheric conditions differ from region to region. Wind speeds tend to be high in maritime and low in continental climates. Van Jaarsveld follows a Gaussian plume approach that applies region-dependent atmospheric conditions (1990 annual statistics mean) in his EUTREND model (Van Jaarsveld 1995, Van Jaarsveld and De Leeuw 1993, Van Jaarsveld et al. 1997). This model is used here to estimate concentration increases at short distances from the source (from 0 to 10km) for different release heights and for sources located in different climates:
The accumulated exposure increase has been calculated for a long-lived substance (benzene; residence time of seven days) and a short-lived substance (hydrogen chloride; residence time of seven hours). These two substances have been selected because the residence time and therewith the accumulated exposure increase from substances will in general be in between those of hydrogen chloride and benzene. The source strength is kept at one gram per second continuous, but the consequence of the release heights as identified in Section 7.2.4 has been analysed (1m, 25m, and 150m). The accumulated human exposure increase from a release on location (i) at each distance is the product of concentration increase times population density integrated over the whole surface: The population density is assumed to be constant (one person·km-2) in the areas local to the source. The result of the calculations is the increase of human exposure (in person·µg·m-3) accumulated over the area from 0 to 10 km from the source. The courses of the accumulated human exposure increase versus distance are depicted in Figures 7.7 to 7.11, and abstracted in Table 7.3. Clear differences exist between the accumulated increase at varying release heights on the one hand, and climate regions on the other hand, as well as between the two substances hydrogen chloride and benzene. Figure 7.7. The increase of accumulated exposure versus distance local to the source (from 0 to 10 km) from one gram benzene released at 1m in four different climatological regions in Europe (population density is one person·km-2). Figure 7.8. The increase of accumulated exposure versus distance local to the source (from 0 to 10 km) from one gram benzene released at 25m in four different climatological regions in Europe (population density is one person·km-2). Figure 7.9. The increase of accumulated exposure versus distance local to the source (from 0 to 10 km) from one gram benzene and hydrogen chloride released at 150m in four different climatological regions in Europe (population density is one person·km-2). Figure 7.10. The increase of accumulated exposure versus distance local to the source (from 0 to 10 km) from one gram hydrogen chloride released at 1m in four different climatological regions in Europe (population density is one person·km-2). Figure 7.11.The increase of accumulated exposure versus distance local to the source (from 0 to 10 km) from one gram hydrogen chloride released at 25m in four different climatological regions in Europe (population density is one person·km-2). Figure 7.11.The increase of accumulated exposure versus distance local to the source (from 0 to 10 km) from one gram hydrogen chloride released at 25m in four different climatological regions in Europe (population density is one person·km-2). Table 7.3. The increase of accumulated exposure from one gram of benzene and hydrogen chloride at different distances from the source (0.5km, 5km and 10km), and released at different heights (1m, 25m and 150m) and in different climate regions in Europe. The accumulated exposures are expressed as the proportion from the accumulated benzene exposure at a height of 25m at 10km distance (20·20km2) in South Europe (69.7 person·µg·m-3). The population density is in all cases one person·km-2.
There is a notable difference in accumulated exposure between the maritime and North European climate regions on the one hand, and the South and Central European climates on the other hand. The climate region becomes more important with lower release heights. This is caused by the considerable difference in wind velocities between the regions. Low wind velocities give slower dilution and subsequently higher concentrations than high wind speeds (direct effect). In addition, low wind velocities go together in a stable and neutral atmosphere with low mixing heights (indirect effect). Decreasing mixing height has an increasing effect on the concentration increase for low sources, but a decreasing effect on the concentration increases for high sources (since they release above mixing height). Wind velocities in the south and central climate regions are on average lower than those are in the maritime and northern climate regions. The direct and indirect effects of wind velocity therefore result in diverging concentrations for low sources, but in converging concentrations for high sources between climate regions. Atmospheric conditions also show variation within regions themselves, as well as over the seasons. However, the subsequent variation in accumulated exposure increase remains within 10% (based on calculations with a similar model that is not further reported here: Van Jaarsveld and De Leeuw 1993). Though benzene and hydrogen chloride have similar accumulated increases at very short distances (< 0.5km), they already show a clear divergence within the first 10km for low release heights. This is mainly due to dry deposition which is considerably higher for hydrogen chloride than for benzene. Emission at 150m will often be above the mixing height and therefore result in increases of accumulated exposures that are nearly the same for both substances. Removal by dry deposition is still very small here since the concentrations at ground levels are small. All accumulated exposures in Table 7.3 and Figure 7.7 to 7.11 relate to a population density of one person·km-2. Tobler et al. (1995) give detailed data about population densities in the European grid. As can be seen from Figure 7.6, which represents this data at regional resolution, population densities vary considerably between European regions (from one to more than five hundred persons km-2). Tobler et al. (1995), EEA (1998) and Stanners and Bourdeau (1995) show that large variation also exists within built-up areas. The population of big cities for instance is distributed rather unequally over the city-area, though generally concentrated in the city-centre. City-centres are mostly limited to an area of at maximum 10 10km (exceptions may be Berlin, London and Moscow). Typical population densities have been defined on the basis of data from Tobler et al. (1995) about Paris:
Hence, the total population of 2.2 million persons in Paris-city (roughly 10 10km2) exceeds by far the total population in most cities and smaller communities. This would even be the case if Paris-city had a population density of 900 person ;km-2 (which would correspond to a total population of 90.000 persons). One thus has to be careful in choosing the population density within the first 10km (or 20 20km2) from the source. While the population density in Figure 7.6 reflects the urbanisation in the relevant region, often no information is available in life cycle assessment about the exact location of a source in relation to built-up areas. We therefore suggest to take the mean population density within the relevant region in Figure 7.6 as the one for those within the first 10km. 7.2.6 Accumulated human exposure increase regional to the source (from 10km to several hundreds to thousand kilometres)At longer distances, the plume from a source in vertical direction is distributed equally in the mixing layer of the atmosphere. Trajectory or one-dimensional Lagrangian modelling is an often-used way to trace concentration increases resulting from substance transport and removal at long ranges (Seinfeld and Pandis 1998). Atmospheric conditions differ from region to region, and notably precipitation and wind speeds tend to be high in maritime and low in continental climates. The Wind rose Model Interpreter (WMI) of the integrated assessment model EcoSense (Krewitt et al. 1997) follows trajectory modelling based on region dependent atmospheric conditions (1990 annual statistics mean). The WMI-module is used here to assess long-range accumulated exposures (see Annex 7.4 for model specifications and adaptations). Similar to the modelling of exposure local to the source, calculations have been performed for hydrogen chloride and for benzene at an emission rate of one gram per second continuous. At the regional scale, release height only has minor importance for a long-lived substance as benzene. However, it will considerably influence the removal by deposition and consequently the amount of a short-lived substance going into long range transport. The calculations for hydrogen chloride have therefore been performed for the release heights 1m, 25m and 150m by correcting for deposition in the source grid-square of respectively 38%, 30%, and 9% of the emissions (see Annex 7.4). Since atmospheric conditions are region-dependent, the patterns of concentration increase will depend on the geographical location of sources. Also population density is region-dependent. The concentration increase in each 150 150km grid-square is multiplied with the total population in that grid-square (derived from the same data as used for Figure 7.6). Next, the products of concentration increase times population in each grid-square are accumulated over the grid (see Formula 7.1). The result is the accumulated exposure increase (in person µg m-3) over a distance of several hundreds to thousands kilometres from a source located in the specified grid-square in the European grid. The procedure is repeated for every grid-square as the source grid-square. The results for hydrogen chloride are represented in Figure 7.12 and the results for benzene in Figure 7.13.
Figure 7.12. The increase of exposure accumulated (in person µg m-3) over the total receiving area (from 10km to several hundred to thousand kilometres) posed by a release of one gram emission hydrogen chloride at 25m in the source grid-square. The mean is 2,460 person µg m-3, the standard deviation is 1600 person µg m-3 (both weighted for population density). The accumulated exposure is attributed to the respective source grid-square. The increase in accumulated exposure from a release at 150m can be obtained by multiplying with a factor 1.30 (s.d. 0.02). The accumulated exposure increase from a release at 1m can be obtained by multiplying with a factor 0.89 (s.d. 0.04).
Figure 7.13. The increase of exposure accumulated (in person µg m-3) over the total receiving area (from 10km to several hundred to thousand kilometres) posed by a release of one gram benzene at 25m in the source grid-square. The mean is 50,000 person µg m-3, and the standard deviation is 33,000 person µg m-3 (both weighted for population density). The exposure increase is extrapolated to transport distances where all benzene is removed from the atmosphere (see Annex). The accumulated exposure is attributed to the respective source grid-square. The patterns of accumulated exposure increase for hydrogen chloride in Figure 7.12 roughly reflect the pattern of population densities in Figure 7.6. The pattern in Figure 7.13 of accumulated exposure increase for benzene is much smoother. Also the variation of exposure increases over the grid is smaller for benzene than for hydrogen chloride. There is a difference of less than a factor 20 for benzene, but almost a factor 100 for hydrogen chloride between highest rating source grid (South-Eastern Netherlands) and the very low rating source grids (in some very sparse populated areas in the far North) of accumulated exposure increase. Accumulated increases are rather close between neighbouring grid-squares. However, the spatial differences are expected to become sharper when exceedance of threshold values would be taken into account (see also Section 7.3). To closer examine the importance of spatial variation in atmospheric conditions and population density, accumulated human exposure increase has been plotted versus distance for seven sites. Four of these sites are similar to the ones selected in Section 7.2.5. The other three sites are the neighbouring grid-squares of the Dutch site. The four Dutch sites experience rather similar atmospheric conditions, but their populations vary considerably (largely explained by the land-water ratio in those grid-squares). The results are represented in Figure 7.14 and 7.15. Figure 7.14. Increase of accumulated exposure for one gram hydrogen chloride released at 25 m at the North, Central, South European and maritime site. To compare dependence of the increase of accumulated exposure increase on climate with dependence on local population, three Dutch sites with similar climate as the maritime site but different population densities have been included. Figure 7.15. Increase of accumulated exposure versus distance for one gram benzene released at 25m at the North, Central, South European and maritime site as a function of distance from the source. To compare dependence of the increase of accumulated human exposure on climate with dependence on local population, three Dutch sites with similar climate as the maritime site but different population densities have been included. Though the residence time of benzene corresponds to a source distance of about 3500km we present model results only up to the distance where the trajectories hit the nearest edge of the model domain. The curves in Figure 7.14 and 7.15 are linearly interpolated due to the accumulated grid-squares each representing a discrete value (being the product of concentration increase and population). The marked sections in the hydrogen chloride diagram illustrate that grid-squares of 150 150km are rather coarse for exposure assessment of a short-lived substance. The model domain as a whole is on the other hand too small to trace a long-lived substance as benzene over its full atmospheric residence time. The residence time of benzene corresponds to a source distance of about 3500km, but the source distance of the nearest edge of the model domain is between 1600km (South and North Europe) and 2400km (Northern Netherlands) for the sites considered here. Due to the longer lifetime, accumulated exposure increase of benzene is less dependent on the population in the source grid-square and more on the regional population than for hydrogen chloride. The curves for the short-lived hydrogen chloride in Figure 7.14 are dominated by the influence of population in the source grid-square (within 80 km from the source), and in the four nearest neighbour grids (step at 150km). The grid-square in the South-Eastern Netherlands extends into Belgium and Germany. It is one of the most densely populated grid-squares with almost 11 million persons. In spite of the high wind speeds that dilute concentrations, the accumulated human exposure increase for emissions in this grid is therefore larger than for the sites in other climate regions. The south-western (maritime) and north-eastern sites in the Netherlands are also densely populated (respectively 6.4 and 4.5 million persons). The next group of curves includes the grid-squares with smaller populations in the North-Western Netherlands and Austria (both 1.5 million persons) and Italy (0.9 million persons). The exposure increase in the North-West Netherlands and Italy source grid squares is almost equal due to high wind speeds in the Netherlands (7 m s-1) that reduce, and low wind speeds in Italy (4 m s-1) that enhance concentration increase. Austria shows a larger exposure increase than Italy, because wind speeds in Austria (5 m s-1) are slightly higher and the population is considerable higher than in Italy. The discussion in the previous section already has shown that atmospheric conditions at the North European site lead to low concentrations. However, in this diagram it is the very low population with 0,2 million persons in the source grid-square and 0.1-0.3 million persons in the adjacent grid-squares that leads to an extraordinarily low human exposure. Accumulated exposure increase of benzene (Figure 7.15) shows to be much less related to the population in the source grid-square but more to regional population than the short-lived hydrogen chloride. Again, the highest accumulated exposure is caused by emissions from the South-Eastern Netherlands. The curve for emissions from the Central Europe grid-square (Austria) is almost catching up though, whereas the population in the source grid-square is by a factor of seven smaller. That is because the Central European site is situated between highly populated areas in the United Kingdom, the Benelux countries, France, Germany, Italy, Hungary, and Poland. The South-Eastern Netherlands has a rather peripheral location on the other hand. Even the South European site is ranking before the North-Western Dutch grid-square. The course of accumulated exposure increase from the four Dutch sites with similar regional populations but different populations in the source grid-square is comparable after several hundred kilometres. Atmospheric conditions have a smaller influence on the pattern of accumulated human exposure increase than expected (not plotted). Even for the short-lived hydrogen chloride, which is rapidly scavenged by precipitation, the wind speed and precipitation show to be of minor importance for human exposure increase accumulated over long distances compared to population. This finding is stronger still for benzene whose small scavenging ratio renders wet deposition negligible and whose atmospheric fate is far less determined by local wind speeds, due to its long transport distances. 7.2.7 Total increase of accumulated exposure from air emissionsTotal exposure increase from an emission is the sum of the accumulated human exposure increase local to the source (from 0 to 10km) and the exposure increase regional to the source (from 10km to several hundreds or thousands kilometres).
Where: ΔAHEs,i(......) = The factor that relates one gram of substance (s) released at a height (rh) by a source located in grid-square (i), to the increase in accumulated human exposure (in person µg m-3). Local refers to increase within 0 to 10km from the source (20 20km), and regional refers to an increase over a distance of several hundreds to thousands of kilometres from the source. ΔAHE(default) = The factor that relates one gram of substance (s) released at default height (25m) and under default atmospheric circumstances (South Europe), to the increase in accumulated human exposure (in person µg ;m-3) within short distances from the source (<10km) (based on a population density of one person per km2). F(rh,ac)s,j=i = The factor modifying the default increase of accumulated human exposure according to the actual height of release (rh) and the actual atmospheric circumstances (ac) in the receiving area (j=i) local to the source, and the substance released (s) PDj=i = The factor modifying the default increase of accumulated human exposure according to the population density in the receiving area (j=i) The accumulated human exposure increase over long distances from the source can be taken from Figure 7.12 for hydrogen chloride and from Figure 7.13 for benzene. The exposure increase of benzene is hardly influenced by release height and thus for all heights similar. For hydrogen chloride, the data have to be adjusted according to the factors for the relevant release heights that are mentioned in the caption to Figure 7.12. The accumulated human exposure increase local to the source can be calculated from the default of 69.7 person µg m-3 for a population density of one person km-2 multiplied by the population density in the grid-square where the source is located. The default of 69.7 person µg m-3 relates to the exposure increase from benzene and from a release height of 25m in the area of 0 to 10km from the source (20 20km). Table 7.3 provides factors to modify this exposure increase according to the substance being emitted (benzene or hydrogen chloride), the height of release (1m, 25m, 150m), and the climate region in which the source is located. The results of applying Formula 7.2 and 7.3 to an emission of one gram hydrogen chloride and benzene are illustrated in Table 7.4. The table shows basically the same trends as discussed in the previous sections, though the dominance of the accumulated human exposure increase local to the source becomes very strong now for hydrogen chloride. Short-lived substances as hydrogen chloride have a large share of their impact within the first 10km from the source and this is obviously intensified for low heights of release and high population densities. The sensitivity for source height and population density local to the source makes the exposure increase from short-lived substances far more uncertain than the exposure increase from long-lived substances. As Table 7.4 illustrates, long-lived substances as benzene usually will have most of their impact regional to the source. Table 7.4: The sum of the local and regional increase of exposure for one gram of benzene and hydrogen chloride release at 25m at seven sites in Europe.
The analysis of release height for different industrial processes as reported in Section 7.2.4 indicates that a release height of 25m can be taken as default for most processes (the majority of sources have a release height around 25m). It is suggested not to deviate from this default release height of 25m unless a deviating release height is strongly probable. Only release heights for energy production and few other industries (see Table 7.2) typically will be relative high compared to the overall mean. A release height of 1m is relevant for emissions from transport. Population densities will usually be higher in build-up areas (900 person km-2) and city-centres (21,500 person km-2). This will especially have large influence on exposure increase when the source is located in the vicinity of a relatively large city and the peak of the concentration increase occurs in the middle of this city. However, the location of a source in relation to a built-up area will in general not be known in life cycle assessment. In addition, a population density of 900 person km-2 over 10km2 corresponds to a population of 90,000 persons. While a city of that size is as such not unusual, this will often already be reflected in the population density of the region where this city is located (see Figure 7.6). The proposed framework in this chapter therefore suggests taking the population density in the relevant region (see Figure 7.6) to quantify the exposure increase local to the source. The number of toxic substances in a typical life-cycle inventory can be considerable. However, the spatial differentiated exposure factors in this chapter relate to two substances only: Hydrogen chloride and benzene. These two substances have been selected on the basis of their lifetimes. Hydrogen chloride represents substances with a very short lifetime, while benzene represents rather long-lived substances. The exposure increase from other substances will thus in general be in between the increase from hydrogen chloride and benzene. The results for these two substances can therefore with reasonable confidence be used to evaluate spatial variation in source locations. Exposure increase within short distance from the source and over long distances has been quantified with two different models. The models have been calibrated to the extent possible such that input and model data were comparable. Nevertheless, both models are based on different mathematics. The results have been added together without extensively checking how fluently the exposure increase from the local model runs over into the one of the regional model. A rough comparison, which is not further reported here, has been made with results from the OPS-model (Van Jaarsveld 1990, Van Jaarsveld and De Leeuw 1993). This model is limited to source grid-squares in the Netherlands, but fluently integrates modelling of local and regional concentration increases. The comparison suggests that it is reasonable to sum local and regional exposure increases as done here. A better calibration is recommended, however, in combination with quantification of uncertainties related to each model as such and from putting them together. The presented site-dependent factors cover accumulated exposure increase from a release, and disregard whether threshold values are exceeded as a result of the exposure. The spatial differences between the factors are expected to become more pronounced if exceedance of threshold values would be taken into account. 7.3 Effect assessment (evalution of threshold exceedance)7.3.1 IntroductionThe impact assessment phase in life cycle assessment initially emerged from the wish to aggregate the large amount of data from inventory analysis to a manageable amount of impact data. For most impact categories and also for human toxicity, rather simple modelling was used at first to establish characterisation factors. Those characterisation factors were all based on equivalency assessment on the basis of intrinsic substance characteristics. For human toxicity, the emission data from inventory were usually converted in dilution volumes by means of dividing emission data by some kind of threshold value. (Potting et al. 1999, Potting 2000) No assessment of threshold exceedance was performed in this simple modelling since the available data did not allow such evaluation. Threshold information was used in toxicity assessment only to express the emission quantity of a given substance in the volume needed in the amount of receiving environment needed to dilute the emission until the threshold value was reached. The so calculated dilution volumes provided the possibility to aggregate substances with different toxic effects. (Potting et al. 1999, Potting 2000) The basis of equivalency in this simple modelling was taken in the toxicity potential of each substance [58]. The impact from an emission quantity equal to the threshold value was put on one, and the impact from any deviating quantity was assessed as the ratio of that quantity divided by the threshold value for that substance. The underlying assumption was that the toxicity impact from an emission quantity at the threshold value for a substance has the same importance as the toxicity impact from the quantity at the threshold value of another substance. To put it more clearly: If the quantities of both substances are at their threshold value, the impacts from a neuro-toxic substance and an irritating substance are regarded as equally important [59] (Potting et al. 1999). The initial human toxicity factors have later been replaced by factors based on more sophisticated modelling. Present typical toxicity factors as from Guinée et al. (1996), Hauschild and Wenzel (1998), Huijbregts (1999) and Hertwich et al. (2001) now also cover modelling of fate and exposure increase. Aggregation of calculated exposure increases from different substances is still based on threshold values. Where initially occupational standards were often used, however, it is now common practice to use no-effect-concentrations or safe doses. As mentioned in Section 7.2, the presently typical methods for the assessment of human toxicity impact in life cycle assessment do still not take into account background exposures, since they calculate exposure increases rather than actual exposures (sum of background exposure and exposure increase). This section discusses the need for threshold evaluation, and provides information for a selection of substances to implement a qualitative evaluation in life cycle assessment. 7.3.2 Need for evaluation of threshold exceedanceThere is ongoing discussion about whether and how to perform an evaluation of threshold exceedance in life cycle assessment. The discussion is relevant in particular with regard to human toxicity. Human toxicity assessment initially focused on quantifying the probability of surpassing threshold values (like no-effect-concentrations or regulatory standards) in the direct vicinity of emission sources. This focus was directly related to the perceived local character of human toxic impact. Contrary to the present situation, increased pollution levels could in general be traced back to a neighbouring single source (usually large point sources like industry of waste processing). The severity of human toxic and eco-toxic impact local to some sources moved environmental regulation to curb the most pressing situations and prevent similar ones in future. Since then, a large body of policy instruments has been implemented (like licenses, levies and subsidies, and anti-pollution taxes). Risky situations local to sources have in this way been prevented by keeping the emissions from the point source under control. Risk was interpreted as exposure levels above a given threshold. (Potting et al. 1999) Meanwhile, the effective implementation of environmental policies in most of the industrialised countries has led to a situation where the emissions of toxic substances from large point sources in general have been reduced considerably. Large reductions have been achieved by add-on emission reducing technologies or by structural technological improvements in production processes. Risk from a single source to its local environment is nowadays prevented in most cases, and the total emissions from all large point sources together have been reduced substantially for many substances over the last decades. (Potting and Hauschild 1997, Wenzel et al. 1997, Hulskotte et al. 1997, EEA 1998) In spite of the remarkable successes in emission reduction, management of risk from toxic substances is more topical than ever. As mentioned above, individual sources rarely by themselves cause an exceedance of threshold values in their local environment. However, the intricate net of sources and the dispersion of many substances over large distances have led to a widespread presence of a broad variety of different substances that result from many sources together (rather than from a single source). Even though the exposure to an individual substance may remain below its no-effect-concentration, the accumulated exposure to a cocktail of substances may pose a risk. The toxic impact of exposure to a mixture of different substances is unknown, but can be additive or even synergistic. In addition, whereas initially no-effect-concentrations were supposed to exist, there is nowadays growing evidence that a substance may have effects below its assumed no-effect-concentration. This concern is raised by a number of observations of effects on human beings and animals that are highly suspected to relate to such combined exposures. Examples of this are the reduced sperm quality in mens, the increase in testicle cancer and the relative increase in the frequency of female trout in some water bodies (Toppari et al. 1995) and the increase in asthma and allergy among humans in Western European countries. In the present environmental situation, it has become relevant to consider exposure situations both above and below relevant threshold values, but still to evaluate exposures above threshold as being more severe compared to the ones below threshold. This was the main incentive to explore the possibilities for a qualitative evaluation of threshold exceedance in life cycle impact assessment as part of the Danish LCA-methodology development and consensus creation project. 7.3.3 Possibilities for including evaluation of threshold exceedance [60]As demonstrated in Section 7.2.3, the concentration increase from an outdoor source may have a considerable contribution local to that source (from 0 to 10km), but it rapidly becomes smaller at longer distances. Neither close to the source nor at longer distances, however, will outdoor emissions nowadays be expected to cause increases that by themselves lift the ambient concentration from below to above threshold values (see also Section 7.2.3). The ambient concentration at a given location, the concentration that one inhales, is the sum of the existing background concentration and the concentration increase from the given source. Information about the background concentration is thus indispensable to perform an adequate threshold evaluation. The background concentration at a given location is often the result of contributions from more sources together, while these sources can each be close to, or more distant from that location. Obviously, background concentrations in densely populated areas will in general be higher than in sparsely populated areas due to differences in societal and economic activity (i.e., source density). The risk for threshold value exceedance by a concentration increase will therefore also be higher in densely populated areas. At the same time, the accumulated exposure increases are obviously also higher for sources located in densely populated areas (exposure being the product of concentration times population). If exceedance of threshold values had been taken into account, the combined effect of this would probably have resulted in more pronounced spatial differences in the site-dependent factors as established in Section 7.2. Integrated assessment models like the RAINS model described and used in Chapters 3, 4 and 6 are an obvious way to combine the estimation of background concentrations from multiple sources and the concentration increases from an individual source, with population densities and with threshold evaluation. Whereas such models already exist for other impact categories, however, they are only of limited availability for the assessment of human toxicity. Important reasons are the infinite number of potential human toxic substances, their considerable differences in fate, and the differences in number and type of their possible sources. Also for human toxicity assessment, integrated assessment models are expected to become more important. At present, developments take place in that direction for several groups of substances. However, the present state-of-the-art in modelling and data availability regarding toxic substances does not yet allow making such threshold evaluation at more than a qualitative level. One solution is to refrain from inclusion of threshold information in the quantification of human toxicity impact. This assumes that similar exposure increases are equally important, regardless whether the background concentration (together with the concentration increase) is below or above the relevant no-effect-concentration. An intermediate approach could be to consider background concentrations as default being below threshold values, unless they are with high probability close to, or above these values. Such intermediate approach is presented in the next sections. This part of the developed framework also covers indoor situations. 7.3.4 Chosen type of threshold valuesOne threshold figure for exposure, above which effects occur and below which no effects are seen, is a crude simplification of reality. Humans differ in terms of age, size, sex, race and other physiological factors, and consequently they react differently upon chemical exposure. Established "threshold" levels are often based on results obtained from animal experiments at relatively high exposure levels. This implies respectively extrapolation from animal-to-human and from high-to-low dose levels when establishing the threshold levels. In some situations it is further necessary to extrapolate from short-to-long term exposure durations. Threshold levels for the external environment are developed to protect even the more sensitive population groups. Therefore, exposure above the threshold levels will not necessarily result in massive toxicological effects. On the other hand, caution should be taken as definitions of threshold levels for regulatory purposes are sometimes not only based on a toxicological/scientific background but also take on technological feasibility, costs of compliance, prevailing exposure levels, social, economic and cultural conditions (WHO, 1998). WHO stresses that a distinction should be made between WHO guidelines - derived from purely epidemiological/toxicological data - and other "quality standards" (like regulatory thresholds) where the above societal factors may have influenced the levels (WHO, 1998). The same principle is applied by the EU, which operates with both EU Guidelines and EU limit values. The guidelines are based on evaluation of scientific data only and are the levels below which only insignificant effects may be expected, whereas the legally binding limit values may be the result of cost-benefit analyses (EEA, 1997). The developed framework will - where possible – compare actual exposure levels with guidance values; i.e. the guidance values will be considered "threshold". These "threshold levels" include air concentration levels and acceptable/tolerable daily intakes (ADI/TDI's) which are assumed to cause no significant acute and also no chronic effects after life-long exposure/intake. The existence of a threshold - meaning that an exposure level exists below which no toxic effect is seen - is controversial. Some scientists argue that no substances have thresholds, whereas others argue that all substances have. However, the more general assumption is that some substances act via a non-threshold mechanism; especially genotoxic substances. Therefore, acceptable exposure levels for these substances are often expressed as a level, which implies a life long risk of 10-5 or 10-6 of obtaining cancer. 7.3.5 Chosen emission/exposure situations and substancesAs argued in Section 7.3.2, single sources in themselves do usually not evoke exceedance of threshold values for human toxicity anymore. It was after all local problems that initiated the extensive body of process oriented environmental policy. For the purpose of impact assessment in LCA, it seems therefore fair to assume that environmental concentration levels resulting from one single source generally remains below the no-effect-level due to the process oriented policy measures. At least this is expected to be the case for the most developed countries. However, there are exceptions from this assumption. The first exception concerns the zone very closely surrounding the discharge point where exceedance of the no-effect-level may still occur. In environmental regulation, the exceeding of the no-effect-level is accepted within a certain defined dilution zone. This zone is in general situated at, or immediately surrounding the domain of the releasing industry, and might thus be considered a part of the technosphere rather than the ecosphere. It can therefore be argued that LCA can disregard this exception from the marginality assumption in characterisation modelling. (Potting and Hauschild 1997) The second exception concerns occupational exposures and exposures resembling occupational exposures. This type of exposure situations are subject of another part of the Danish LCA-methodology development and consensus creation project. (Schmidt et al. 2000) and not further addressed here. Other human exposure situations, which typically might be near or above thresholds are listed below. The list is a further development of a list prepared at the first workshop of the Danish LCA-methodology development and consensus creation project carried out 15 May 1998.
Inhalation will be the primary exposure route for many of the described exposure situations, but also indirect exposure via drinking water and foodstuffs can be relevant as well as direct skin contact (mainly allergic reactions in the consumer phase). Table 7.5 Overview of situations where guidance values may be exceeded regularly.
¤ To be used with caution. Has been derived from the TDI assuming: 64 kg body weight, inspiration of 22 m³ per day and the same bioavailability/uptake via oral and inhalation exposure. Especially the latter assumption may be questioned. * `bw': Abbreviation for `body weight'. # In relation to LCA, attention must be paid to substances with non-threshold mechanisms, e.g. benzene and particles. For these substances, any elevation in exposure will result in an elevated risk. The recommended threshold levels are therefore less relevant in relation to site characterisation. & For industrialised countries, regulation of point sources will often aim at protecting the surrounding area from above threshold exposure situations. This assumption may often be interpreted as default, but exceptions may occur. It is beyond the scope of this methodology development and consensus project to outline all substance specific exposure situations that may be "near or above threshold". A number of substances have therefore been selected. These substances are those contributing significantly to the EDIP97 normalisation factor for human toxicity (Hauschild and Wenzel, 1998). They are supplemented by a number of other typical and well-known environmental pollutants known as potentially harmful to human health, including particles and some aromatic and/or halogenated substances. The prevailing exposure levels will be outlined and where possible, they will be quantified and compared to threshold levels. An extensive description for each substance is given in Annex 7.6, but the results are summarised in Table 7.5. Background concentrations in the outdoor environment have high probability to be close to or above threshold values on a regional scale in industrialised regions with a highly intricate net of sources emitting the given substance. Maps of regional exposure levels over Europe have been found for mercury (Ryabonshapko et al. 1998), lead and cadmium (Pekar et al. 1998a), lindane, polychlorinated biphenyls and benzo(a)pyrene (Pekar et al. 1998b), photochemical oxidants (Simpson et al. 1997), and nitrogen and sulphur (EMEP/MSC-W 1998). Berdowski et al. (1997) give emission projections over the European grid for a number of persistent organic pollutants and heavy metals. Annex 7.5 gives an overview of addresses where the several reports can be obtained. Annex 7.6 gives a more qualitative description or regional exposure levels together with the exposure levels in those other situations as identified above. It is difficult to give general recommendations on whether or not a specific exposure situation encountered in LCA can be assessed to be near or above "threshold". However, table 7.5 presents a crude summary of the findings of the present paper. The table should not be regarded as bare facts, but as a best attempt to summarise the previous paragraphs, which are again based on the level and extent of the consulted literature. Furthermore, it should be observed that many of the data of the consulted references originate from the beginning of the 1990'ies, since when the situation has been rapidly changing due to for instance new abatement technology and changes in the traffic load. 7.4 ConclusionsThis chapter explores the influence of spatial differences in atmospheric conditions and source characteristics (location, release height, location in relation to built-up area) on the accumulated human exposure increase and subsequent effect from atmospheric emissions. The spatial differences in exposure increase are thoroughly quantified for two substances, the effects of possible exceedance of threshold values are covered in a more qualitative way. Some remarks about the established site-dependent exposure factors and about threshold exceedance have already been made in the previous section. Here, some main conclusions are drawn. There is a difference of less than a factor 20 for benzene, but almost a factor 100 for hydrogen chloride between highest rating (South-Eastern Netherlands) and low ratings (in very sparse populated areas in the far North) of the (regional) accumulated exposure increase. The spatial differences are expected to become sharper when exceedance of threshold values would be taken into account. The population density shows to have the largest influence on the differences in regional exposure increase from sources at different locations. The importance of population density becomes stronger for a short-lived substance like hydrogen chloride that has its increase of exposure predominantly within the first hundred km from the source. Also variation in atmospheric conditions (region-dependent) and source height have an influence on the regional exposure increase. However, their impact is smaller compared to population density. Accumulated exposure increase local to the source is very dominant for short-lived substances as hydrogen chloride. Their impact occurs largely within the first 10km from the source. The sensitivity for source height and population density local to the source makes the (total) exposure increase from these substances far more uncertain than the exposure increase from long-lived substances that have most of their impact regional to the source. The majority of industrial sources appear to have a release height around 25m and it is therefore suggested to take this as default unless a deviating release height is strongly probable. Only release heights for energy production and few other industries will typically be higher (see Table 7.2), and those for transport will be typically lower compared to the overall mean. It is suggested to take the population density in the relevant region (see Figure 5.4) to quantify the exposure increase local to the source (from 0 to 10km). Population densities will usually be higher in build-up areas (900 person km-2) and city-centres (21000 person km-2). However, the location of a source in relation to built-up area is in general not known in LCA. The number of toxic substances in a typical life cycle inventory can be considerable, while the spatially differentiated exposure factors in this chapter relate to two substances only. The exposure increase from other substances will thus in general be in between the increase from hydrogen chloride and benzene. These two substances can therefore with reasonable confidence be used to evaluate the effect on accumulated human exposure increase from spatial variation in source locations. It is difficult to give general recommendations on whether or not a specific exposure situation encountered in LCA can be assessed to be near or above "threshold". Table 7.5 presents a crude summary of the findings of the present paper, however, that can be used for a qualitative evaluation of possible exceedance of threshold values. This chapter does unfortunately not result in a set of factors integrating exposure and subsequent effect (from threshold exceedance), that can be used to characterise the contributions of various substances and emission situations to human toxicity in life cycle assessment. The state-of-the-art in modelling human toxicity does not allow such integrated factors, however. The material produced in this chapter can merely be used for sensitivity analysis in life cycle assessment. Hauschild and Potting (2003) describe in detail how this can be done. The work in this chapter focuses on direct exposure and effect from atmospheric emissions. Spatial differences with regard to other exposure routes were regarded less important and not addressed (see Section 7.2.2). Huijbregts et al. (2003) assessed in a recent study the effect of using different generic environmental conditions (typical conditions in Australia, Western Europe, and the United States of America). It was found that the uncertainty in fate and exposure factors for ecosystems and humans due to choices of different sets of environmental conditions is between a factor 2 and 10. Particularly, fate and exposure factors of emissions causing effects in fresh water ecosystems and effects on human health have relatively high uncertainty. This uncertainty is mainly caused by the continental difference in the average soil erosion rate, the dimensions of the fresh water and agricultural soil compartment, and the fraction of drinking water coming from ground water. These results encourage to closer look into spatial issues of these exposure routes as well. 7.4.1 AcknowledgementThe authors like to express their gratitude to Burkhard Wickert (IER, University of Stuttgart) for processing of the population data on the Geographical Information System. The work for this chapter was also supported by a Training & Mobility grant from the European Commission, by means of the European Commission under the JOULE Program, and under the EUREKA project LCAGAPS by means of the Danish Agency for Industry and Trade. 7.5 ReferencesATSDR (1997a). Toxicological Profile for Benzene (Update). Agency for Toxic Substances and Disease Registry, Public Health Service, U.S. Department of Health and Human Services. ATSDR (1997b). Toxicological Profile for Chlorinated dibenzo-p-dioxins. (Draft for Public comment). Agency for Toxic Substances and Disease Registry, Public Health Service, U.S. Department of Health and Human Services. ATSDR (1997c). Toxicological Profile for Lead (Draft for Public comment). Agency for Toxic Substances and Disease Registry, Public Health Service, U.S. Department of Health and Human Services. ATSDR (1997d). Toxicological Profile for Cadmium (Draft for Public comment). Agency for Toxic Substances and Disease Registry, Public Health Service, U.S. Department of Health and Human Services. ATSDR (1997e) Toxicological Profile for Mercury (Draft for Public comment). Agency for Toxic Substances and Disease Registry, Public Health Service, U.S. Department of Health and Human Services. ATSDR (1998). Toxicological Profile for Sulfur dioxide (Draft for Public comment). Agency for Toxic Substances and Disease Registry, Public Health Service, U.S. Department of Health and Human Services. Berdowski, J.J.M., W.J. Jonker, P. Verhoeve. Emissions in the Netherlands. Industrial sectors and regions in 1993 and estimates for 1994. No. 27 in publication series emission registration (In Dutch). The Hague (the Netherlands), Dutch Ministry of Housing, Spatial planning and the Environment, 1995. Berdowski, J.J.M., J. Baas, JP.J. Bloos, A.J.H. Visschedijk and P.Y.J. Zandveld. The European Atmospheric Emission Inventory of Heavy Metals and Persistent Organic Compounds. Forchungsbericht 104 02 672/03. Umweltbundesamtes and TNO Institute of Environmental Sciences, Energy Research and Process Innovation, 1997. Berge, E. Six-hourly data in the EMEP 150 km grid. Written communication and data on magnetic tape. Oslo (Norway), EMEP Meteorological Synthesising Centre – West, 1994. Derwent, R.G., and K. Nodop. Long-range transport and deposition of acidic nitrogen species in north-west Europe. Nature, Vol. 324 (1986), November Issue, pp. 356-358, 1986 EC. Risk assessment of existing substances. Technical guidance document in support of the commission regulation (EC) no. 1488/94 on risk assessment for existing substances in accordance with council regulation (EEC) no. 793/93 (XI/919/94-EN). Brussels (Belgium), Directorate General Environment, Nuclear Safety and Civil Protection from the European Commission, 1994. EC. Technical guidance document in support of the commission directive 93/67/EEC on risk assessment for new notified substances and the commission regulation (EC) no. 1488/94 on risk assessment for existing substances. Ispra (Italy), European Bureau of Chemicals, 1996. EEA. Europe's environment. Statistical compendium for the second assessment. Copenhagen (Denmark), European Environment Agency, 1998. EEA (1997). European Environmental Agency. Air pollution in Europe 1997. EEA Environmental Monograph No. 4. Copenhagen. ISBN 92-9167-059-6. ETCAE (1997). Corinair 94. Summary report. Final Version. Report to the European Environment Agency from the European Topic Centre on Air Emissions. European Council (1998). Common position (EU) No. 40/98 outlined by the council 16 June 1998 regarding limitation of the emission of volatile organic compounds from the application of organic solvent in certain activities and facilities. Guinée, J., R. Heijungs, L. van Oers, D. van de Meent, T. Vermeire and M. Rikken. LCA impact assessment of toxic releases. Generic modelling of fate, exposure and effect for ecosystems and human beings with data for about 100 chemicals (series of publication concerning product policy, no. 1996/21). The Hague (the Netherlands), Dutch Ministry of Housing, Spatial planning and the Environment, 1996. Harssema, H. Calculating and measuring air quality (H100-204). Wageningen (the Netherlands), Wageningen Agricultural University, 1995. Hauschild, M.Z. and H. Wenzel: Environmental assessment of products. Vol. 2 - Scientific background, 565 pp. Chapman & Hall, United Kingdom, 1998, Kluwer Academic Publishers, Hingham, MA. USA. ISBN 0412 80810 2 Hauschild, M. and J. Potting. Spatial differentiation in life cycle impact assessment – the EDIP2003 methodology. Guideline from the Danish Environmental Protection Agency, Copenhagen, 2003. Hertwich E.G., S.F. Mateles, W.S. Pease, T.E. McKone. Human toxicity potentials for life-cycle assessment and toxics release inventory risk screening. Environmental Toxicology and Chemistry, 20:928-939, 2001. Huijbregts, M.A.J. Priority assessment of toxic substances in the frame of LCA. Development and application of the multi-media fate, exposure and effect model USES-LCA. Amsterdam (the Netherlands), Interfaculty dept. of Environmental Science, 1999. Huijbregts, M.A.J., S. Lundi, T.E. McKone and D. van de Meent. Geographical scenario uncertainty in generic fate and exposure factors of toxic pollutants for life-cycle impact assessment. Chemosphere, Vol. 51, issue 6, pp. 501–508. Hulskotte, J.H.J., G.P.J. Draaijers, J.J.M. Berdowski, W.L.M. Smeets and C.S.M. Olsthoorn. Recalculations of emissions to air for the period 1990-1996 (in Dutch). Publication series Emission Registration no. 41. The Hague (the Netherlands), Dutch Ministry of Housing, Spatial planning and the Environment, 1997. Jensen B, Wolkoff P (1996). VOCBASE - odour and mocous membrane irritation thresholds and other physico-chemical properties. 2.1. National Institute of Occupational Health, Copenhagen. Jensen B, Wolkoff P (1997). VOCBASE – a database of odour thresholds, irritation of mucous membranes and chemical parameters for volatile organic compounds (in Danish). Dansk Kemi; 5: 16-17, 20-26. Krewitt, W., P. Mayerhofer, R. Friedrich, A. Trukenmüller, T. Heck, A. Greßmann, F. Raptis, F. Kaspar, J. Sachau, K. Rennings, J. Diekmann, B. Praetorius. ExternE – Externalities of energy. National implementation in Germany (EUR 18271). Directorate-General XII for Science, Research and Development of the European Commission, 1997. Krüger, O., J.P. Tuovinen. The effect of variable sub-grid deposition factors on the results of the Lagrangian long-range transport model of EMEP. Atmospheric Environment, Vol. 31 (1997), Issue 24, pp.4199-4209. Metcalfe, S.E., J.D. Whyatt, R.G. Derwent. A comparison of model and observed network estimates of sulphur deposition across Great Britain for 1990 and its likely source attribution. Quarterly Journal of the Royal Meteorological Society, Vol. 121 (1995), Issue 526 (July), Part B, pp.1387-1411. NYSDEC (1995). Ambient air monitoring report for volatile organic compounds. Division of air resources - Bureau of Air Quality Surveillance. Summary of Toxic Monitoring Data from 1990 to 1993. New York State Department of Environmental Conservation. Reports can be obtained from: Bureau Director - BAQS, 50 Wolf Road, Albany, NY 12233-3256. Information in this paper has been retrieved from the Internet: http://www.dec.ny.us/website/pollution/voc_rpt.html. Potting J, Hauschild M (1997). Predicted environmental impact and expected occurrence of actual environmental impact. Part II: Spatial differentiation in life-cycle assessment via the site-dependent characterisation of environmental impact from emissions. Int. J. LCA, 4: 209-16. Potting, J., M. Hauschild, and H. Wenzel. "Less is better"and "only above threshold"; Two incompatible paradigms for human toxicity in life-cycle assessment? International Journal of Life Cycle Assessment, Vol. 4 (1999), Issue 1, pp.16-24. Potting, J. A. Trukenmüller, H. van Jaarsveld and M. Hauschild. Human exposure from air emissions. In: Potting, J. Spatial differentiation in life cycle impact assessment. Ph.D. thesis. Utrecht (the Netherlands), 2000. Potting, J. Spatial differentiation in life cycle impact assessment. Ph.D. thesis. Utrecht (the Netherlands), 2000. Schmidt, A., Rasmussen, P. B., Andreasen, J., Fløe, T. and K. E. Poulsen. Integration of the working environmennt in life cycle assessments (LCA) – a new methodology. Guidelines from the Danish Environmental Protection Agency No. xxx 2000, Copenhagen (2000) Seinfeld, J.H. and S.N. Pandis. Atmospheric chemistry and physics: From air pollution to climate change. New York (United States of America), John Wiley & Sons, 1998. Stanners, D. and P. Bourdeau. Europe's environment. The dobris assessment. Copenhagen (Denmark), European Environment Agency, 1995. Tobler, W., U. Deichmann, J. Gottsegen and K. Maloy. The Global Demography Project (technical Report TR-95-6 [online]). Santa Barbara (United States of America), dept. of Geography, University of California of the National Centre for Geographic Information and Analysis, 1995 [cited 11 August 1999]. Available from Internet http://www.ciesin.org/datasets/gpw/globldem.doc.html. Toppari, J., J.C.Larsen, P. Christiansen, A. Giwercman, P. Grandjean, L.J. Guillette, B. Jégou, T.K. Jensen, P. Jouannet, N. Keiding, H. Leffers, J.A. McLachlan, O. Meyer, J. Müller, E. Rajpert-DeMeyts, T. Scheike, R. Shapre, J. Sumpter and N.E. Skakkebæk. Male reproductive health and environmental chemicals with estrogenic effects (Miljøprojekt no. 290). Danish Environmental Protection Agency, Copenhagen, 1995. Trukenmüller, A. Using WMI: Implementing atmospheric trajectory models with the Wind rose Model Interpreter. Stuttgart (Germany), IER of the University of Stuttgart, 1998. van der Ven B (1995). Default values. A discussion paper for the meeting of the SETAC workgroup "Data handling" on Sep. 4th 1995 at Gothenborg. Van Jaarsveld, H. An operational atmospheric transport model for priority substances; specification and instructions for use. Report number 222501002. Bilthoven (the Netherlands), RIVM, 1990. Van Jaarsveld, J.A. Modelling the long-term atmospheric behaviour of pollutants on various spatial scales. Utrecht (the Netherlands), PhD thesis, Utrecht University, 1995. Van Jaarsveld J. A., W. A. J. van Pul and F. A. A. M de Leeuw. Modelling transport and deposition of persistent organic pollutants in the European region. Atmospheric Environment Vol. 1997, Issue 31, pp. 1011-1024 Van Jaarsveld, J.A. and F.A.A.M de Leeuw. OPS: An operational atmospheric transport model for priority substances. Environmental Software, Vol. 1993, Issue 8, pp. 91-100. Van Leeuwen and J.L.M. Hermens. Risk assessment of chemicals: An introduction (ISBN 0-7923-3740-9). Dordrecht (the Netherlands), Kluwer Academic Publishers, 1995/1996. Wenzel, H., Hauschild M.Z. and Alting, L.: Environmental assessment of products. Vol. 1 - Methodology, tools, techniques and case studies, 544 pp. Chapman & Hall, United Kingdom, ISBN 0 412 80800 5, Kluwer Academic Publishers, Hingham, MA. USA., 1997. Wolkoff P, Clausen P A, Nielsen G D (1998). Volatile organic compounds (VOCS) in the indoor environment. State-of-the-science report. AMI Dokumentation 2. Arbejdsmiljøinstituttet, Copenhagen. In Danish. Wolkoff P, Nielsen P A (1993). Statens Byggeforskningsinstitut. Indeklimamærkning af byggevarer. Del 2: Faglig og teknisk dokumentation af en prototypeordning. SBI-rapport 233. Statens Byggeforskningsinstitut. In Danish. WHO/UNEP (1992). Urban air pollution in Megacities of the World. World Health Organisation, United Nation Environment Programme, Blackwell, Oxford. WHO (1992). Environmental Health Criteria 134. Cadmium. International Programme on Chemical Safety (IPCS), World Health Organisation, Geneva. WHO (1993). Environmental Health Criteria 150. Benzene. International Programme on Chemical Safety (IPCS), World Health Organisation, Geneva. WHO (1994a). Environmental Health Criteria 163. Chloroform. International Programme on Chemical Safety (IPCS), World Health Organisation, Geneva. WHO (1994b). Environmental Health Criteria 170. Assessing human health risks of chemicals: Derivation of guidance values for health-based exposure limits. International Programme on Chemical Safety (IPCS), World Health Organisation, Geneva. WHO (1995). Environmental Health Criteria 165. Lead. International Programme on Chemical Safety (IPCS), World Health Organisation, Geneva. WHO (1997a). Environmental Health Criteria 188. Nitrogen Oxides (second edition). International programme on chemical safety (IPCS), World Health Organisation, Geneva. WHO (1997b). Environmental Health Criteria 195. Hexachlorbenzene. International Programme on Chemical Safety (IPCS), World Health Organisation, Geneva. WHO (1998). Air Quality Guidelines for Europe, World Health Organisation, Regional Office for Europe, Copenhagen, WHO Regional Publications, European Series. (NB! Information for this paper has been retrieved via the Internet addresses: http://www.who.dk and http://www.who.org/peh/specprg.htm.) Williams, M. (ed.). Exposure assessment. Report number 1. Cost 613/2 Report series on air pollution epidemiology. Directorate-General XII for Science, Research and Development of the Commission of the European Communities. Annex 7.1 Characterisation factors for human toxicity assessment from emissions to air
Annex 7.2 Characterisation factors for human toxicity assessment from emissions to water
Annex 7.3 Characterisation factors for human toxicity assessment from emissions to soil
Annex 7.4 Specifications for the Wind rose Model InterpreterDerwent and Nodop (1986) introduced a climatological concept to trace concentration increases resulting from substance transport and removal in the long range. This concept has been used since then in several acid deposition models (e.g. Metcalfe et al. 1995). This concept is also the basis of the Wind rose Model Interpreter (WMI) of the EcoSense integrated assessment model (Krewitt et al. 1997). The modelling approach is receptor oriented and differentiates between twenty-four sectors of the wind rose, such that from each sector a straight-line trajectory arrives at the receptor point. Concentrations at that point are obtained by averaging over the results from these trajectories, suitably weighted by the frequencies of winds in each 15º sector. WMI provides a chemical kinetics interpreter that supports stationary trajectory modelling with arbitrary mass balance and following straight lines on a variety of model grids (Trukenmüller 1998). For the present study WMI has been employed to set up a single layer model with a horizontal resolution of 150 150 km2 on the EMEP [63] grid. Input data of our model have been taken from the EcoSense data-base and include 1990 annual amounts of precipitation and 945 hPa wind roses at each grid-square. These data are aggregated values of six-hourly output of the LAM50E (Limited Area Model, 50 km, Europe) numerical weather prediction model at the Norwegian Meteorological Institute (Berge 1994). Our model uses a constant mean mixing height, which is derived from meteorological statistics for long-distant transport. Those statistics, and parameterisation of removal by dry and wet deposition and chemical conversion of hydrogen chloride and benzene have been taken from Van Jaarsveld (1990): Table A: Parameter-values used to model dry and wet deposition, chemical conversion and vertical mixing in the single layer trajectory model employed for the assessment of regional accumulated human exposure.
The assumption of instantaneous vertical mixing in the single layer model leads to an underestimation of near surface concentrations at distances less than some 10 km from the source. The underestimation of near source concentrations has no direct influence on the assessment of this local human exposure in this study where we use EUTREND results at short distances. However, it entails an underestimation of dry deposition in the source grid-square and therewith an overestimation of the transport to other grid-squares and thus accumulated exposure (Krüger and Tuovinen 1997). The `local dry deposition' has significant influence on the mass balance for hydrogen chloride. We have evaluated local deposition of hydrogen chloride by comparing dry deposition of the trajectory model in the source grid-square with deposition estimates obtained from the Gaussian OPS model (Van Jaarsveld 1990) for the various release heights. Based on this comparison, release height and wind speed dependent local deposition factors have been derived, assuming that a corresponding fraction of emissions is deposited directly to the source grid-square. High wind speeds dilute concentrations and thus decrease human exposure close to the source, but increase the distances over which a substance is transported. Transport over larger distance results in more people being exposed to the (albeit lower) concentration. The direct net effect of high wind speed on accumulated exposure is therefore usually small. There are also indirect effects of wind speed, which tend to decrease concentration and thus accumulated exposure. Wind speed influences mixing height (as discussed in the section 7.2.5), as well as the aerodynamic resistance and the pseudo-laminar layer resistance, which are relevant for removal by dry deposition. These indirect effects, unlike the direct ones, do not cancel out each other, but they are small (second order) and not taken into account in the simple trajectory model here. Dutch mixing height and dry deposition statistics from Van Jaarsveld (1990) are assumed valid for the whole of Europe. Spatial variability of precipitation is considered in the trajectory model here. While wet deposition is of minor importance for benzene, hydrogen chloride is removed from the atmosphere with every shower due to its great scavenging ratio (see Table A). Mean annual removal rates by wet deposition in the Netherlands have been derived for both substances from the empirical relation and statistics of probability, amount and duration of rainfall presented in Van Jaarsveld (1990). Those Dutch removal rates are extrapolated to the whole model domain by linear scaling with the amount of annual precipitation in each grid-square. The implicit assumption behind this scaling exercise is that the number of precipitation events per year is proportional to the amount of annual precipitation, or that the long-term mean of rainfall amount per shower is approximately the same in each grid-square. Precipitation varies largely over the grid. Annual rainfall amounts to 2000–3200 mm a-1 in grid-squares at the Norwegian coast around Bergen. Between 1500-2000 mm a-1 are found at the Western Irish and Scottish coast, in North-West Spain and some alpine grid-squares in Switzerland and Austria. However, precipitation is less than 200 mm a-1 in the Sahara Desert, parts of Turkey, South-East Russia and Kazakhstan. As a consequence, the assessment of annual wet deposition equals that of dry deposition of hydrogen chloride in the Bergen grid-square, while wet deposition is negligible in the low precipitation regions. The calculated minimum atmospheric residence time of hydrogen chloride is 4.4 h for emissions in the Bergen grid, while the maximum is 8.5 h in the Sahara Desert and Kazakhstan. For 80% of the model domain the residence time is almost constant at 6.9 h with a standard deviation of only 14%. These numbers apply to high sources (150 m) and do not include the increased dry deposition (and thereby further reduced importance of wet deposition) local to the source from low releases. Thus, even though precipitation varies largely, its impact on lifetime and accumulated exposure shows little variation and that is therefore not visible in Figure 7.13, because the grey scale in that map only resolves differences greater than a factor 2. Due to the longer lifetime of benzene, accumulated exposure to that substance is less dependent on local and more on regional population density. The model domain is too small to trace benzene concentrations over their full residence time. Approximately 40% of the benzene emitted at the Central European site and almost 60% of the benzene emitted at the North European site is subject to atmospheric transport beyond the edges of the model grid. Those figures have been calculated from the difference of the emitted and the removed substance in the WMI report concerning removal by both deposition and chemical conversion. Similar mass balances have been derived for emissions from each grid-square and used to extrapolate accumulated exposure until all emitted substance is removed. This extrapolation roughly doubles the `raw' exposure assessment of the model. It gives an overestimate by assuming European population density also for the adjacent areas of the Atlantic, the polar sea, Siberia, and the Sahara Desert. So the actual accumulated exposure is expected to be in the range of 50–100% of Figure 7.13, provided model estimates within the grid are accurate. Annex 7.5 Addresses of reports about regional emissions and exposuresFurther information on regional emissions, deposition patterns and/or background concentrations for several of the substances treated may be found in the following literature/references: "Transboundary acidifying air pollution in Europe. Part 1: Estimated dispersion of acidifying and eutrophying compounds and comparison with observations". EMEP/MSC-W. Report 1/98. July 1998. "Transboundary acidifying air pollution in Europe. Part 2: Numerical Addendum". EMEP/MSC-W. Report 1/98. July 1998. "Photochemical oxidant modelling in Europe: multi-annual modelling and source-receptor relationships." EMEP/MSC-W. Report 3/97. July 1997. "Atmospheric supply of nitrogen, lead, cadmium, mercury and lindane to the Baltic Sea." EMEP/MSC-W. Note 3/98. July 1998. "Long-range transport of selected persistent organic pollutants" Part I (EMEP/MSC-E 2/98) "Mercury in the atmosphere of Europe: concentrations, deposition patterns, transboundary fluxes" (EMEP/MSC-E 7/98) "Modelling of long-range transport of lead and cadmium from European sources in 1996." (EMEP/MSC-E 5/98) Berdowski, J J M et. al (1997). "The European atmospheric emission inventory of heavy metals and persistent organic pollutants for 1990". Forshungsbericht 104 02 672/03. EEA (1997), se general reference list. ftp://info.rivm.nl/pub/lae/edgarv20/ For future information retrieval on background levels, the following contacts are valuable: EMEP Meteorological Synthesizing Centres West (MSC-W) Det norske meteorologiske institutt (DNMI) EMEP Meteorological Synthesizing Centres East (MSC-E) Sergey Dutchak Meteorological Synthesizing Centre- East Keldrova U1. 8-1 117 321 Moscow (msce@sovam.com) TNO - Department for Ecological Risk Studies 7.6 Annex 7.6 Exposure situations "near or above threshold"Author: Frans Møller Christensen 1 Introduction The aim of this annex paper is to describe typical "near to, or above threshold human exposure situations" in terms of typical/rule-of thumb magnitude and duration, as an input to the development of a site factor framework for assessment of human toxicity in LCA. Exposure situations covered by this annex paper: Human exposure in or via the external environment (mainly inhalation of ambient air and intake of foodstuffs and water) Indoor exposure (excluded are occupational exposure, and exposure situations resembling occupational exposure, for instance painting) Consumer exposure (excluded are exposure situations resembling occupational exposure, for instance painting) 2 Threshold One threshold figure for exposure, above which effects occur and below which no effects are seen, is a crude simplification of reality. Humans differ in terms of age, size, sex, race and other physiological factors, and consequently they react differently upon chemical exposure. Further, established "threshold" levels are often based on results obtained from animal experiments at relatively high exposure levels, which implies respectively extrapolation from animal-to-human and from high-to-low dose levels when establishing the threshold levels. In some situations it is further necessary to extrapolate from short-to-long term exposure periods. Threshold levels for the external environment are developed to protect even the more sensitive population groups. Therefore, exposure above the threshold levels will not necessarily result in massive toxicological effects. On the other hand, caution should be taken as definitions of threshold levels for regulatory purposes are sometimes not only based on a toxicological/scientific background but also take on technological feasibility, costs of compliance, prevailing exposure levels, social, economic and cultural conditions (WHO, 1998). WHO stresses that a distinction should be made between WHO guidelines - derived from purely epidemiological/toxicological data - and other "quality standards" (like regulatory thresholds) where the above societal factors may have influenced the levels (WHO, 1998). The same principle is applied by the EU, which operates with both EU Guidelines and EU limit values. The guidelines are based on evaluation of scientific data only and are the levels below which only insignificant effects may be expected, whereas the legally binding limit values may be the result of cost-benefit analyses (EEA, 1997). Throughout this paper, actual exposure levels will - where possible - be compared with guidance values; i.e. the guidance values will be considered "threshold". These "threshold levels" include air concentration levels and acceptable/tolerable daily intakes (ADI/TDI's) which are assumed to cause neither significant acute nor chronic effects after life-long exposure/intake. The existence of a threshold - meaning that an exposure level exists below which no toxic effect is seen - is controversial. Some scientists argue that no substances have thresholds, whereas others argue that all substances have. However, the more general assumption is that some substances act via a non-threshold mechanism; especially genotoxic substances. Therefore, acceptable exposure levels for these substances are often expressed as a level, which implies a life long risk of 10-5 or 10-6 of obtaining cancer. 3 Typical high concentration exposure situations Human exposure (except occupational) situations, which are believed to be typically near or above thresholds are listed below. The list is a further development of a list prepared at the first workshop in sub-project V of the LCA-methodology project carried out 15 May 1998. Urban areas, especially with high traffic intensity [64] Long range transport of some air pollutants which may elevate regional background concentrations considerably Areas around point sources [65] Accidents (accidental risk is usually considered separately in LCA) Indoors Direct consumer exposure to products; for instance cosmetics (containing heavy metals, preservatives, perfumes and oxidised tensides which may all cause allergic reactions), textiles (containing for instance azo dyes, some of which may provoke allergic reactions), toys (which have appeared to contain potentially carcinogenic and endocrine acting substances) and a variety of products containing chromium and/or nickel (both being allergens) Inhalation will be the primary exposure route for many of the described exposure situations, but also indirect exposure via drinking water and foodstuffs can be relevant as well as direct skin contact (mainly allergic reactions in the consumer phase). 4 Typical substances involved in high exposure situations It is beyond the scope of this paper to outline all substance specific exposure situations which may be "near or above threshold". A number of substances have been selected for a narrower study. The substances selected are those contributing significantly to the 1990 EDIP normalisation factor for human toxicity (Hauschild and Wenzel, 1998). They are supplied with a number of other typical and well-known environmental pollutants known as potentially harmful to human health, including particles and some aromatic and/or halogenated substances. The prevailing exposure levels will be outlined and where possible, they will be quantified and compared to threshold levels. References in this paragraph will mainly be WHO/IPCS (World Health Organisation/ International Programme on Chemical Safety) and ATSDR (Agency for Toxic Substances and Disease Registry) criteria documents. Further information and original references can be found in these documents. 4.1 NO2/NOx NOx covers both NO and NO2 with NO2 being the more toxic. Often just NOx is measured and/or given in tables. In the present EDIP methodology, NOx is considered to be NO2. This assumption is also used here and is partly justified by the fact that NO often will be oxidised to NO2 by ozone (EEA, 1997). The main outdoor NOx sources are combustion processes. According to Corinair94 (ETCAE, 1997), about 50% of the European emission comes from "Road transport" [66] , a little less than 20% from "Combustion in energy and transformation industry" and 10-15% from "Other mobile sources and machinery". The mobile source contribution to NOx emission is estimated to be between 20 and 80% in different megacities of the world (WHO/UNEO, 1992; see also WHO, 1997a). Indoor NOx-sources comprise gas stoves, unvented (i.e. unventilated) gas space heaters and water heaters, Kerosene space heaters, wood stoves and tobacco products (WHO, 1997a). WHO (1997a, 1998) suggests the following health-based guidance values for NO2: Short term: 200 µg/m³ (0.11 ppm) as a one-hour average daily maximum. Long term: 40 µg/m³ (0.023 ppm) as an annual average. EEA (1997) cites another WHO guidance value of 150 µg/m³ (0.086 ppm) as a 24-hour average. Average annual concentrations in large American, European and Asian cities often exceed 40 µg/m³ (WHO, 1997a). In Europe it is estimated that about 40% of the urban population (27 mio. individuals) is exposed to an annual average above 50 µg/m³ (EEA, 1997). In many major cities one-hour average exposure levels of above 400 µg/m³ occur regularly (WHO, 1997a). Maximum 24-hour concentrations regionally may reach 60-70 µg/m³ in most of central Europe (EEA, 1997). These exposure levels are well below the indicated WHO 24-hour guidance value and as such judged not to pose a health risk to the population outside urban areas (EEA, 1997). Indoor NOx-concentrations rely on a number of factors including outdoor concentration, ventilation rates and indoor sources. In homes without indoor sources, the NOx-level has been measured to 26-80% of the outdoor level (WHO, 1997a). Thus indoor concentrations even in homes without indoor sources may in some situations exceed the WHO annual average guidance value of 40 µg/m³. In about 45% of American homes and up to 100% in some other countries, gas is used for cooking, heating water or drying clothes. Indoor NO2-concentrations in excess of 100 µg/m³ (average over one-two weeks) have been measured. Data on indoor short term concentrations, suggest levels several times higher than the average values, i.e. situations with short term concentrations above 200 µg/m³ may frequently occur. Concentrations are higher during the wintertime with high heat production and reduced ventilation, and concentration gradients usually exist from the indoor point source to other parts of the home (WHO, 1997a). Indoor NO2-concentrations may be even higher in homes with unvented gas space heaters and Kerosene heaters. One study indicates NO2-concentrations of 100 µg/m³ (average over one-two weeks) for 50% and above 480 µg/m³ for 8%, respectively, in homes with Kerosene heaters. A peak value of 847 µg/m³ was measured. A large field study showed concentrations above 100 µg/m³ in 70% and above 480 µg/m³ in 20% of homes with unvented gas space heaters (WHO, 1997a). 4.2 SO2 The major source of SO2 in the environment is fuel combustion in connection with energy & transformation industries as well as manufacturing industries. These sources contribute to 80-85% of the SO2 emission (ETCAE, 1997; ATSDR, 1998). Other sources are the chemical and allied product manufacturing, metal processing, petroleum and related industries, other industrial processes, and on road vehicles (ATSDR, 1998). SO2 is mainly formed by combustion of coal or oil with a high sulphur content (ATSDR, 1998). WHO guidance values are (ATSDR, 1998; WHO, 1998): 10-min exposure limit: 500 µg/m³ (0.2 ppm). 1-hour exposure limit: 350 µg/m³ (0.13 ppm). 24-hour exposure limit: 125 µg/m³ (0.05 ppm). Annual average: 50 µg/m³ (0.019 ppm). The SO2 air concentration is about 1-5 µg/m³ in very remote clean areas, whereas highly industrialised areas may reach 6000 µg/m³ (ATSDR, 1998). Areas with above background SO2-levels are found in or close to urban areas, in the proximity to sites where SO2 is produced or where SO2 was disposed of and in some situations in proximity to hazardous waste sites (ATSDR, 1998). An air pollution study in megacities of the world showed short term peak values above 700 µg/m³. Annual averages above 150 µg/m³ were found in some cities (e.g. Beijing, Mexico City and Seoul) and levels of 50-100 µg/m³ were reported for some other (e.g. Rio de Janeiro and Shanghai) (WHO/UNEP, 1992). For European conditions it is estimated that about 70% of the total population of all cities (about 37 mio. individuals) is exposed to levels above 100 µg/m³ as a 24-hour mean. Furthermore, maximum 24-hour concentrations regionally may reach 100-150 µg/m³ in several areas of Europe (Central/East Europe and the UK) indicating that during episodic "winter smog" situations in Central and North-western Europe, a large part of the population is exposed to SO2 concentrations which pose a certain health risk (EEA, 1997). 4.3 Particles The principal man-made source for traditional air pollutants, including particles, is combustion (WHO/UNEP, 1992). Particles include both primary particulates in the form of fly ash and soot and secondary particulates, sulphate and nitrate aerosols formed in the atmosphere following gas to particle conversion (WHO/UNEP, 1992). Diesel-fuelled engines emit significant quantities of particles (WHO/UNEP, 1992). Particles in the air environment are often termed SPM (suspended particulate matter). In terms of measuring values, quite a few parameters can be measured. Traditionally, suspended particles have been measured as TSP (total suspended particulates) or as Black Smoke (BS). BS represents the black soot particles from combustion, and is dominated by coal smoke and diesel soot. BS is a relevant indicator for the assessment of health effects, but the measurement technique provides fairly inaccurate results (EEA, 1997). More recent, the indicator for suspended particles has become PM10 (particles with diameter below 10 µm) measuring the particles believed to cause the major health concerns as these may penetrate deeply into the airways. However, measurement data of this parameter are still very incomplete (EEA, 1997). Recent evidence suggests that there is no lower limit for health effects connected to particle exposure. Therefore WHO has not set a guidance value. Instead an exposure-reponse model has been set up from which regulators/decision makers can define an "acceptable" exposure level [67] . The EU is presently considering how to handle PM10. The UK has recommended a 24-hour average PM10 guideline of 50 µg/m³ (EEA, 1997). In a study of air pollution in megacities of the world (WHO/UNEP, 1992), it was concluded that in 12 cities (mainly Asian, but also Mexico City and Cairo) there was a serious problem with annual average SPM concentration levels of 200-600 µg/m³ and peak concentrations frequently above 1000 µg/m³. In five cities (São Paulo, Rio de Janeiro, Moscow, Los Angeles and Buenos Aires) the air concentration of particulates was assessed to cause moderate to heavy pollution; WHO guidelines (prevailing at that time!) were exceeded by up to a factor two and short term guidelines exceeded on a regular basis. For the remaining three cities (Tokyo, New York and London) WHO-guidance values were, by and large, being met. In Europe the PM10 guideline recommended by the UK was exceeded extensively in most of the cities for which data are available (EEA, 1997). Regional PM10 concentrations can reach 25 µg/m³ as an annual average in certain parts of Central/North Western Europe. Urban contribution to the annual average is most often smaller than the regional component. Therefore, to control long-term average PM10, abatement at regional scale contributions is very important. For maximum short-term (24 hour) episodes, the urban contribution is more important (EEA, 1997). In relation to LCA, the linear relationship proposed is interesting as it implies a non-threshold mechanism. The relevance of site characterisation in relation to above or below threshold concentration therefore becomes obscure. However, differences in environmental fate of different particle emissions should be taken into account when considering site characterisation. 4.4 CO According to Corinair 1994, the main CO emission source is road traffic which accounts for about 60% (ETCAE, 1997). WHO guidelines (WHO, 1998): 15-min average: 100 mg/m³ 30-min average: 60 mg/m³ one-hour average: 30 mg/m³ 8-hour average: 10 mg/m³ The WHO megacity study (WHO/UNEP, 1992) concludes that CO exposure levels are a serious problem [68] in one city (Mexico City – one-hour averages up to 67 mg/m³), a moderate to heavy problem [69] in seven cities (Cairo, Jakarta, London, Los Angeles, Moscow, New York and Sao Paulo; one hour averages of 30-60 mg/m³ and 8-hour averages of 10-20 mg/m³), a minor problem [70] in six cities, and for six cities there were no data. In most of the 15 EU cities with the highest CO concentrations, WHO 8-hour guidance values are exceeded - in some cities up to a factor three - and the one-hour values are exceeded in a few (EEA, 1997). Problems with CO exposure seem to be restricted to areas close to main road networks (EEA, 1997). 4.5 NMVOCs NMVOCs [71] cause both direct and indirect toxic effects. The direct effects are connected to the toxic properties of the individual VOC substances, whereas the indirect effects on humans and ecosystems are the result of the VOC contribution to photochemical ozone formation. Ozone will not be included here, as it is covered in LCA by the effect category on photochemical ozone formation. 4.5.1 Outdoors According to Corinair94 (ETCAE, 1997), the major European outdoor emission sources of NMVOC are: approx. 30% from road traffic, 30% from solvent and other product use, about 20% from agriculture, forestry, land use and wood stock exchange and approx. 10% from different energy production and manufacturing combustion's. VOCs as a group is difficult to assign a specific toxicity as the group covers a wide range of substances. VOCs from industrial point sources usually consist of one or a number of specific substances, whereas VOCs from combustion processes are a very complex mixture, usually containing a major fraction of alkanes, medium amounts of methane, olefines, ethene and monocyclic aromatics, minor amounts of aldehydes (some being formaldehyde), and trace amounts of PAHs (between others benzo(a)pyrene and naftalene) (van der Ven, 1995). In a measurement program carried out by the New York State Conservation (NYSDEC, 1995), a number of VOCs were measured at different sites, mainly in urban and industrialised areas. On the basis of these results, it was concluded that especially benzene was problematic. Furthermore, three chlorinated compounds (carbon tetrachloride, 1,2-dichlorethane and 1,1,2-trichlorethane) were potentially problematic. However, the measured concentrations were close to the detection limits and the results therefore uncertain. Benzene will be discussed separately in a later paragraph. The results also showed that point sources may elevate background concentrations in the surrounding area considerably. These findings are not surprising when compared with the European figures showing that about 30% of the VOC emission originates from solvent and other product use (ETCAE, 1997). As a general rule-of-thumb, concentrations of specific VOCs can therefore be assumed to be considerably higher than background concentrations close to facilities manufacturing or applying solvents. As mentioned above, different VOC's have different toxicities. Therefore and because of the different nature of point sources (type and amount of VOC manufactured or applied as well as the degree of emission abatement techniques and stack heights), it is not possible to give a general statement about VOC exposure levels close to point sources. Despite the previously mentioned assumption that point sources at least in industrialised countries are often regulated in order to reduce human health impact close to the sources [72], a case-by-case approach is recommended. Remarkable elevated concentration levels of VOCs resulting from combustion processes are believed to result mainly from traffic emissions. A significant compound in this respect is benzene which is discussed in a later paragraph. VOC emitted from high stacks are to a high degree prone to photochemical reactions prior to potential human exposure. VOCs reaching the water or soil environment (leaks/spills of organic solvents, emission via sewer systems and deposition of VOCs) may potentially contaminate drinking water and bio-accumulate in foodstuffs leading to an oral intake. VOCs in drinking water may further cause indoor inhalation exposure, see below. 4.5.2 Indoors VOCs in the indoor environment originate from a number of sources: office machines, cleaning agents, tobacco smoke, microbial formation (e.g. building materials attacked by fungis), human activity (bio effluents and cosmetics), building materials and from outdoor, with the latter three being the major contributors (Wolkoff et al., 1998). See also Gustafsson (1992), who describes various exposure situations for different indoor VOCs. VOCs may cause several toxic effects (e.g. eye and upper airways irritation, direct toxic effects to the lungs, asthma and carcinogenicity) as well as odour problems (Wollkoff et al., 1998). It has been proposed that indoor air quality guidelines be established as 1/40 of the occupational threshold limit values (Wolkoff et al., 1998). The factor 1/40 is introduced to account for continuous indoor exposure (contrary to an 8-hour working day) and to protect sensitive population groups (including children) which are not present in working environmental situations. With the exposure levels present, irritation and smell are usually considered and observed as the relevant VOC indoor effects, the latter being the more sensitive as smell limits are generally lower compared to irritation thresholds (Wolkoff and Nielsen, 1993; Wolkoff et al., 1998). Smell and irritation are symptoms which belong to the so-called "sick-building syndrome". Whether smell is just a temporary inconvenience or has a real health impact is uncertain (Wolkoff and Nielsen, 1993). Furthermore, field studies do not show a systematic connection between VOC levels and "sick-building syndrome" complains (Wolkoff et al., 1998). However, it is hypothesised that the dominating factor is the extent of the incoming VOCs' reaction with oxidants to form more aggressive compounds (Wolkoff, 1998). VOCs from building materials are divided into primary and secondary VOCs. Primary VOCs originate from the chemicals used in the building materials, and as a rule-of-thumb these are emitted within one year. Secondary emissions result from chemical and/or physical processes which result in VOC emission from the material, for instance oxidation of the material or adsorbed VOCs, other chemical influence and mechanical impact/wear. Emission of primary VOCs dominates in the beginning of a building material's lifetime, whereas secondary emission becomes important in time and may continue in the entire life time of the material. Secondary VOCs as well as VOCs formed by reaction with for instance ozone may have very low thresholds and thus contribute to a permanent bad indoor climate. The scientific foundation for the assessment of secondary emissions needs to be fully developed (Wolkoff et al., 1998). The Danish indoor climate labelling scheme for building materials (Wolkoff and Nielsen, 1993) focuses on primary VOC emission only. However, this approach is believed also to reduce problems with secondary emissions (Wolkoff et al., 1998). The lower threshold for irritation and smell is used as the critical "toxic" effect [73]. Threshold levels may be found in the "VOCBASE" (Jensen and Wolkoff, 1996, 1997). Smell is usually the determining factor. 50% of the threshold value (the lower of smell and irritation) is used as an acceptable indoor climate concentration. The 50% level is chosen, as more VOCs are present simultaneously (Wolkoff et al., 1998). When testing a material, it is placed in a climate chamber and the VOC emission is measured during a period. Based on the result, it is calculated how the emission would take place in a standard room, and an emission profile is plotted. From this profile, it is determined when the emission will be below the acceptable indoor level - the "time value". As a check, this value is compared with a sensoric assessment carried out as a smell test on a human panel (Wolkoff and Nielsen, 1993; Wolkoff et al., 1998). When developing the labelling scheme, a number of tests were carried out. The tests included three sealants, three paints and three carpets, and the results are presented in Wolkoff and Nielsen (1993). In these tests, time values were both calculated and determined with sensoric assessment. The sealants emitted many and very different VOCs. Calculated time values ranged from 8 to 102 months. The sensoric assessment showed one month for the better alternative, whereas an acceptable level was not reached for the worse alternative within the time period tested (approx. two months). The paints emitted 6-8 VOCs each. Time values ranged from about two weeks for all alternatives when calculated to 2 - 20 days in the sensoric assessment. The carpets did not reach acceptance in the sensoric assessment within the time period tested. The calculations showed time values of 61 to 98 months primarily due to the content of 4-phenylcyclohexene in the rubber back cover (Wolkoff and Nielsen, 1993). It is difficult to set up general recommendations in relation to extent and duration of exposure to indoor VOCs, as these are very dependent on the substance, the source (e.g. the building material), and the physical circumstances (e.g the extent of ventilation). Furthermore, work still needs to be done in order to assess indoor exposures in LCA. The present EDIP characterisation factors are calculated based on environmental processes in the external environment. One of the issues to face is whether "smell" should be considered a toxic effect and if so its estimated weight. Although not further discussed in the paper, it should also be mentioned that the direct application of VOC-containing consumer products like paints, may result in significant human exposures. 4.6 Benzene Automobile petrol contains about 1-2% benzene (ATSDR, 1997a). The major general source of benzene in ambient air is non-combusted benzene in automobile exhaust. Thus, elevated benzene concentrations may be found in areas with high traffic intensity. Point sources resulting in elevated levels of benzene are gas-filling stations and other facilities handling fuels as well as industrial facilities manufacturing or applying benzene (ATSDR, 1997a; WHO, 1993). Major indoor sources are cigarette smoke and off-gassing from building materials (WHO, 1993). Consumer exposure is high in connection with handling of products containing benzene (solvents, paint, etc.). Benzene is a carcinogen and the health risks at low-level exposure are not clearly established (WHO, 1993). Exposure should, therefore, be avoided as much as possible. WHO has set a lifetime 10-5 unit risk guideline level of 6 µg/m³ (WHO, 1998). EU has not yet established guidance values, but the UK, the Netherlands, Italy and Germany have recommended guidance values within the range of 3-16 µg/m³ as annual averages (EEA, 1997). Drinking water may be contaminated locally, for instance if retrieved in the vicinity of underground petroleum tanks. The WHO drinking water guidance value is 10 µg/l (ATSDR, 1997a). Outdoor environmental levels range from 0.5 µg/m³ or less in remote rural areas and up to 100 µg/m³ or even higher in urban areas with high traffic intensity. General urban area levels are 5-30 µg/m³ (WHO, 1993). Levels of 0.4 to 16 µg/m³ have been reported in an industrial area, with many organic chemical and petroleum producer, user, and storage facilities (ATSDR, 1997a). Levels of up to 102 µg/m³ have been measured in connection with industrial refineries (WHO, 1993). During refuelling of automobiles, levels of 3.2 and up to 10 mg/m³ (10,000 µg/m³) have been measured (WHO, 1993; ATSDR, 1997a) [74]. However, this exposure is rather short term and it is estimated that an average person uses 70 min. per year refuelling a car (ATSDR, 1997a). Recent European measurements show that city background levels are presently in the same range as the nationally recommended guideline levels of 3-16 µg/m³ (EEA, 1997). It is estimated that on average more than 99% of the human benzene exposure occurs via inhalation (ATSDR, 1997a). However, the drinking water may be contaminated locally. In a contaminated drinking water well on the USA East coast, levels of 330 µg/l - significantly above the WHO guideline - have been measured (ATSDR, 1997a). It can be seen that ambient air benzene exposure levels may exceed guidance values in cities and industrialised areas. However, in relation to LCA, this may be assessed less relevant when considering the non-threshold mechanism of benzene. As for particles, any elevation in exposure may cause an elevated risks of adverse effects when assuming non-threshold. The major indoor exposure results from smoking. It has been estimated that for US conditions about half of the benzene exposure (outdoor and indoor) results from cigarette smoking. Thus, for smokers this is the main exposure route. Passive exposure to cigarette smoke is responsible for 5% of the total intake in non-smokers. VOC-contaminated drinking water may indirectly cause inhalation exposure indoors. VOCs may evaporate, for instance during showering. Benzene air concentrations of 758-1670 µg/m³ have been measured in the shower stall and levels of 366-498 µg/m³ in the bathroom during and immediately after showering in water containing about 300 µg benzene/l (ATSDR, 1997a). 4.7 Chloroform Chloroform is a technical chemical and is emitted to the environment during manufacture and further processing. In 1987, the annual production worldwide was 440 kilotonnes. Emission factors for controlled facilities have been reported to vary between 0.51 kg (controlled facilities) and 3.35 kg (uncontrolled facilities) per ton chloroform processed. These are the major emission sources of chloroform (WHO, 1994a). However, chloroform is also formed in different (industrial) processes by the reaction between chlorine and organic matter: Paper bleaching, chlorinating of drinking water, chlorinating of cooling water and chlorinating of waste water or by degradation of other chlorinated compounds; including traffic exhaust (decomposition of 1,2-dichlorethane, which is a gasoline additive), and decomposition of trichloroethene and probably 1,1,1-trichloroethane in the atmosphere (WHO, 1994a). WHO has defined Tolerable Daily Intakes (TDIs) of 15 µg per kg body weight per day and 8-10 µg per kg body weight per day for non-cancer effects and carcinogenicity, respectively, (WHO, 1994a) [75] Ambient air concentrations in the US range from levels of 0.1-0.25 µg/m³ in remote areas to 0.3-9.9 µg/m³ in urban areas. Levels of 4.1-110 µg/m³ have been found in point source dominated areas (WHO, 1994a). Measurements from 1976 showed levels of 1-15 µg/m³ for cities in Japan and Europe. More recent measurements have shown levels <1 µg/m³ for Dutch and German conditions (WHO, 1994a). Indoor air concentrations are typically in the range of 1-10 µg/m³ (WHO, 1994a), probably originating from drinking water evaporation and from outdoor. In swimming baths, levels of 100 g/m³ are common (WHO, 1994a). Chlorination of drinking water may contribute significantly to chloroform exposure. In the US, levels of 60 µg/l have been found in drinking water from surface water supplies. German values range from 0.1 to 14.2 µg/l (9 µg/l for Rhine water), and Japanese values of 18 and 36 µg/l have been reported (WHO, 1994a). Chloroform has been found in many foodstuffs. Especially high levels have been found in de-caffinated coffee, olive oil, pork, sausages and soft drinks (WHO, 1994a). General US population intake has been assessed to be 1 µg/kg body weight/day from food, 0.5 µg/kg/day from drinking water, 0.3 µg/kg/day from indoor air and 0.01 µg/kg/day from outdoor air, i.e. well below the TDIs. However, it is obvious that population groups close to significant point sources may reach daily intakes above the TDIs. 4.8 Hexachlorobenzene (HCB) Previously, HCB was used extensively as a seed dressing, but this use was discontinued in most countries in the 1970'ies. Present HCB releases to the environment are the use of some chlorinated pesticides, incomplete combustion, old dump sites and inappropriate manufacture and disposal of wastes from the manufacture of chlorinated solvents, aromatics and pesticides (WHO, 1997b). WHO has defined Tolerable Daily Intakes (TDIs) of 0.17 µg per kg body weight per day and 0.16 µg per kg body weight per day for non-cancer effects and carcinogenicity, respectively, (WHO, 1997b) [76]. Distant from point sources, environmental concentrations of HCB are low; a few ng/m³ or less in ambient air and a few ng/l or less in drinking and surface water. Higher levels have been measured near point sources. HCB is persistent and bioaccumulating in the environment and the major source of HCB is exposure via foodstuffs. Estimated average daily intake in the general population is 0.0004-0.003 µg per kg body weight per day, well below the TDIs (WHO, 1997b). However, in some Asian and European countries the levels of HCB in foodstuffs - and thus the daily intake - is higher (WHO, 1997b). Population groups with a diet consisting mainly of wild life animals, for instance marine fish, may have a considerably higher daily intake. HCB accumulates considerably in breast milk. The daily intake for babies may reach 0.018-5.1 µg/kg/day (WHO, 1997b). 4.9 Dioxins Chlorinated dioxins (CDDs) are a family of compounds with several congeners that differ considerably in toxicity. The most toxic congener in mammals is the 2,3,7,8-TCDD (ATSDR, 1997b). Usually the toxicity of a dioxin mixture is indicated by a number of 2,3,7,8-TCDD-equivalents (TEQ). Some of the dioxin congeners are very persistent and bioaccumulating in the environment (ATSDR, 1997b). CDDs usually occur concurrently with other chemicals such as chlorinated dibenzofurans (CDFs) in the environment. Major sources are: combustion processes (municipal, medical and industrial hazardous waste, as well as fossil fuels and wood combustion), during the production, use and disposal of certain chemicals (e.g. chlorinated pesticides and benzenes), during the production of bleached pulp and during the production and recycling of several metals (ATSDR, 1997b). The WHO TDI is 10 pg/kg/day measured as TEQ (ATSDR, 1997b). No air guidance value has been encountered. Consumption of food (including human milk) is by far the most important CDD exposure route for the general population. The second most important source is inhalation of CDDs from municipal and industrial waste incinerators and other incineration and combustion processes (ATSDR, 1997b). For the general US population it is estimated that about 98% of the daily human 2,3,7,8-TCDD intake (0.047 ng/day on average; lower bound 0.008 ng/day; upper bound 0.3 ng/day) originates from food, especially meat and dairy products. Fish contribute with about 10%. Residents in the Great Lake region, regularly consuming fish from the great lake, have an estimated daily intake of 0.39 to 8.4 ng/day (ATSDR, 1997b). The total CDD intake is estimated to be 18 to 192 pg TEQs/day, equalling 0.3 to 3.0 pg/kg/day assuming a 65 kg body weight. Studies conducted in other industrialised countries have reported similar values to those obtained for the United States (ATSDR, 1997b). It can be seen that the levels for the general population are below the WHO TDI, whereas specific population groups may have a considerably higher intake. CDDs accumulate in breast milk. The daily intake by nursing infants in the United States has been estimated to be 83.1 pg TEQs/kg/day. Other studies have reported intakes of 35-53 pg TEQ/kg/day for infants within the first year (ATSDR, 1997b). These values are above the WHO TDI guidelines and must be assumed to be even higher in extreme situations (e.g. mothers eating many fish). Several studies indicate that state-of-the-art incinerators with appropriate air pollution devices should not pose a significant health hazard regardless of the incinerator location (ATSDR, 1997b). This issue is however still a big discussion. Individuals smoking 20 cigarettes a day have a daily intake of about 0.26 pg TEQ/kg (ATSDR, 1997b). CDDs have been found in various consumer products such as plastic packaging for coloured candle wax, textiles for air filters for home heating systems and paper products. CDD is also found in coffee filters. In a worst case scenario, where four small coffee filters are used per day and it is assumed that CDD leaches into the coffee, a daily intake of 10 pg/day is estimated (ATSDR, 1997b). 4.10 Lead Lead is a naturally occurring element. It has been estimated that about 19,000 tonnes/year are emitted to the air in connection with volcanic emissions and geological weathering. Air emissions from mining, smelting, and consumption of over 3 mio. tonnes of lead per year result in an estimated emission of 126,000 tonnes/year (WHO, 1995). Lead and its compounds may enter the environment at any step during mining, melting, processing, use, recycling or disposal. Major uses are in batteries, cables, pigments, petrol (gasoline) additives, solder and steel products (WHO, 1995). Lead is also emitted from oil and coal combustion where it occurs naturally. WHO has set a guidance value of 0.5 µg/m³ as annual average (WHO, 1998), a drinking water guideline of 0.05 mg/l (equalling 100 g/day assuming intake of 2 litres/day) and because lead is accumulating extensively in blood, a blood lead level of concern of 20 µg/dl (0.2 mg/l) (WHO, 1995). EU operates with an air limit value of 2 µg/m³ (EEA, 1997). The major source of lead emission to the air has been the use of leaded additives in gasoline. However, the levels have been reduced dramatically as a consequence of the widespread substitution in automobile gas (ATSDR, 1997c). For US conditions the major lead emission source shifted from transportation exhausts to industrial releases in 1988. In 1995 the emission from transportation was only about half the emission from industrial sources (ATSDR, 1997c). For urban sites the air concentration levels dropped from 0.8 µg/m³ in 1979 to 0.1 µg/m³ in 1988. And it must be assumed to be much lower today. In the same period (1979-1988) the industrial releases also dropped significantly (nearby concentrations dropped from 2.9 µg/m³ to 0.4 µg/m³) due to the introduction of emission controls. However, the industrial releases have not dropped significantly since the mid 1980'ies (ATSDR, 1997c). Ambient air levels of above 10 µg/m³ have been reported for urban areas near a smelter (WHO, 1995). Thus, lead emitting point sources are significant exposure sources for the local environment. As for 1990-93 European conditions, it has been stated that lead may still represent an air pollution problem near roads with intense traffic in countries with high lead content in gasoline. However, it is believed that more recent measurements will show further reduced levels due to the ongoing lead substitution in gasoline (EEA, 1997). As lead is an element, it may not be degraded in the environment and as such deposits in soil and on water surfaces. Thus, even though the lead air emissions have been reduced dramatically, considerable amounts can still be found in water, soil and foodstuffs. For the general population, the major lead exposure route is via food and drinking water (WHO, 1995). Drinking water concentrations are usually below 5 µg/l, but water taken from taps where lead is used in the plumbing may contain more than 100 µg/l. Lead is present in many foodstuffs. Dairy products, meat, fish, poultry, grain & cereal products, vegetables, fruits and beverages all contribute with between 4 to 10 µg/day of the average US adult intake of 56.50 µg/day (ATSDR, 1997c). Air exposure may be a major exposure route, depending on factors such as tobacco, occupation, proximity to motorways and lead smelters (WHO, 1995). Young children may ingest significant lead amounts when playing at contaminated soil sites either during "soil-eating" or via hand-to-mouth activities. Lead levels in soils beside roadways are typically 30-2000 µg/g; soil adjacent to a smelter had lead levels of above 60,000 µg/g and soil around houses painted with lead based paints may have lead levels >10,000 µg/g (ATSDR, 1997c). 4.11 Cadmium Cadmium is a naturally occurring element. About 10% of the total world-wide air emission (3900-12800 tonnes/year) originates from natural sources (weathering of minerals and volcanic emissions), whereas man-made sources are responsible for the remaining parts with industrial metal mining and production (zinc, cadmium, copper and lead) being the major parts. Other significant sources are fossil fuel combustion, refuse incineration, phosphate fertiliser manufacture, cement manufacture and wood combustion (ATSDR, 1997d; WHO, 1992). WHO has set an air guidance value of 5 ng/m³ (WHO, 1998). The WHO guideline for drinking water is 0.005 mg/l and the provisional tolerable weekly intake is 0.4-0.5 mg (ATSDR, 1997d). Exposure routes for cadmium are similar to those of lead described above. For non-smokers, the general major exposure route is via food (WHO, 1992; ATSDR, 1997d). There may be significant differences in cadmium intake, as point sources (e.g. metal processing industry and incineration plants) may contribute significantly to the cadmium exposure. Near sources of cadmium pollution, individuals may inhale 1-75 µg/day, which is a considerable contribution to the total daily intake (ATSDR, 1997). In the US, the adult intake of cadmium from food has been estimated to be about 0.21-0.23 mg/week, resulting mainly from grain, cereal products, potatoes, and other vegetables (ATSDR, 1997d). Variations in these figures indicate that a significant part of the population may consume more than the recommended WHO provisional guideline. Smokers are exposed to about 1.7 µg cadmium per cigarette smoked. The amount of cadmium absorbed from smoking one pack of cigarettes per day is about 1-3 µg/day. This roughly equals the dietary intake, as cadmium absorption from the lungs is considerably higher than from the gastrointestinal tract (ATSDR, 1997d). 4.12 Mercury Mercury is a naturally occurring element. Mercury is emitted to the environment from both natural (volcanic activity and weathering of Hg-containing rocks) and anthropogenic sources. Major anthropogenic sources are mining, industrial processes involving the use of mercury (including chlor-alkali manufacturing facilities), combustion of fossil fuels (primarily coal), production of cement; and medical and municipal waste incinerators (ATSDR, 1997e). Mercury has three valence states and is found in the environment in the metallic form and in the form of various inorganic and organic complexes. The most common form in the air is the metallic form. In soil and surface waters, metallic mercury may be converted to organic mercury with methyl mercury being the most common. This is particularly problematic as this form is soluble, mobile, and rapidly enters the aquatic food chain (ATSDR, 1997e). WHO has set an annual air guidance value of 1 µg/m³ (WHO, 1998). WHO guidelines for intake are (ATSDR, 1997e): Drinking water guideline for health-related organics (applies to all forms of mercury): 0.001 mg/l Permissible tolerable weekly intake: 5 µg/kg (total Hg); 3 µg/kg (CH3Hg). Mercury is a ubiquitous part of the environment, and human exposure may be inhalation in ambient air and ingestion of drinking water and foodstuffs. Furthermore, human exposure may occur through dental (mercury is a part of amalgam) and medicinal treatments (ATSDR, 1997e). The major non-occupational exposure for the general population is via food, especially fish and fish products (ATSDR, 1997e). Levels between 2 and 8 µg per day (0.22 to 0.86 µg/kg/week), which are below the WHO recommended guidance values have been estimated (ATSDR, 1997e). However, it is obvious that population groups consuming considerable amounts of marine mammals may exceed the WHO guideline values. Another major mercury source is dental amalgam. A survey of different studies has shown that the daily intake via that route must be considered to be below 10 µg/day (equals about 1 g/kg bw/day) (ATSDR, 1997e), which is in the same range as the dietary intake. Mercury uptake from drinking breast milk may also be a significant exposure route (ATSDR, 1997e). Inhalation of ambient air contributes to less than 1% of the total intake (ATSDR, 1997e). 5 Summary It is difficult to give general recommendations on whether or not a specific exposure situation encountered in LCA can be assessed to be near or above "threshold". However, Table 7.5 presents a crude summary of the findings of the present paper. The table should not be regarded as bare facts, but as a best attempt to summarise the previous paragraphs, which are again based on the level and extent of the consulted literature. Furthermore, it should be observed that many of the data of the consulted references originate from the beginning of the 1990'ies, since when the situation has been rapidly changing due to for instance new abatement technology and changes in the traffic load. Further information on regional emissions, deposition patterns and/or background concentrations for several of the substances treated may be found in the literature/references given in Annex 7.5. 8 Ecotoxicity8.1 Introduction Authors: Jens Tørsløv [77] Michael Hauschild [78]Dorte Rasmussen77 8.1 IntroductionEmissions of chemical substances to the environment may cause effects on organisms as well as on the structure and functioning of the ecosystem through their toxic action. Different types of chemicals act through different toxicological mechanisms, and compared to other environmental impact categories in LCIA, the cause-effect relationship in ecotoxicity is very complex. Also the number of substances in a product system that may contribute to ecotoxicity is higher, and often includes a range of substances with different characteristics. Toxicity is a relative concept, and paraphrasing the ancient Swiss physician Paracelsus, all substances are toxic if the dose ingested is large enough. The ecotoxicological impact of a substance can be regarded as a result of its toxicological properties on the one hand and the exposure to the target on the other hand. The exposure depends not only on the released quantities but also on the fate of the substance in the environment, e.g. distribution between environmental compartments, biodegradation and ability to accumulate in biota. Characterisation modelling for ecotoxic chemicals in LCA seeks its inspiration from environmental risk assessment but there are important differences. Risk assessment is often performed in a legislative context where the purpose can be to help ensure that there is no unacceptable risk to the environment, rather than to provide the best estimate of the actual risk. Therefore, a conservative approach is often followed, whereas a detailed risk assessment is conducted only if a preliminary assessment indicates a risk. Life cycle impact assessment (LCIA), on the other hand, attempts to address all relevant environmental impacts associated with a product, not just the impacts from toxic and persistent chemicals. To avoid an unintentional bias in the treatment of the different impacts, LCIA thus aims at providing the best estimation for each type of impact. Therefore, a conservative estimate of the ecotoxic effect of a substance is unwanted in the context of LCA - we must aim for the best estimate of the actual effect properties of the substance. 8.1.1 Ecotoxicity modelling in LCIAThe SETAC Europe's First Working Group on Life Cycle impact assessment (WIA1) proposed the following framework for characterisation of ecotoxic or human toxic impacts in the environmental impact assessment component of life cycle assessment (LCIA) (Jolliet et al., 1996): (8.1)
The environmental impact potential, EP is presented as the product of an effect factor E, a fate factor F, and the total emitted quantity Q. The index i represents the chemical, n the environmental compartment to which the emission is released, and m the route of exposure of the ecosystem The difference between existing LCIA characterisation models for ecotoxicity lies in the fate factor for which different models have been developed or adopted (e.g. Guinée et al., 1996, Jolliet and Crettaz, 1997, Wenzel et al., 1997, Huijbregts, 1999, Hertwich, 1999). In all these methodologies, the effect factor is the same, taken as the inverse of the predicted environmental no effect concentration, PNEC (8.2)
Assumption of linearity The underlying assumption is that the effect depends on the properties of the substance only, not of the receiving environment. The impact is proportional to the quantity that, upon the reductions and modifications included in the chosen fate modelling, reaches the target system and the proportionality (the slope of the quantity-impact curve) is always the same. This linearity is also assumed in current risk assessment as performed using generic tools like EUSES (RIVM, 1998) or CalTOX (McKone, 1993) but nonetheless it contradicts the experience that thresholds of effects exist and that the curve is not linear in reality (see e.g. Potting and Hauschild, 1997a and b). A novel approach to LCIA, the EcoIndicator99, aiming at damage modelling, attempts in a very generic form to consider the non-linearity by taking into account the background situation of the exposed ecosystem and uses a different effect factor (Goedkoop and Spriensma, 1999). This approach holds a strong potential for spatial characterisation of ecotoxicity since it allows taking the spatial variation in the state of the exposed ecosystems into account. However, sufficient information about background concentrations of manmade chemicals in the different ecosystems is not available today, and in order to make the approach operational for LCIA, the authors generalise their approach by taking out the spatial differentiation. At present, there is thus no alternative to assuming linearity between quantity of substance reaching the ecosystem and impact on the ecosystem in life cycle impact assessment of ecotoxicity. This means that the only fate processes that are relevant to include in spatial characterisation modelling are those processes altering the quantity of substance reaching the different parts of the environment – processes like biological degradation, chemical transformation, evaporation, deposition and sedimentation etc. Processes which influence concentration through dilution and dispersion without altering the total quantity of substance will not influence the characterised ecotoxic impact. When linearity is assumed, exposing 1 m³ of water to a substance concentration of 100 mg/m³ is equivalent to exposing 100 m³ of water to a concentration of 1 mg/m³. 8.1.2 Ecotoxicity modelling in EDIP97In the EDIP97 methodology (Wenzel et al., 1997, Hauschild and Wenzel, 1998), ecotoxicity is considered in aquatic ecosystems (acute and chronic), in terrestrial ecosystems (chronic exposure) and in wastewater treatment plants. For each endpoint, a simplified fate modelling is applied based on a modular approach where redistribution between the environmental compartments and potential for biodegradation are represented as separate factors. The equivalency or impact factor for chronic ecotoxicity in environmental compartment n is determined as: (8.3)
Where the redistribution factor, fmn expresses the fraction that is redistributed from the initial compartment m to the final compartment n, in which the ecotoxicological impact is modelled. BIO represents the potential for biodegradation as determined from standardised tests for ready and inherent biodegradability and the effect factor is the inverse PNEC value for the ecosystems of compartment n as described in expression (8.2). The "site-generic" EDIP methodology is prepared for inclusion of spatial differentiation for all non-global impact categories through site factors SF intended to modify the site-generic characterisation factors: (8.4)
The site-generic impact potential in EDIP97 is interpreted as the largest impact to be expected from the emission and the site factor is seen as the spatially determined probability that the full impact will occur, i.e. SF ranges between 0 and 1. Wenzel and co-authors give guidance on the quantification and use of the SF without making the site factor really operational. The EDIP methodology also involves some other possibilities for spatial differentiation. For the fraction of airborne emissions that deposits, the redistribution factor, fmn is set at "a" when n is the aquatic compartment and 1-a when n is the terrestrial compartment. EDIP97 allows "a" to be chosen according to the conditions of the region where the emission takes place. For Danish conditions, a=0.5 is proposed while a global default is set at a=0.2. Furthermore, in EDIP97, spatial information in the form of initial dilution data for waterborne emissions, is suggested to be included as technical information in the weighting of the potential contribution to acute aquatic ecotoxicity in order to reflect the differences in dilution potential (and hence the probability of acute effects) for different types of aquatic systems. For further background on the EDIP97 methodology, the reader is referred to the description in Wenzel et al. (1997) and Hauschild and Wenzel (1998). 8.1.3 Determining the physical level at which the spatial variation must be quantifiedParameters that influence the fate of substances in the environment often show large spatial variations. This variation depends on the scale, at which they are observed. For soil, the variation in e.g. the content of organic material can vary orders of magnitude on the 10-3 m scale (from rock material to a humus particle). The variation may stay within one order of magnitude between fields (102-103 m scale) and even less between countries or regions (105-106 m scale). At the extreme end, the scale of the whole planet earth (4107 m scale), the spatial variation is zero (which is why spatial characterisation modelling is only discussed for the non-global impact categories). This general relation between spatial variation and physical scale is illustrated in Figure 8.1 a and b Figure 8.1a +b. The degree of spatial variation of different environmental parameters depends on the physical or geographical scale at which it is studied. Examples of physical scales are: 10-2 m between particles of stone, clay and organic material and between water and particles in soil and water systems 10-1 m between small lumps of soil 1 m between patches of soil or water 10-102 m between parts of field, forest or lake 103-104 m between fields/habitats/lakes-streams/estuaries/ocean 105 m between small countries or regions of larger countries, types of climate, lakes, ocean 106 m between larger countries or regions of continents (like Europe), climatic zones 107 m between hemispheres, groups of climatic zones >4·107 m between Earths (only one Earth → no variation) In general, the relevant scale at which to determine the degree of spatial variation must be the scale that is influenced by the substance, i.e. the scale at which the substance is dispersed as part of its transfer and transformation (fate). This means that the relevant scale depends on the lifetime and the dispersion velocity of the substance in the environment. Consequently, removal processes and the type of the receiving environment are important factors (dispersion is quick in air, slow to medium velocity in water and very slow and mainly vertical in soil). The spatial variation is attempted quantified at two scales throughout this chapter
For the further discussion the following levels of spatial variation are defined for the fate modelling of a substance:
The aim of this chapter on ecotoxicity is to analyse the actual spatial variation for the relevant scale(s) (if possible under consideration of its dependence on the lifetime of the substance), to provide background for the best spatial characterisation modelling deemed possible and discuss how large a part of the actual spatial variation, it covers. Finally, to suggest a framework for calculating site factors to modify the existing site-generic EDIP characterisation factors for ecotoxicity and implement this framework to derive site factors for Europe. 8.2 The cause-impact chain and its descriptorsFigure 8.2 presents the cause-impact chain from the emission of a substance through a number of possible fate and exposure processes to the impact on the target – the part of the environment where the impact is preferably measured for the impact assessment. Each of the links in the chain can be characterised by a set of descriptors, which are important for the processes covered by the link. These descriptors are discussed in Section 8.2.
Figure 8.2. The cause-impact chain. Examples of descriptors for site characterisation are indicated. 8.2.1 The emitted substanceThe inherent physico-chemical and biological properties of a substance have a strong influence on its interaction with the environment. The following physico-chemical and biological properties are examples of parameters that are commonly used in modelling of the environmental fate of chemical substances:
Recommendations The substance-specific properties (descriptors) mentioned above are site-independent and have no direct relevance for the spatial characterisation of the ecotoxicological impact. 8.2.2 The emissionSeveral characteristics of the emission may influence the fate of the emitted substances and therefore also the extent to which an exposure eventually takes place. Important parameters are:
The emitted quantity to the different environmental media Emission to atmosphere, water and soil is given as mass emitted per functional unit in the inventory of the LCA. Generally, no information is included about how large the emission from one functional unit is compared to the total emission output from the process. The characterisation can therefore not be based on the full impact of the process. Emissions to waste treatment facilities are not terminal and the modelling of the relevant waste treatment processes (i.e. their resulting emissions to air, surface water, ground water or soil) is considered part of the inventory analysis. In case of emission to air, the dispersion modelling performed for human exposure reveals that the location of the emission point is not significant since the long transportation distances tends to even out local differences in the fate and exposure except for very short-lived substances. For the ecotoxicity impact category, the target is terrestrial and aquatic ecosystems, and the main descriptors of relevance to spatial characterisation modelling are: the density of ecosystems within the deposition area as described by the fraction that is covered by `natural areas'. Cultivated agricultural land is considered part of the technosphere rather than the environment, and natural ecosystems within urban areas are disregarded due to their insignificant contribution to the total area of natural ecosystems. the distribution of the deposited fraction between land systems and water systems (the relative prevalence of the latter being much lower in continental than in coastal areas) Annex 8.4 shows the relative frequency of natural areas within the countries of Europe. If this information is combined with atmospheric dispersion modelling as performed in e.g. Chapter 3 it is possible to determine the fraction of the emitted substance that deposits to ecosystems. This work has not yet been performed but as a first approximation, Europe is divided into four regions and the relative share of natural areas within each region calculated. Table 8.1. Relative share of natural areas within the four regions of Europe. More information on definition of the regions and the calculations is given in Annex 8.4.
If it is assumed that the major part of an atmospheric emission deposits within the region where it is emitted, Table 8.1. allows introducing a spatial differentiation for airborne emissions in the fraction deposited to ecosystems on land. The sea is considered entirely to be a `natural area' and all emissions deposited to sea are regarded as exposure of a natural area. Annex 8.5 suggests average factors for the distribution between sea and land for deposition originating from the same European regions. The data are based on calculations of the relative fractions of emissions of NH3 and NOx that deposit to land and sea for air-borne emissions from different European countries. Table 8.2. Relative share of emissions depositing to sea and land. For definition of the regions and calculations see Annex 8.5.
* The share of emissions deposited to freshwater systems is considered irrelevant here, due to the relative uncertainty of the deposition factor Used in combination, Table 8.1 and 8.2 allow deriving spatially differentiated factors for the share of emissions deposited from air that exposes aquatic or terrestrial ecosystems. Table 8.3. Relative share of emissions potentially contributing to aquatic and terrestrial ecotoxicity for different regions of Europe.
The temporal course of the emission The emission flux may be continuous or discontinuous, depending on the type of the source. For continuous emissions, there may be a broad variation in the duration from normal industrial processes (lasting from hours to days) to emissions from landfills (lasting from years to millennia). The temporal pattern will have an influence on the environmental impact as organisms respond to the concentrations they are exposed to rather than to the emitted amounts. Intermitted emissions with long intervals where no emission occurs may allow the exposed organisms to overcome the stress from sub-lethal exposures. On the other hand, emissions that come in pulses, e.g. from a batch production, may reach much higher transient environmental concentrations compared to a situation where the same amount of substances is discharged continuously over a longer period of time. A main entrance route for chemical substances to the aquatic environment is via municipal wastewater treatment facilities. Due to the mixing and the hydraulic retention time, emissions from municipal treatment facilities can be regarded as relatively continuous regardless the temporal variation of the original source. Temporal changes in the composition of wastewater from industries with own wastewater treatment facility may, however, be considerable due to variations in production activities. The temporal course of release may thus be important for the environmental concentrations, but tends to lose its importance with increasing distance from the source, as dilution, different fate related processes as well as contributions from other sources contribute to level out temporal variations. Substances emitted to air, that reach the aquatic or terrestrial environment through deposition, can be considered as coming from a continuous source regardless the nature of the emission, with the possible exclusion of targets in the immediate vicinity of the source. This is due to the efficient dispersion in the atmosphere. For characterisation modelling of ecotoxicity, it is therefore assumed that the contribution to atmospheric deposition from a functional unit is infinitesimal and that temporal variations can be disregarded. Apart from atmospheric deposition, the main route for most substances to reach the terrestrial environment are through organic waste products, e.g. sewage sludge, which may be used as an agricultural fertiliser and applied once a year or with longer intervals. The ecotoxicity impact categories are concerned with impacts on natural ecosystems, and the farm fields would normally be considered part of the technosphere, in which case, emission via sewage sludge is without relevance to ecotoxicity (but still a relevant exposure route for human toxicity). As a special group of substances, pesticides, are unintentionally emitted directly to the natural environments in the immediate surroundings of the sprayed field through wind drift and surface run-off. Like the application of the pesticides, this form of emission to the terrestrial environment is highly discontinuous. Pesticides may also reach the surrounding environment through evaporation followed by atmospheric deposition or through leaching to surface water (Hauschild, 2000). Both of these emission routes are considered to be continuous. The extreme end of the spectrum of continuous emissions is represented by leaching of persistent substances, notably metals, from landfills. Here, the duration of the emission may be centuries. Recommendations The emitted quantity to the different environmental media For emissions to air spatial variation is taken into account using the deposition factors in Table 8.3 for that fraction of the emission which deposits. The temporal course of the emission As discussed above, the temporal course of the emission may at the local level have a considerable influence on the environmental concentrations of toxic substances and thus the impact on biota. Consequently, a discontinuous or very quick emission of a substance will be more severe than a slow, continuous emission of the same quantity. Temporal variations in the emission pattern can, however, be disregarded for impacts occurring at a regional level, i.e. at distances from the emission point where the temporal variation are insignificant due to mixing and fate processes. In the aquatic environment temporal variations are relevant only for assessment of the local effects, i.e. acute aquatic ecotoxicity. Here,
Consequently, only in cases of emission with highly temporal variations the emission pattern has relevance for spatial characterisation, and only in the assessment of the acute aquatic ecotoxicity in the area near the emission point. This is not given further treatment here but it is suggested that such emissions be flagged for a more detailed evaluation if relevant. In the terrestrial environment we are only concerned with impacts at the regional level where temporal variations are considered of little importance. Situations with discontinuous emissions are emission through wind drift or surface run-off of pesticides to environment surrounding agricultural areas (other emission routes for pesticides can be regarded as continuous) and emission through adsorption to sludge and application to agricultural areas. 8.2.3 Fate - dispersion and distributionA substance's environmental fate includes transport, distribution between compartments and transformation from the emission point to the target system, a biological community, population or an organism, where it may exert its toxicity. The following fate-descriptors may be relevant for site characterisation and are therefore discussed in details:
8.2.3.1 Dilution and evaporationIn the vicinity of the discharge point to the aquatic environment, dilution is the most important process determining the level of exposure. Locally, acute as well as chronic effects can be observed, but normally, only chronic effects are expected at larger distances from the source where other processes than dilution determine the exposure. Dilution is an important descriptor in the spatial differentiation for acute ecotoxicity effects whereas it has no significance for the spatial differentiation for chronic ecotoxicity. For the present purpose, the "initial dilution zone" of an emission is encompassing the medium from the discharge point to the point where the emission leaves the technosphere and enters the ecosphere (or in other words where it becomes an emission). Particularly for waterborne emissions, the immediate surroundings of the discharge point are often considered to be part of the technosphere in the sense that even acute ecotoxic effects are accepted here. Outside the initial dilution zone, the further dilution is determined by diffusion and mixing due to the turbulence of the receiving water. Examples of the dilution capacity of different types of water courses are presented in Annex 8.1 and summarised in Table 8.4. Table 8.4. Typical dilution factors obtained 1000 m from the discharge point in different types of aquatic environments.
For emissions to soil, the initial dilution depends on the type of the emission, e.g. an application of pesticides through spraying or granules, an application of sewage sludge or a deposition of air-borne pollutant. Regardless the emission type, no spatial differentiation is expected in the dilution or dispersion pattern for emissions to soil. Some substances emitted to the soil or water environment will eventually evaporate. Substance-specific properties as well as environmental factors determine the evaporation rate. The evaporation is represented in the site-generic fate model but will show a spatial variability, which depends among other factors on the temperature. Calculations have been performed in Annex 8.8 for different values of Henrys constant and at three temperatures: 10oC (reference temperature), 6 oC (Northern European countries) and 15 oC (Southern European countries). A site factor is expressed as the ratio between the evaporation in the reference situation and the evaporation in the Southern European and the Northern European situation respectively in Table 8.5. For East and Western European countries, the reference temperature is assumed most descriptive and the correction factor assumes the value 1. Table 8.5. Site factors, SFevap for volatilisation from water for two European regions
Recommendations For the impact categories chronic aquatic ecotoxicity and chronic terrestrial ecotoxicity, dilution is not considered relevant for inclusion in spatial characterisation modelling at the spatial scale considered.
The spatial variation in equilibrium partitioning to air from water or soil for semi-volatile chemicals can be modelled based on knowledge of the annual average temperature as made operational by the site factors in Table 8.5. 8.2.3.2 DispersionIn addition to lowering the concentration to which the environment is exposed, the dispersion that leads to the dilution discussed under 8.2.3.1 also has as a consequence that the emitted substance is spread over a much larger area down the river or in the lake, estuary or ocean as well as potentially allowing different types of ecosystems to be exposed to the substance when the emission takes place to an early part of the hydrological cycle (e.g. a stream). The dispersion is primarily caused by the advective flow of the water and hence by the residence time of the substance in the free water mass as determined by removal processes like degradation, evaporation and immobilisation. Persistent toxicants like heavy metals and chlorinated organic compounds may thus be transported between different environmental compartments and have an impact on several types of organisms and ecosystems before they are degraded or irreversibly incorporated into an organic or inorganic matrix. This mechanism is determined by the ability of the substance to persist in the environment rather than by the point of discharge into the environment. On the effect side, all aquatic ecosystems are generally represented by just one PNEC value for chronic aquatic effects. Within life cycle impact assessment, the impact of a toxic substance can not be modelled as more severe when the release point is at the `upper' part of the hydrological cycle compared to a release directly to the marine environment. The ability to persist in the environment, which strongly influences dispersion potential, is represented as a substance-specific characteristic in the site generic impact potential (see expression 8.4) Recommendation For emissions directly to soil, dispersion is very small and it is considered that the spatial variation at the actual level of the substance's fate as well as at the level relevant for spatial characterisation modelling is negligible. For emissions to aquatic systems the dispersion will depend on the type of receiving system – river, lake, estuary or ocean. As discussed in Section 8.1 and in Section 8.2.3.1 under Dilution, processes that influence exposure without reducing the total quantity of substance available for exposure can not be taken into account in current life cycle impact assessment because the impact factors are the same regardless of the concentration at which exposure occurs. Furthermore, no distinction is made between different types of aquatic systems in calculating the effect factor – the same PNEC is used for all aquatic systems. The ability of a substance to act on several targets from the release into the environment until it is irreversible trapped or degraded is mainly governed by the lifetime of the substance rather than the point of release. Consequently, this issue is addressed through the inclusion of biodegradation and other removal mechanisms in the site-generic characterisation factor. This means that there is no possibility for representing the effects of dispersion and no need to represent the position of the release point in the hydrological cycle for waterborne emissions in the spatial characterisation model. 8.2.3.3 Adsorption and immobilisationAdsorption is an important process that strongly influences the bioavailability and toxic impact of many chemical substances. In Annex 8.2 it is substantiated, that only the dissolved and freely available fraction of a substance will interact (bioaccumulate and exert toxicity) with biological organisms in the environment. Sorption processes (which include adsorption and its reverse process de-sorption) determine the distribution of a chemical between a liquid and a solid phase like pore water and soil or water and particulate material. Adsorption is primarily governed by the fraction of particles that represents the largest surface area. In the water column this is often the small organic particles below 0.2 m, which are regarded as dissolved organic carbon (DOC). The adsorption efficiency indicated by the sorption coefficient Kd depends on the affinity of the substance to these particles. For non-polar organic substances, the octanol-water partition coefficient, (Kow) is often used to indicate the potential for adsorption to organic matter. For substances that dissociate, including most metals, the adsorption efficiency depends strongly on the pH in the receiving environment and on the presence of other types of sorbents, e.g. ionic sites of organic matter and minerals. A review of sorption processes and mechanisms is given in Annex 8.2 while Annex 8.3 exemplifies with the influence of sorption on biodegradation of LAS in sediments. In aquatic systems, sorption may lead to a more permanent removal of the substance from the aquatic compartment when particles become integrated in the sediment. In sedimentation areas, the organic content in of the sediment is often high and may act as a trap for adsorbed substances, e.g. metals and lipophilic organic compounds. Sedimentation is the net result of deposition and re-suspension of particles. The rate of sedimentation depends on the hydraulic conditions of the aquatic system and on the nature of its particulate material. The net sedimentation can be regarded as the product of the net sedimentation in the different types of water environments which the substances pass from the emission point to the sea. In Annex 8.9 the removal of substances through the combined effect of biodegradation and sedimentation is calculated for a standard river, lake, estuary and sea for a readily biodegradable, an inherently biodegradable and a not biodegradable substance with log Kow varying in the interval from -3 to 6. By combination of the fractions left, a site factor SFsed for the removal through adsorption and sedimentation can be calculated for three standard scenarios reflecting different positions of the emission point in the hydro-geological cycle:
For each scenario, the SFsed factor is calculated by multiplication of the substance fractions fi remaining after passing the water systems involved in the scenario. For scenario 1) SFsed is thus calculated as the f-values for river, lake, estuary and sea. The f-values are calculated and shown in Annex 8.9 and the resulting SFsed-values are shown in Table 8.6. Table 8.6. Site factors (SFsed) representing removal by the combined effect of sedimentation and biodegradation for readily biodegradable, inherently biodegradable and not biodegradable organic substances of different lipophilicity for the three emission scenarios: emission to river and from there through lake to estuary and sea, emission through estuary to sea and emission directly to sea.
N.B.: Not biodegradable Immobilisation of metals may take place through chemical precipitation of compounds with low water solubility. In particular, anaerobic sediments act as a trap for some metals e.g. through formation of sulphides. For most metals, the adsorption and precipitation are strongly influenced by the pH of the medium as shown in Annex 8.2. pH also influences the function of micro-organisms and thereby the biodegradation of organic substances in the environment as discussed in the section on biodegradation below. Recommendations Sorption is relevant for characterisation modelling because it facilitates removal of the substance through sedimentation. Sorption is a reversible process and the decreased bioavailability of a substance that sorbs can be seen as a "dilution of the substance in time" – the concentration at any given time will be lower if the substance sorbs. This is not reflected in the characterisation modelling. For the spatial characterisation modelling, it seems reasonable to assume that the variation in sorption and sedimentation among different types of water recipients is more important than the geographically determined variation within one type of recipient across Europe. It is thus the former that should be addressed in the model, i.e. for the aquatic environment the differences between streams, lakes, estuaries and ocean in sedimentation velocity (and hence removal due to adsorption). The removal from the water column through the combination of sedimentation and biodegradation can be represented by the site factors in Table 8.6 For metals that form insoluble salts under environmental conditions, this will be the process that determines the actual propagation of the metal in the environment. This means that the metal will disperse until it interacts with anions and precipitates. The conditions allowing immobilisation of metals through precipitation will, however, always be represented within the actual fate area, and there will thus be no spatial variation in this aspect. Spatial variation of the precipitation of metals will therefore not be considered in the spatial characterisation model. Annex 8.2. gives a more detailed discussion of the mechanisms of adsorption and immobilisation. 8.2.3.4 BiodegradationThe dominating transformation process for organic substance in soil and aquatic systems is biodegradation. Given the right conditions, most organic compounds can be transformed to other substances and ultimately mineralised through microbial degradation. Hereby, the potential for exposure to the mother compound is reduced or eliminated. The potential for biodegradability is one of the most important factors when the environmental impact of substances is assessed, and it is already represented in the site-generic characterisation factor developed under EDIP. Here, the biodegradation potential is treated as a purely substance-specific property derived from the data on the behaviour in standardised biodegradability test according to OECD or EU guidelines (OECD, 1992, EEC, 1987). Results from such standardised screening tests for biodegradability are obtained under laboratory conditions and used as indicator for the potential of a substance to degrade in the environment as discussed further below. A number of factors may have a significant influence on the degradation of substances in the environment:
Organism-related factors Biodegradation of organic substances depends on the presence of competent micro-organisms in sufficient numbers. Natural microbial communities consist of a very diverse biomass and when a `new' substance is introduced in a sufficient high concentration the biomass may adapt to degrade the substance. The mechanism of adaptation includes growth of specific degraders that by nature are competent to degrade the substance, but also enzyme induction, exchange of genetic material and development of tolerance to toxicity of the substances may be involved. Both readily degradable substances and inherently degradable substances may require an adaptation. Biodegradation studies often show an initial lag phase where no or only little mineralisation of the substances is observed, but the environmental significance of adaptation is probably more pronounced for inherently biodegradable substances than for readily biodegradable substances. The adaptation of a microbial community to degrade a substance thus depends on the history of the community, i.e. whether it has been exposed to the substance or a structurally related substance previously. This means that when a xenobiotic substance has been used and emitted for a period of time, the likelihood of finding competent degraders will increase. There is thus a higher chance of adaptation in environments that are exposed to chemicals more or less continuously. Substrate-related factors In the laboratory tests that are used for evaluation of the degradation of chemical substances the substances are applied in high concentrations (mg/l - range) compared to the levels that are expected to be found in the environment (lower g/l range). In general, growth of micro-organisms is not supported when the substrate is present at concentration below about 10 g/l. The reason for this threshold is possibly a lack of sufficient stimulus to initiate an enzymatic response. Consequently, the environmentally relevant concentrations of chemicals are mostly at levels where the chemicals can not be the primary substrate for degrading micro-organisms. In standard tests, the substrate (the tested chemical) is applied as the sole carbon source, while in the environment a large number of other substrates are available. In aquatic environments, the concentration of dissolved organic carbon is often found in the range 1 – 10 mg/l, i.e. around a factor of 1000 higher than the chemical pollutants. Although only a fraction of this pool of organic substance is readily degradable by micro-organisms, it is likely to be more attractive as a carbon source than the much lower concentrations of chemicals present. On the other hand, the natural carbon sources may facilitate a degradation of the chemicals present at lower concentrations by co-metabolism. The co-metabolised substrates may then be available for further degradation. Environmental factors The redox potential, i.e. the availability of oxygen is probably one of the most important environment-related factors. Under normal circumstances aerobic conditions are prevailing in soil, in the water phase of aquatic systems and in top sediments. Anaerobic conditions are present in sediments, soil micro-environments and in some treatment steps in biological treatment plants. Low oxygen concentrations may moreover be present in eutrophic (nutrient rich) lakes and sediments. Thus anaerobic conditions are found in many natural (and technical) sub-compartments. Another important parameter for degradation is the temperature. Most laboratory tests are performed at room temperature (20-25 °C). Under environmental conditions, the temperatures typically range from below 0 to 50 °C and in rare cases even higher. Considering, that the optimum range is probably from 20 to 30 °C for most micro-organisms and that degradation rates on average increase roughly by a factor of 2 for every 10 °C increase of the average ambient temperature within this range, it is clear that the temperature plays an important role in degradation processes. The effect of the temperature on degradation rates is discussed further in Annex 8.3. Annex 8.7 presents annual average temperatures for the European countries and for the four European regions introduced in Section 8.2.2. For the four European regions, the estimated annual average temperatures are given in Table 8.7. Table 8.7. Estimated annual average temperatures for the four European regions.
The biodegradation half-life can be assumed to increase roughly by a factor of 2 for every 10oC decrease in temperature. This gives the following correction factors for the influence of the temperature on biodegradation (reference temperature = 10°C):
The pH in different types of environments may range from 5 – 8 and microbial organisms can be active within this range. However, for bacteria as a group, slightly alkaline conditions favour the activity and the optimum pH range is 6-8, and at pH levels below 5 the activity is decreased significantly. Fungi have in general a lower pH-optimum in the range 5-6. The bioavailability of the substances is critically important also to degradation, which primarily takes place in the aquatic phase of the ecosystems, i.e. the pore water in soil and sediments and in the aquatic compartment. The bioavailability is determined by sorption processes and is discussed under 8.2.3.3. An illustration of the influence that some of the environmental descriptors discussed above may have on the environmental biodegradation is given in Annex 8.3 for the detergent LAS. Recommendations Biodegradation has a major influence on the impact of chemicals in the environment. It is influenced by several factors related to the microbial organisms performing it, the availability of substrate and the environmental conditions. Most of these factors show no variation in their distribution at the spatial scale at which the substance's fate occurs, except for very short-lived substances. This is the case for the redox potential (occurrence of anaerobic conditions) and the pH, except for emissions to lakes where conditions may vary strongly from lake to lake within a region. For temperature, there is a clear geographical trend going from north to south in Europe and therefore temperature-dependence shall be included in the spatial characterisation model. An operational approach may be based on the division of Europe into three to four regions and the assumption that the substance stays within the region that it is emitted in. Site factors for the temperature influence on biodegradation in northern, mid-, and southern Europe are proposed. Also for adaptation of micro-organisms there is a differentiation according to whether the ecosystem is frequently exposed to industrial or municipal wastewater discharges or not. This differentiation will not be included in the spatial characterisation model because of a lack of concrete knowledge of the significance of this parameter. 8.2.3.5 Other transformationFor some substances, transformation may also occur through physical or chemical interactions like photolysis, hydrolysis or chemical reactions. Photolysis Photolysis is the cleavage of light-sensitive molecules under the influence of light. Photolysis may be an important degradation process for some chemicals in water. The rate of photolysis in the water depends on a number of environmental conditions such as latitude, cloudiness, time of day and year, concentration of suspended matter in the water column, and to some extend on the temperature. Apart from the concentration of suspended matter, all of these parameters are related to the geographical location and it is thus possible to include their influence on the photolysis rate in a spatial characterisation model. Since the extinction of light occurs rather quickly down through the water column (and even more quickly in soil), it is in the present context only considered relevant only for substances emitted to air. For air-borne emissions the dispersion will be on a scale that for practical purposes averages out geographically determined differences in light intensity. Photolysis is thus not included in spatial characterisation. Hydrolysis Hydrolysis is the cleavage of bonds in the molecule of many organic substances due to reaction with water molecules. It is only considered relevant for substances present in water, where it may be the predominant degradation route for some organic substances. The rate of hydrolysis depends on the pH and the temperature. Hydrolysis is normally represented in the biodegradation potential since normal test conditions for biodegradability also facilitate hydrolysis. Its temperature dependency is hence considered included under the temperature dependence of biodegradation (8.2.3.4). As for biodegradation, the pH dependence of hydrolysis is not considered relevant to include in the spatial characterisation model. Chemical interaction Once emitted, the substance may interact with the multitude of other chemical substances present in the environment as pollutants or natural compounds. One of the most important reactions with relevance for nearly all volatile organic compounds is the photochemically driven oxidation by hydroxyl radical in the atmosphere under the influence of light and in the presence of nitrogen oxides, NOx. Photochemical oxidation will vary with the intensity of sun light (like photolysis) and the background concentration of hydroxyl radical. The latter will vary locally with the occurrence of organic substances e.g. from other pollution sources, i.e. it may be lower in industrialised and urban areas. For air-borne emissions, however, it is anticipated that the dispersion will be on a scale that averages out such differences so variations in the hydroxyl radical concentration can be disregarded in site characterisation. The highest rate of transformation through reaction with other substances is expected to occur where emissions of man-made pollutants are frequent. Recommendations Apart from hydrolysis, none of the transformation mechanisms show important variations at the actual spatial scale of the fate of the substance except for very short-lived substances. For hydrolysis, the rate will be temperature-dependent and thus follow the geographical temperature gradient mentioned earlier. It is however expected that data on the removal through biodegradation also include removal through hydrolysis and no separate treatment is therefore given to hydrolysis. 8.2.4 ExposureFollowing the transport and transformations of the emitted substance as discussed in 8.2.3, an exposure of one or more target systems occur. The effect elicited by the exposure is a response to the overall exposure experienced by the target system and it can be difficult to isolate the contribution from the product system alone. Concentration increase caused by the emission is in principle modelled or at least represented by the fate model. Some of the fate models used in LCIA actually attempt to estimate a concentration increase in the environment caused by the product system using modified environmental models like EUSES or CalTOX as discussed in Section 8.1.1. Given the constraints of life cycle assessment where the inventory result is an emitted mass corresponding to the functional unit, the value of such concentration estimates is debatable. In EDIP it was chosen to represent the most important fate properties in a transparent way in the fate model rather than adopt already existing models that were developed for something else to arrive at a quite intransparent fate model of questionable relevance. The background level of chemical exposure of the ecosystem is crucial to the quantification of the impact from the concentration increase caused by the product system. Normally, the contribution from a product system to the mixture of chemicals present in the environment is small. Since different chemicals often interact in their toxicity, it is the overall level of exposure that is relevant here, not just the background concentration of a specific substance. As discussed in Section 8.1.1, it is a general assumption in characterisation modelling of ecotoxicity that the concentration-impact curve of the ecosystem is linear. The background level of pollution is thus not taken into account. Even though a high degree of spatial variation in the background level can occur at the relevant scale, this is also ignored in chemical risk assessment today except in very site-specific studies. EU is currently implementing environmental monitoring programmes, which in the future may enable assessment of background levels of toxic pollutants. Bioconcentration of the chemical is attempted represented in a generic way in the PNEC when this is estimated from chronic data generated in tests with a duration that allows the bioconcentration to occur and influence the test results. In the EDIP97 method (in contrast to other LCIA methods for ecotoxicity), a correction for bioconcentration is introduced when the PNEC value is based on acute test data. No spatial variation is anticipated in the bioconcentration potential. Biomagnification, i.e. food chain concentration is not represented in the EDIP97 methodology where exposure is considered through contact to the surrounding medium and there is no specific representation of the top predators that are susceptible to biomagnification. This aspect is not included in the other models for ecotoxicity characterisation modelling that are known to the authors but it should be included in future revisions of the EDIP method. For the current context, it is not likely to show spatial variation at the relevant scale. Duration of exposure depends on the overall removal rate for the substance in that environmental compartment and it should in principle be represented in the site generic fate modelling, i.e. the spatial variation should be taken into account through the considerations presented in 8.2.3. Recommendations The background level of pollution of the exposed ecosystem is expected to be an important site-dependent descriptor, since the species of the ecosystem are exposed to the combination of the chemicals from the product system and the chemicals emitted by other activities exposing the same ecosystem. It is, however, at present not possible to incorporate site-dependent information about the background level of pollutants in the exposed ecosystems in characterisation of ecotoxicity. 8.2.5 Target systemThe target system is the ecosystem or ecosystems that are exposed to the emission from the product system. The relevant descriptor for this link of the cause-impact chain is the sensitivity of the exposed ecosystem as expressed by the concentration-effect curve of the substance. Also the aspect of potential exposure of several consecutive aquatic ecosystems is relevant here. 8.2.5.1 Sensitivity of ecosystemDifferent aquatic ecosystems show different sensitivities depending on their species composition. Ecosystems that are already exposed to man-made pollutants may have lost the most sensitive species so the species sensitivity distribution curve is shifted towards higher values. This means that the already exposed ecosystems are less sensitive and hence that the concentration that protects the function of pristine ecosystems is lower than the concentration protecting already exposed systems. Normally, only one PNEC is derived representing all aquatic (or terrestrial) ecosystems. Only for very few substances, there will be adequate toxicity data to allow estimating a PNEC value based on information for species that are actually representing the ecosystem that is exposed. A review and statistical evaluation of published aquatic mesocosm studies (Petersen 2000) showed a variation in sensitivity of 2.5 orders of magnitude between different zooplankton communities and that the variation among individual groups of macroinvertebrates is 1- 2 orders of magnitude. Such studies indicate that there is a high variability among individual aquatic communities and ecosystems and that the size of this variation may be comparable to the fate based variation in exposure. The results were estimated from actual measured concentrations and can therefore be attributed to biological differences among the exposed organisms. In generic LCA as well as in risk assessments, it is however not possible to include the variation in sensitivities among the individual aquatic systems and in general, the estimated PNEC values are believed to represent a level where the ecotoxicological impact on the system is acceptably low. Recommendations The sensitivity distribution curves and the ecosystem no effect concentrations will vary between aquatic ecosystems, but this is a variation not taken into account in current methods for generic risk assessment. Development in risk assessment tools aims at an incorporation of site-dependent information on background exposure and possibly also ecosystem sensitivity (Feijtel et al., 1997, ECETOC, 1999) but until such tools become available it is not possible to include these aspects in site-dependent characterisation modelling of ecotoxicity. 8.2.6 ImpactIn the characterisation of ecotoxicity, no distinction is made between the different types of effect caused by the chemical (lethality, endocrine disruption or other reproductive effects, avoidance or other). The effect parameter PNEC is determined as the concentration at which no effects are observed in a test system. The effect that is modelled is thus the effect expected to occur if the PNEC value is exceeded. No spatial variation is expected for this part of the cause-impact chain. Recommendations No spatial differentiation is relevant. 8.2.7 Descriptors in the spatial characterisationSummarising Sections 8.2.1-8.2.5, the following descriptors have been identified as potentially relevant to the spatial characterisation modelling in LCIA 8.2.7.1 Emissions to airThe air compartment is treated exclusively as a dispersion medium and therefore, the issue is the spatial variation in exposure of aquatic and terrestrial systems through deposition from the atmosphere. Table 8.8. Descriptors of relevance to the spatial characterisation of emissions to air
8.2.8 Emissions to waterFor emissions to the water compartment, a distinction is made between acute and chronic ecotoxic effects. Acute aquatic ecotoxicity The only descriptors considered of relevance are related to the emission. They concern the temporal course of the emission (pulse or continuous) and the dilution immediately after discharge. Due to the assumption of linearity between concentration and impact according to the current ecotoxicity characterisation modelling including the EDIP97, these descriptors can not be represented in the spatial characterisation modelling (see Section 8.1.1). Chronic aquatic ecotoxicity For chronic aquatic ecotoxicity, several descriptors are identified as relevant to spatial characterisation modelling. These are presented in Table 8.9. Table 8.9. Descriptors of relevance to the spatial characterisation of emissions to water
8.2.9 Emissions to soilFor chronic terrestrial ecotoxicity several descriptors are identified as relevant to spatial characterisation modelling. They are presented in Table 8.10.
Table 8.10. Descriptors of relevance to the spatial characterisation of emissions to soil 8.3 Development of spatial characterisation factorsThree levels of spatial differentiation have been defined:
The existing characterisation factors for ecotoxicity from EDIP97 or similar are considered to represent the full site-generic impact potential and are in principle not modified in the present context. The ambition is to develop site-factors that represent the extent to which exposure allows this full potential to be reached as discussed in Section 8.1.2. 8.3.1 Inclusion of spatial variation of descriptorsFrom the analysis of the causality chain for ecotoxicity in soil and water in Sections 8.2.1-8.2.5, the descriptors relevant to spatial characterisation modelling were identified in Tables 8.8-8.10. Among these, the following are judged possible to include in life cycle impact assessment: Emissions to air: Included:
Not included:
Emissions to water: Included:
Not included:
Emissions to soil: Included:
Not included:
8.4 A framework for spatial characterisation modellingThe already existing site-generic characterisation factor is interpreted as representing the full (in the case of EDIP97, but indeed, for some other LCIA methodologies, it is rather the average) impact from the substance. The spatial characterisation factor is the product of the site-generic characterisation factor and a site factor, which is seen as a modifier expressing the degree to which the full impact comes through:
... as already expressed in equation (3). In contrast to the expression in (8.3), the site factor, SF depends both of the substance properties and the spatial characteristics of the process. Hence, in equation (4), SF has been given the index "I" in addition to the index "p". The modelling of SF must represent the spatial variation in the descriptors listed in Section 8.3.1, and this is attempted through expressing SF as a product of the following variables:
(8.5)
8.4.1 The emission componentFor emissions to air or emissions to air or soil which are found to evaporate, the SFemis factor reflects the fraction of the deposited part of the emission that will expose water or soil ecosystems. In connection to EDIP97, SFemis is defined as (8.6) Chronic aquatic ecotoxicity: (8.7) Chronic terrestrial ecotoxicity: (8.8.) Acute aquatic ecotoxicity: The fractions deposited to water and soil natural areas are found for the four European regions in Table 8.3. The approach may later be sophisticated to allow country-specific modelling by combining the information on the relative frequency of natural areas within the countries of Europe (Annex 8.4) with atmospheric dispersion modelling as performed in e.g. Chapter 3. Based on Table 8.3, SFemis is calculated assuming a global default value of 0.2 for a: Table 8.11 SFemis for emissions occurring in different regions of Europe.
8.4.2 The evaporation componentThe temperature dependency of evaporation is substance specific. In Section 8.2.3.1 the influence of the temperature on evaporation is expressed in generic formulas. Based on some rough assumptions regarding the average temperature in different European regions values for SFevap are proposed in Table 8.5 which is repeated below: Table 8.5. Site factors, SFevap for volatilisation from water for two European regions
SFevap was developed for evaporation from water but it is proposed used also for evaporation from soil. 8.4.3 The biodegradation and transformation componentThe SFbio factor reflects the variation of biodegradability with the average temperature of the region where the fate of the substance takes place. It is relevant for both aquatic and terrestrial systems. The annual average temperature over Europe varies around 10 C between the Nordic region and the Southern region with the Western and Eastern regions in-between. If it is assumed, that the current site-generic fate modelling (in EDIP97 or other LCIA methodology) corresponds to an average mid-European situation, the factor is determined in Section 8.2.3.4 as:
The approach may later be sophisticated to allow country-specific modelling through combining the information on the annual average temperature of the countries of Europe (Annex 8.7) with atmospheric dispersion modelling as performed in e.g. Chapter 3 In a future development of the site-generic EDIP97, it would be relevant to modify the BIO factor representing the substance-specific biodegradation potential based on results of standardised biodegradability tests. In EDIP97, the ratio between BIO for a not biodegradable substance and BIO for a readily biodegradable substance is only a factor 5. This should be increased e.g. seeking inspiration in the suggested biodegradability rates in the EU TGD (1996) for natural terrestrial environments and natural waters based on the same test results. 8.4.4 The sorption and sedimentation componentThe SFsed factor must reflect the spatial variation in the relative importance of sedimentation as a removal process for substances adsorbing to particulate material in different aquatic systems. The SFsed factor is only relevant for substances emitted to or ending in the aquatic compartment of the environment. In EDIP97, there is no consideration of removal due to sedimentation. This is equivalent to operating with a removal factor with the value 1 (just like no potential for biodegradation is represented by a BIO factor value of 1). Site factors for sorption and sedimentation of organic substances are developed in section 8.2.3.3. They include the biodegradability of the substance in the calculation of the fraction, which is removed from the water phase of the aquatic system by sedimentation. Thus, the removal by sedimentation depends on:
These parameters are included in the values for SFsed presented in Table 8.6 and repeated below. Table 8.6. Site factors (SFsed) representing removal by the combined effect of sedimentation and biodegradation for readily biodegradable, inherently biodegradable and not biodegradable organic substances of different lipophilicity for the three emission scenarios: emission to river and from there through lake to estuary and sea, emission through estuary to sea and emission directly to sea.
N.B.: Not biodegradable 8.5 Spatial characterisation factors for ecotoxicityIn the framework laid out for calculation of site factors, SF in Section 8.4 and expressed in Expression (8.5), the determining parameters are Spatial
Using Expression (8.5) and the values given for the factors of SF in the previous sections, the ranges (min-max value) of SF are calculated and shown in Table 8.12. Table 8.12. Ranges (min-max value) of the site factor SF
8.6 Discussion and recommendationsAnalysing the extreme values of the site factors shown in Table 8.12, it is found that the largest variation which can be introduced by using this framework for spatial characterisation of aquatic ecotoxicity is a factor 33 (between the value for a semi volatile and not biodegradable substance emitted directly to the sea in Northern Europe and a strongly lipophilic substance emitted to a river in Western Europe). For substances of less extreme lipophilicity(logKow<4), the largest variation is a factor 7.8, and it is found between the same two situations. For terrestrial ecotoxicity, the largest variation is a factor 5.6 (between the value for a semi volatile and not biodegradable substance emitted to soil in Northern Europe and any substance emitted to soil in Southern Europe). Except for extremely lipophilic substances, this is a rather modest spatial variation, and it is indeed expected to represent only a minor part of the actual spatially determined variation in the fate and exposure of ecosystems to chemicals within Europe. Several reasons can be given for this:
It is the authors' judgement that the site factors developed for ecotoxicity are accompanied by considerable uncertainties, and that these uncertainties may well exceed the variation given by the factors. Application of the factors in life cycle impact assessment may thus introduce a model uncertainty which is larger than the reduction of the spatially determined uncertainty. On this background, the authors do not find it recommendable to apply the developed site factors in an attempt to perform spatial characterisation of ecotoxicity in LCIA. Currently, work is underway in the OMNIITOX project under the fifth Frame Programme of EU on development of a European consensus method for characterisation of ecotoxicity in LCA (http://www.omniitox.net). This method involves a comprehensive multimedia fate model with the option of spatial differentiation at the level of countries for Europe. The reader with interest in spatial characterisation of ecotoxicity is referred to the results of this work which will be available towards the end of 2004. 8.7 ReferencesAnkley, G. T., D. A. Benoit, J. C. Balogh, T. B. Reynoldson, K. E. Day & R. A. Hoke: Evaluation of potential confounding factors in sediment toxicity tests with three freshwater benthic invertebrates. Envir. Toxicol. Chem. 13: 627–635, 1994. Borkar, M.D. and Pillia, K.C.: Sedimentation rate in the Trombay bay, west coast of India. Indian Journal of Marine Sciences, 20(2), 115-117, 1991. Brugmann, L.: Metals in sediments and suspended matter of the river Elbe. Sci.Tot.Env., 159(1), 53-65, 1995. Chakrapani, G.J. and Subramanian, V.: Rates of erosion and sedimentation in the Mahanadi river basin, India. J. of Hydrology, 149(1-4), 39-48, 1993. Chenhall, B.E.: Anthropogenic marker evidence for the accelerated sedimentation in Lake Illawarra, New South Wales, Australia. Oceanographic Literature Review, 43(3), 305, 1996. Christensen T.H.: Cadmium Soil sorption at low concentrations: VIII. Correlation with soil parameters. Water, Air and Soil Pollution 44, 71-82, 1989. Das B.K., and Singh, M.: Rate of sediment accumulation in lakes of Udaipur, Rajasthan, India using the 210Pb method. Environmental Geology (New York), 24(1), 28-33, 1994. ECETOC: Environmental Exposure Assessment. Technical Report No. 61. ECETOC, 1994. ECETOC: GREAT-ER User Manual. Special Report no. 16, European Centre for Ecotoxicology and Toxicology of Chemicals, Brussels, 1999. EEC: 87/302/EEC: Test for potential biodegradability, equivalent to OECD 302 B-C (1992/1981) , 1987. Egebart, C.L.: Modelling aquatic fate of neutral organic pollutants for LCIA. KPD-20-99, Department of Manufacturing Engineering, Technical University of Denmark, 1999. EU-TGD: Technical Guidance Document in support of the commission directive 93/67/EEC on risk assessment of chemical substancesBrussels, 1996. Farmer, J.G.: The determination of sedimentation rates in Lake Ontario using 210Pb dating method. Canadian Journal of Earth Sciences, 15(3), 431-437, 1978. Feijtel, T., Boeije, G., Matthies, M., Young, A., Morris, G., Gandolfi, C., Hansen, B., Fox, K., Holt, M., Koch, V., Schroder, R., Cassani, G., Schowanek, D., Rosenblom, J. and Niessen, H.: Development of a geography-referenced regional exposure assessment tool for European rivers – GREAT_ER, Contribution to GREAT_ER1. Chemosphere, 34(11), 2351-2373, 1997 Georgiyevski, A.Ye. and Kuptsov, V.M.: Determination of sedimentation rates in the Gulf of Ria by the non-equilibrium 210Pb method. Okeanologiya, 26(2), 1986. Goedkoop, M and Spriensma, R: The Eco-indicator 99 A damage oriented method for life cycle impact assessment. Methodology report, preliminary internet version 5 October 1999, PRé Consultants B.V., Amersfoort, the Netherlands, 1999. Guinée, J., Heijungs, R., van Oers, L., van de Meent, D., Vermeire, T. and Rikken, M.: LCA impact assessment of toxic releases. Generic modelling of fate, exposure and effect for ecosystems and human beings with data for about 100 chemicals. VROM, no. 1996/21, The Hague, 1996. Haitzer M., S. Hoss, W. Traunspurger, C. Steinberg: Effects of dissolved organic matter (DOM) on the bioconcentration of organic chemicals in aquatic organisms - a review -. Chemosphere 37(7), 1335-1362, 1998. Harremöes P. & A. Malmgren-Hansen: Lærebog i Vandforurening. Polyteknisk Forlag, 1989. Hatzinger, P.B., M. Alexander: Effect of aging of chemicals in soil on their biodegradability and extractability. Environmental Science and Technology 29(2), 537-545, 1995. Hauschild, M.: Estimating pesticide emissions for LCA of agricultural products. In Weidema, B.P. and Meeusen, M.J.G. (Eds.): Agricultural data for life cycle assessments, volume 2, pp. 64-79, ISBN 90-5242-563-9, Report no. 2.00.01, Agricultural Economics Research Institute (LEI), The Hague, 2000. Hauschild, M.Z. and Wenzel, H.: Environmental assessment of products. Vol. 2 - Scientific background, 565 pp. Chapman & Hall, United Kingdom, 1998, Kluwer Academic Publishers, Hingham, MA. USA. ISBN 0412 80810 2 He, Q., Walling, D.E. and Owens, P.N.: Interpreting the 137Cs profiles in several small lakes and reservoirs in the Southern England. Chemical Geology, 29(1-2), 115-131, 1996. Hertwich E.G.: Toxic Equivalency: Accounting for Human Health in Life-Cycle Impact Assessment. University of California, Berkeley, 1999. Houx N.W.H. & W.J.M. Aben: Bioavailability of pollutants to soil organisms via the soil solution. Sci. Total Environ. Suppl., 387-395, 1993. Huijbregts, M. and J. Seppälä: Towards region-specific, European fate factors for airborne nitrogen compounds causing aquatic eutrophication. Int. Journal of Life Cycle Assessment, 5(2), 65-67, 2000. Huijbregts, M.A.J.: Priority assessment of toxic substances in the frame of LCA. Development and application of the multi-media fate, exposure and effect model USES-LCA. Milieukunde, Universiteit van Amsterdam, Amsterdam, 1999. Jolliet, O. and Crettaz, P.: Critical surface-time 95. A life cycle impact assessment methodology including fate and exposure. École Polytechnique Fédérale de Lausanne, Switzerland, 1997. Kleier, D.A.: Phloem mobility of xenobiotics. Plant Physiol. 86, 803-810, 1988. Knaebel, D.B., T.W Federle, D.C. McAvoy & J.R. Vestal: Effects of Mineral and Organic Soil Constituents on Microbial Mineralization of Organic Compounds in Natural Soil. Appl. Environ. Microbiol. 60(12) 4500-4508, 1994 Kristensen, P. and Hansen, H.O.: European rivers and lakes. Assessment of their environmental state. EEA Environmental Monographs 1, European Environmental Agency, Copenhagen, 1994 Lesuers, P., Weber, O., Marambat, L., Tastat, J.-P., Jouanneau, J.-M. And Turon, J.-L. : Age of an Atlantic shelf mud path supplied by an estuary : the west Gironde example (France) was deposited in historic times. Comptes Rendus de l'Academie des Sciences, Seri II (Mecanique, Physique, Chimie, Sciences de l'Univers, Sciences de la Terre), 308(10), 935-940, 1989. Lyman W.J., W.F. Reehl W.F. & D.H. Rosenblatt : Handbook of chemical properties estimation methods; Environmental behaviour of organic compounds. McGraw-Hill, 1982. Madsen, T., M. Winther-Nielsen, & D. Rasmussen: Studies on the fate of linear alkyl benzene sulfonates (LAS) in sludge and sludge-amended soil. The CLER Review, 5(1), 14-19, 1999. Matthijs E. & H. De Henau: Adsorption and Desorption of LAS. Tenside Detergents 22(6), 299-304(1988). McKone T.E.: CalTOX, A Multimedia Total Exposure Model for Hazardous-Waste Sites UCRL-CR-111456PtI-IV. Lawrence Livermore National Laboratory, Livermore, CA, http://www.cwo.com/~herd1/caltox.htm, 1993. OECD: OECD 302 B-C: Test for potential biodegradability, equivalent to 87/302/EEC, 1992. Pedersen F., A. Damborg & P. Kristensen: Industrispildevands miljøfarlighed. Miljøprojekt, 260, 1994. Petersen S.: Model based tool for evaluation of exposure and effects of pesticides in surface water. Draft report to the Danish Environmental Protection Agency, 2000. Potting, J. and Hauschild, M.: Predicted environmental impact and expected occurrence of actual environmental impact. Part I: The linear nature of environmental impact from emissions in life cycle assessment. International Journal of Life Cycle Assessment 2(3), 171-177, 1997a. Potting, J. and Hauschild, M.: Predicted environmental impact and expected occurrence of actual environmental impact. Part II: Spatial differentiation in life cycle assessment via the site-dependent characterisation of environmental impact from emissions. International Journal of Life Cycle Assessment 2(4), 209-216, 1997b. R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental organic chemistry. Wiley Interscience, New York, 1993. Ravichandran, M., Baskaran, M, Santschi, P.H. and Bianchi, T.S.: Geochronology of sediments in the Sabine-Neches estuary, Texas, USA. Chemical Geology, 125, 291-306, 1995. RIVM: Uniform System for the Evaluation of Substances 2.0 (USES 2.0) 679102044. National Institute of Public Health and the Environment (RIVM), Ministry of Housing, Spatial Planning and the Environment (VROM), Ministry of Health, Welfare, Sport (VWS), The Hague (NL). Rostan, J.C., Juget, J. and Brun, A.M.: Sediment rate measurements in the former channels of upper Rhône river using Chernobyl 137Cs and 134Cs as tracers. Sci.Tot.Env., 193(3), 251-262, 1997. Santschi, P.H., Allison. M.A., Asbill, S. and Perlet, B.: Sediment transport and Hg recovery in Lavaca Bay as evaluated from radionuclide and Hg distribution. ES&T, 33, 378-391, 1999. Stober, J.C. and Thompson, R.: Palaeomagnetic secular variation studies of Finnish lake sediment and the carriers of remannence. Earth and Planetary Science Letters, 37(1), 139-149, 1977. van Leeuwen C,H, & J.L.M. Hermens: Risk Assessment of Chemicals: An Introduction, 1995. Veerkamp and Berge: Hazard assessment of chemical contaminants in soil. Revised appendix 3. ECETOC Technical Report No. 40, 1992. Verschueren K.: Handbook of Environmental Data on Organic Chemicals. Environmental Sciences. Electronic Resources. 3rd edition, 1998. Vogel, R.L., Kjerfve, B. and Gardner, L.R.: Inorganic sediment budget for the North Inlet saltmarsh, South Carolina, USA. Oceanographic Literature Review, 44(6), 579, 1997. Wenzel, H., Hauschild, M.Z., and Alting, L.: Environmental assessment of products. Volume 1. Methodology, Tools and Case Studies in Product Development. Chapman and Hall, London, 1997. Yu, K.N., Young, E.C.M., Stokes, M.J. and Guan, Z.J.: Determination of sedimentation rates in eastern sea areas of Hong Kong with gamma-ray spectrometry. Nuclear Geophysics, 9(1), 73-81, 1995. Zuo, Z., Eisma, D, Gieles, R. And Beks, J.: Accumulation rates and sediment deposition in the Northwestern Mediterranean. Deep-Sea Research II, 44(3-4), 597-609, 1997. Annexes: Annex 8.1: Initial dilution of waterborne emissions Annex 8.2: Adsorption and immobilisation Annex 8.3: Biodegradation of LAS under different temperature and sorptive conditions Annex 8.4: Relative share of natural areas within the regions of Europe Annex 8.5: Relative share of emissions depositing to sea and land within the regions of Europe Annex 8.6: Sedimentation velocities in different aquatic systems Annex 8.7: Annual average temperatures of European countries Annex 8.8: Rate constants for evaporation of substances from water Annex 8.9: Removal of substances from water through the combined effect of biodegradation and sedimentation Annex 8.1 Initial dilution of waterborne emissionsThe initial dilution of waterborne emissions is taken as the average dilution in the cross sections of the wastewater plume at the point where the impulse of the plume equals the impulse of the surrounding water masses or where the plume reaches the water surface. The initial dilution is independent of the substance characteristics and governed by the following parameters: The type of discharge The characteristics of the receiving water body (water exchange rate, depth, difference in salinity of the outlet and the receiving water body) The position of the discharge point in the water column (at the bottom or the surface of the receiving water) The geometry of the discharge pipe The discharge flow velocity (both the volume and the linear flow velocity) Each of these parameters may vary substantially within Europe. Examples of resulting and applied dilution factors are shown in Figure 8.3.
Figure 8.3. Distribution of the dilution factors for Dutch waste water emissions 1000 m downstream of the treatment plants. The type of receiving water is indicated - boezem and polder water refers to flooded marsh areas (Leeuwen and Hermens, 1995). Figure 8.3. shows the estimated dilution factors for municipal wastewater treatment plants in the Netherlands 1000 m downstream from the emission point. The data shows a considerable variation with a median dilution factor of 30. A dilution factor of this size at a distance of 1000 meters indicates a small initial dilution at the discharge point. Examples of default values applied as initial dilution factors in the environmental administration are:
The overall average dilution of wastewater emitted to rivers can be estimated roughly for an area by dividing the wastewater emission per capita (representing all wastewater within a specific region) by the excess precipitation. This approach will underestimate the true average value in countries that discharge wastewater directly to the sea or which receive less polluted river water from neighbouring countries. Assuming an average wastewater volume of 200 l per capita per day (person equivalent) this approach gives an average dilution factor for wastewater discharged to aquatic environments excluding marine waters ranging from 12 (the Netherlands), 15 (Belgium and Luxembourg), and 17 in the UK to 134 in Ireland (ECETOC, 1994). Annex 8.2 Adsorption and immobilisationAdsorption is an important parameter that often has a major influence on the bioavailability and toxic impact of chemical substances. Because of the importance of adsorption to environmental fate and impact of toxicants it is discussed in details below. Sorption (adsorption and desorption) are processes that determine the distribution of a chemical between a liquid and a solid phase in a multiphase system like pore water - soil or sediment - water. Adsorption includes interaction with small organic particles below 0.2 µm, which normally is regarded as dissolved organic matter. Sorption processes involve different types of chemical binding from weak and reversible interactions like hydrogen bonds or van der Waals forces to stronger and less reversible ionic or covalent bonds. Sorption is an equilibrium process, the position of which reflects the relative affinities of the substance to the solid and liquid phases. The weaker the bonds to the solid phase, the more the substance will desorb and be present in the liquid phase. The position of the equilibrium is described through the adsorption coefficient, Kd, defined as the ratio between the concentration on the solid phase and the concentration in the liquid phase:
Kd can be determined experimentally but often, it is estimated from knowledge about the substance's molecular characteristics (through structure-activity relationships). Using Kd, the concentration in the liquid phase (Cl) can be determined from the total concentration of the substance in the compartment. Relevance of sorption In general it can be assumed that only the dissolved and freely available fraction of a substance will interact (bioaccumulate and exert toxicity) with biological organisms in the environment. Several studies support this theory. Ankley et al. (1994) showed that the effect of the pesticide chlorpyrifos on the midge Chironomus tentans was highly dependent on dissolved organic carbon (DOC) in the pore water of sediments. DOC is normally defined as the fraction passing a 0.20 or 0.45 µm filter and includes colloids as well as true dissolved matter. LC50 value in pore water corresponded with the LC50 value obtained in pure water only if the fraction adsorbed to DOC was considered to be unavailable for the animals. The nominal pore water concentration was about 10 times higher than the LC50 value. Houx & Aben (1993) exposed nematodes to chlorpyrifos and pentachlorophenol in different soil types. The toxicity of the spiked soil samples was negatively correlated to the soil organic content and the results indicated that the toxicity could be explained by the dissolved fraction of the substances alone. Knaebel and co-workers measured the degradation over time for the detergent LAS adsorbed to different solid materials. The initial rates of degradation were negatively correlated with the adsorption coefficient indicating that the initial degradation rate decreases with increasing strength of the adsorption (Knaebel et al., 1994). It should, however, be mentioned that in dilute media like the water phase of natural aquatic ecosystems, bacterial growth preferentially takes place on solid surfaces. This observation has inspired the theory that growth may be enhanced by the adsorption of organic matter at liquid-solid interfaces. In addition, there is some evidence that the microbial degradation of certain surfactants in environmental samples is faster in the adsorbed state than in solution. Painter et al. 1992 reported the half-life of LAS in river water without sediment to be 1.4 d and 0.7 d in the presence of sediment. In a recent review Haitzer and co-workers studied the effects of dissolved organic carbon (DOC) on the bioconcentration of different organic substances in aquatic organisms. Hydrophobic organic substances like polychlorinated dioxins, PAHs, pyrethroid insecticides, tributyltin and selected surfactants showed a reduced bioconcentration at DOC levels, which can be found in freshwater environments (0- 10 mg/l). For example a 51% reduction of the uptake of benz(a)pyrene was observed going from 0 to 6 mg DOC/l, and a 23% reduction of the uptake of trichlorobenzene by increasing DOC from 0 to 10 mg/l (natural DOC was used, isolated from a stream water). The reduction in the bioconcentration at low DOC levels (0-10 mg/l) was from 0 - 50% for different hydrophobic substances, although higher levels were reported for cationic tensides (Haitzer et al., 1998). Although the general trend is a reduced uptake at increasing levels of DOC, a stimulated uptake has been reported in specific cases where an increased uptake was observed for highly lipophilic compounds as DDT and polychlorinated biphenyls. The mechanism may be explained by an increased uptake via food. It can be concluded from a large number of experiments, including the reports cited above, that DOC has a considerable influence on the availability of hydrophobic substances in the aquatic environment. As mentioned earlier, dissolved organic carbon includes colloids as well as true dissolved matter. DOC thus includes organic substances with a high molecular weight, which act as a sorbent for lipophilic substances. Because of the size of the sorbent-substance complex the uptake and hence the bioavailability is reduced. Some general conclusions can be drawn from the discussion above regarding site characterisation in LCIA:
Mechanism of sorption Depending on their properties, substances sorb through different types of interactions with the solid phase. Non-ionic organic substances are rather hydrophobic and they sorb through weak van der Waals forces and hydrogen bonds to the organic fraction of the solid phase of soil or sediments. The adsorption is often described by Koc, which expresses the adsorption to the organic carbon content of the solid phase. Koc is defined as:
Where foc is the relative content of organic carbon in the solid phase. The degree of sorption of non-ionic organic substances to soil and sediment matrices (Kd) is thus proportional to the content of organic carbon. The sorption capacity is normally not regarded as a limiting factor because of the high concentrations of organic material in soils and sediments and the relatively low environmental concentrations of the released organic toxicants. The frequently used plasticizer DEHP (di(ethylhexyl)phthalate) is an example of a non-ionic hydrophobic organic substance that sorb readily to organic material in aquatic and terrestrial environments. Estimates of the dissolved fraction of DEHP in an aquatic system show a strong dependency on the amounts of suspended organic matter. The dissolved (available) fraction of DEHP may vary by a factor of 5-6, corresponding to a range of suspended organic carbon from 1 – 10 mg C/l observed in aquatic ecosystems. The relationships between dissolved DEHP and the concentration of suspended organic carbon is shown in Figure 8.4.
Figure 8.4. Estimated fractions of DEHP dissolved in the water phase as a function of the organic carbon content in suspended matter. The default value used in the EU-risk assessment guideline (TGD) is indicated with an asterisk. Ionic substances interact with electrically charged groups on the solid phase. The sorption of cationic substances like metals occurs as ion exchange on negatively charged sites on the solid phase of soils and sediments. Negative charges occur predominantly on the surface of clay minerals and on organic material containing dissociated acid and phenolic hydroxyl groups. The pool of dissolved ionic substances competes about these sites. Hydrogen ion has the strongest positive surface charge, and is naturally present in all systems containing water. Consequently, the sorption of cationic substances is strongly pH dependent. As an example, the sorption of cadmium in soils is suggested to depend on the pH according to the following general expression (Christensen, 1989):
The equation predicts a variation in Kd of cadmium in soil between 73 and 2600 (corresponding to a factor of 35), when the pH of the soil is varied within values naturally occurring in different soils (pH 5-8). A similar pH-dependency can be expected in sediments in the aquatic environment although the natural pH variation here is smaller (6.5-8.3) corresponding to a variation of Kd by a factor of 9 using the same expression. A correction for that the bioavailability of chemicals in soils depends on the soil type is introduced; a standard soil containing 3.4% organic matter is introduced, and the bioavailability of non-ionic organic compounds is assumed only to depend on the organic matter content, only. Ministry of Housing, Spatial Planning and the Environment (1994) (the Netherlands) has suggested a correction term for adsorption of heavy metals in soils, which includes the content of clay and organic matter. The correction term defines a standard soil or sediment containing 25% clay and 10% organic matter is defined here. The correction term reads:
Where a, b and c are constants depending on the compound in question. CStandard is the concentration in standard soil or sediment CMeasured is the measured concentration in soil or sediment fCorrection is the correction term The above expression can in principle be extended to any compound, which adsorbs to the clay and organic matter in the soil and sediment. The coefficient a, b and c should be found from experimental investigations. In Danish soils the clay content varies between 0 and 40% and the organic matter content varies between 1 – 8%. In the table 8.13 below, the equation is used to illustrate the importance of the soil type. For a metal like nickel the composition of the soil has a dramatic effects on the bioavailability, whereas the effect is less significant for other heavy metal as mercury, lead and cadmium. Table 8.13 Influence of soil type on fCorrection for a number of metals
Adsorption of anionic organic substances occur through ion-exchange with positively charged sites on the solid phase as hydrous oxides and partial hydroxides of Al, Fe and Mn –ions (e.g. Al(OH)2(H2O)4+). Anionic adsorption is of particular importance to the sorption of dissociated organic acids. This is illustrated by the adsorption of anionic surfactants, which may be described by the Freundlich equation as: Cs = Kd Cl nf, where `Cs' is the concentration of substance in the solid phase and `Cl' the concentration in the pore water, `Kd' is the distribution coefficient and `nf' the slope of the adsorption isotherm. For dissociating organic substances, the adsorption of the ionic form is normally much smaller than the (non-ionic) adsorption of the undissociated form. pH therefore strongly influences the adsorption of such substances. This is demonstrated in Figure 8.5 where empirically determined Kocs for pentachlorophenol is presented as a function of the undissociated fraction of pentachlorophenol (at different pH values). The correlation between the undissociated fraction and the observed Koc is linear starting at a value around 100 l/kg for the fully dissociated pentachlorophenol (predominantly anionic adsorption). A similar type of linear correlation between the dissociated fraction and the Koc value can be expected for other anionic compounds.
Figure 8.5. Measured Koc for pentachlorophenol as a function of the degree of dissociation (Koc values from Verschueren, 1998) In general, an overall Koc for a dissociating compound may be expressed by:
where, The organic carbon partitioning function of the undissociated part of the compound in expressed by:
K*OW is the octanol-water coefficient for the dissociated part of the compound. This coefficient is approximated with (Kleier, 1988):
The estimated KOC is shown as a function of pH for different values of logKOW and pKa in figure 8.6.
Figure 8.6. Estimation of KOC for organic acids. Immobilisation Sediments often contain high concentrations of organic material, especially in sedimentation areas, where they may act as a trap for pollutant sedimenting together with matter. The sedimentation and binding of pollutants in sediments are governed by the mechanisms discussed above and adsorbing substances (e.g. metals and lipophilic organic compounds) may therefore be present in the sediments in high concentrations. Although the distribution of pollutants between the dissolved and adsorbed fraction is normally regarded as a reversible equilibrium, there is often a net transport of pollutants bound to sedimenting material into the sediment. The initial adsorption is a relatively fast process, which may be followed by a slower process where the fraction of adsorbed substance is increased continuously in periods of months or even longer. From experiments performed in soil it is known that availability of a substance is reduced with increasing incubation time indicating an increasing strength of the chemical bonding involved in the adsorption processes (Hatzinger & Alexander 1995). The behaviour of metals in soils and sediments can be characterised by complex chemical reactions. Heavy metal ions present at trace levels in solution are adsorbed highly selectively in soils. Heavy metal adsorption in soil includes different type of adsorption sites (organic constituents, oxides, clay minerals, etc). The organic constituents determine the mobility of heavy metals in the soil matrix mainly because of the size of the organic constituents involved. The interaction of heavy metals with clay minerals includes adsorption, cation exchange as well as irreversible adsorption mechanisms (i.e. non-extractable with neutral salts or weakly acidic solutions), and will in general result in immobilisation of the metals. The inorganic constituents themselves can be considered as immobile apart from resuspension of particles in turbulent water. In soil the mobility of heavy metals tend to be low. Inclusion of adsorption and immobilisation in site characterisation. It is evident that adsorption and immobilisation of chemical substances in the environment have a strong influence on their bioavailability and hence on the potential for exposure. To a large extent, the sorptive properties and possibility of precipitation depends on inherent substance-specific properties and should as such be considered in the calculation of the site-generic characterisation factor. In the EDIP method adsorption is only considered in the modelling of terrestrial ecotoxicity, while no modification of the laboratory data is performed in modelling the aquatic ecotoxicity potential. This aspect should be considered in future revisions of the model for site-generic characterisation factors. As seen throughout Section 1.3.3, the adsorption and immobilisation processes are strongly influenced by environmental parameters among which the most important are :
Annex 8.3 Biodegradation of LAS under different temperature and sorptive conditionsFigure 8.7 shows the degradation of LAS in a surface sediment, which is assumed to be aerobic. The degradation is modelled as a function of temperature and the water content of the sediment (introducing the influence of bioavailability since LAS adsorbs to the solid phase), using a degradation half-life of 1 day for LAS in water at 15C and assuming that the degradation of LAS takes place only in the water phase. 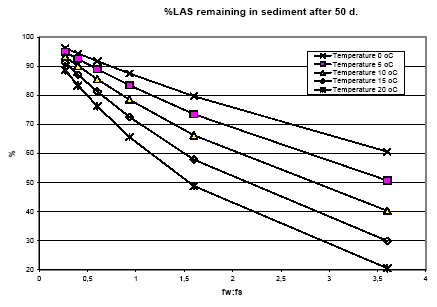
Figure 8.7. Model simulation of the degradation of LAS in aerobic sediment at different temperatures assuming a t½ at 1 day at 15 C. The figure shows moreover the effects of the water content of the sediment (fw:fs: fraction of water : fraction of solids in sediment). Temperature dependency It is well-known that the biodegradation rate increases with increasing temperature within the range tolerated by the degrading micro organisms. The influence of the temperature on the different kinds of micro organisms varies. Investigations on the biodegradation processes in the soil have shown that changes in temperature changes the composition of the microflora. A sudden change in temperature can therefore changes the biodegradation rate immediately, where after a certain adaptation period for the new flora, the biodegradation rate is increases again to a new level, which may not be so far below the initial level. Normally, the rates of biodegradation are described by the Arrhenius equation, which also is used to describe chemicals reaction rates dependency of temperature:
where k(T) is the degradation rate at temperature T k(T0) is a reference degradation rate R is the gas constant T is the absolute temperature Ea an activation energy In Table 8.14 values for Ea/R is given for the degradation of different compounds in water. Table 8.14. Estimated values for Ea/R. In K-1. Data from Verschueren 1998 and Lyman 1982.
It is seen that the dependency of the degradation rate on the temperature varies for the different compounds. With a temperature decrease from 20 oC to 10 oC, the degradation rate may decrease to around 50-75% of the degradation rate at 20 oC. A default value for the temperature dependency on the biodegradation rate in water could be Ez/R = 5000 K-1. With this value the biodegradation rate will be lowered with approximately 50% for every 10 oC decrease in temperature. If a degradation rate (kRef) at a temperature (TRef) is known, the degradation rate at temperature T can be expressed by:
where = Ea/(R•T•T•Ref) For the degradation of pesticides in soil, an value of = 0.08 K-1 have been suggested (Boesten & Van der Linden, 1991). This is the average value of some 50 experiments with a range of pesticides and soils. This coefficient is suggested to be suggested for the description of the temperature influence on the degradation rate in soil. An increase in temperature of 20 oC gives an approximately 5 times higher degradation rate in soil. Bioavailability. Normally it is assumed that the biodegradation primarily takes place in the water phase, meaning that only the dissolved part of the substance in question will biodegrade. Several studies have been carried out to support this theory. Knaebel et al. (1994) measured the degradation (by measuring CO2 evaluation) over time for LAS adsorbed to different solid materials. The desorption coefficient was negatively correlated with the initial rates of degradation – indicating that the initial degradation rate decreases with increasing strength of the adsorption. It should however be mentioned that a general observation is the bacterial growth in dilute media preferentially occurs on solid surfaces. This observation has given rise to the concept that growth may be enhanced by the adsorption of organic matter at liquid-solid interfaces. In addition, there is some evidence that the microbial degradation of certain surfactants in environmental samples occurs faster in the adsorbed state than in solution. It can for example be mentioned that the half-life of LAS in river water without sediment has been measured to be 1.4 d and with sediment the half-life was decreased to 0.7 d (Painter, 19xx). In Figure 8.8 the calculated degradation of LAS in a surface sediment, which is assumed to be aerobic, is shown as a function of temperature and the fw:fs ratio (to illustrate the influence of bioavailability).A degradation half-life in water of LAS is set to 1 day at 15 oC. Even though it may not the case for the degradation for LAS in sediment systems (see the discussion above) the degradation of LAS is in this example is assumed only to take place in the water phase. As seen from this figure the degradation is strongly dependent on the temperature and the fw:fs ratio in the sediment.
Figure 8.8. Degradation of LAS in sediment. Annex 8.4 Relative share of natural areas within the regions of EuropeBased on information from Table 8.15 the relative shares of natural areas and hence the share of air deposited material that exposes natural ecosystems is calculated for the four regions of Europe, the Nordic, the western, eastern and southern countries. The borders of the four regions is taken from Kristensen and Hansen, 1994 (except for Denmark which is considered a Nordic country here). Table 8.15. Frequency of natural areas per European country and for the four European regions
Annex 8.5 Relative share of emissions depositing to sea and land within the regions of EuropeBased on information from Huijbregts and Seppälä (2000), the relative shares of emissions depositing to sea and land is calculated for NH3 and NOx for individual countries and aggregated for the four regions of Europe, the Nordic, the Western, Eastern and Southern countries. The borders of the four regions is taken from Kristensen and Hansen, 1994 (except for Denmark which is considered a Nordic country here). Table 8.16. Fractions of the emissions of NH3 and NOx that are deposited to sea and land for the different European nations and averaged over European regions (based on information from Huijbregts and Seppälä, 2000)
NH3 is a relatively short-lived substance in the atmosphere while NOx is more long-lived. From the average deposition patterns shown in Table 8.16 it is clear, that the difference between the two substances is of little importance except for the Nordic countries. As an average estimate, the following factors are proposed for deposition to sea and land of air-borne emissions originating within the different regions: Table 8.17. Estimated average fractions of emissions deposited to sea and land for the four European regions
8.8 Annex 8.6 Sedimentation velocities in different aquatic systemsBased on information compiled in Egebart (1999), an overview of sedimentation velocities in different aquatic systems is given in Table 8.18. Table 8.18. Deposition velocities in different aquatic systems.
Annex 8.7 Annual average temperatures of European countriesTable 8.19. Monthly and average temperatures for European countries and averaged for European regions.
Annex 8.8 Rate constants for evaporation of substances from waterThe amount of substance evaporating from a water phase is expressed by MEvaporation = A•kT•CW Where kT is the total mass transfer coefficient:
Where: H is Henrys law constant R is the gas constant T is the absolute temperature kW is the liquid phase transfer coefficient, which can be estimated by (Schwarzenbach et al., 1993):
where DO2(Ti) is the diffusion coefficient of oxygen in water at temperature Ti (K) µW(Ti) is the viscosity of water at temperature Ti (K) U10 is the wind velocity at a height of 10 m. U10 is set to 5 m/s. MO2 is the molecular weight of oxygen = 32 g/mole. M is the molecular weight of the substance in question. M is set to 150 g/mole in the present calculations. kA is the air phase transfer coefficient, which can be estimated by (Schwarzenbach et al., 1993):
Where DH2O(Ti) is the diffusion coefficient of water in air at temperature Ti (K) The temperature dependency of Henry's constant can be estimated from (Veerkamp & Berge): H(T2)=H(T1)•e0.024(T2-T1) Calculations have been performed for different values of Henrys constant and at three temperatures: 10oC (reference temperature), 6 oC (Northern European countries) and 15 oC (Southern European countries). The results are given in Table 8.20 below as a reduction factor determined as the ratio between the reference situation and the Northern or Southern European situation. Table 8.20. Temperature dependent evaporation of semi volatile compounds.
Annex 8.9 Removal of substances from water through the combined effect of biodegradation and sedimentationThe net removal from the water phase depends on the sedimentation rates in the different hydro-geological compartments which the substances pass from the emission point to sea, and the time they spend in each of them (which again is determined by the biodegradation rate and the hydraulic retention time for the different compartments). In other words, the fraction of the substance that ends up in the sediments depends on the location of the emission point, and the hydraulic retention time of water in different sections of the hydrological cycle (river, lake). As a reference situation, the chemical is assumed to be emitted directly to the sea. The retention time in the sea is set at 35 days, corresponding to the degradation time of 80% of a readily biodegradable substance with a half-life of 15 days (according to the EU TGD, 1996). In order to derive at the 50% degradation of an inherent biodegradable substance within 35 days, the half-life of an inherently biodegradable substance is set to 35 days in agreement with the application factors for biodegradation (BIO) used in the EDIP. This is lower than the recommended biodegradation half-life of 150 days defined by EU TGD (1996). The biodegradation half-life of a non-biodegradable substance is set to 2500 days corresponding to 1% degradation within 35 days. Biodegradation Biodegradation is treated as a first-order process with the rate constant kBio. The amount removed by biodegradation (MBio) from a water body during the time interval dt is thus expressed as: MBio = kBio•VCW•dt where V is the volume of water body (m³) kBio is the first order biodegradation constant (d-1) Cw is the concentration of the substance in the water phase (mg/m³) Sedimentation Sedimentation is also treated as a first-order process. The amount removed from the water phase by net-sedimentation (MSed) (i.e. sedimentation minus re-suspension) during the time interval dt is: MSed= Vs•A• KSS•CW•dt where Vs is the linear sedimentation velocity (kg solid/m2/d) KSS is the sediment-water partition coefficient (m³/kg) estimated by: Kss = fOC•KOC KOC is the organic carbon partition coefficient here estimated by KOC = KOW A is the area of the water body (m2) Four types of surface waters are considered: river(1), lake(2), estuary(3) and sea(4). For each type of surface water (i), a correction factor fi is calculated representing the fraction of substance which is left after the combined removal through sedimentation and biodegradation:
... since the rate constant of the sedimentation process can be expressed as Vs •KSS/Z Where Z is the water depth of surface water i (m) Ti is the retention time in surface water i (river, lake, estuary, sea) (days) If the sedimentation rate is neglected then f4 = 1, corresponding to the reference state. Table 8.21. The default characteristics of the surface waters are given in Table 8.22.
Table 8.22. Estimated reduction factors (fi) representing removal by the combined sedimentation and biodegradation for readily biodegradable, inherently biodegradable and not biodegradable organic substances in a river, a lake, an estuary or a sea. It is seen that for biodegradation outweighs sedimentation as removal mechanism for the readily and inherently biodegradable substances, particularly when the residence time is long as it is in lakes. The temperature dependence of biodegradation is treated in the next section. Only for extremely lipophilic substances does removal by sedimentation play a substantial part. 9 Integration of external noise nuisance from road and rail transportation in lifecycle assessment9.1 Introduction Authors: Per H. Nielsen [79], Jens E. Laursen [80] 9.1 IntroductionNoise nuisance from transportation of goods is a well-known environmental impact, which has received a lot of attention in the literature (Vejdirektoratet, 1998) and in practical noise control projects (Vejdirektoratet, 1999). However, it is a general impression of noise reduction activities that the existing noise producing processes are taken for granted and/or that the products or services that are provided by the noise producing processes are taken for granted. The consequence is that following two means are primarily used in addressing the noise nuisance problem:
Only few activities attempt at modifying the products and services and hereby reducing or eliminating the noise generating processes. The purpose of this chapter is to analyse noise nuisance from a product-oriented point of view and to make some first attempts to integrate noise nuisance from transportation processes as an impact category in LCA. This will allow an integration of noise nuisance as an environmental consideration in product development and environmental comparison of products and services based on quantitative LCA, so that noise can be evaluated and accounted for at the same level as other environmental impact categories in the future. The focus of the present report is merely on noise that is annoying human beings during transportation of products. Nuisance, perceived by employees during the production process is not dealt with since this is an issue of working environment that is already addressed separately, see (Wenzel et al., 1997). The disturbance of sensitive areas by noise (e.g. nature parks and recreational areas) and the disturbance of animals are not addressed in this report. These aspects could, however, be addressed in future studies to provide a more complete representation of the noise problem associated with production of services and goods. As will become clear in the following, several simplifications with respect to e.g. noise distribution and quantification of the noise nuisance, are required to make generic noise models applicable for LCA. However, these simplifications are necessary and justified by the general need for a quantitative relation between the production of goods and generation of noise nuisance, otherwise the omission of noise nuisance data in LCA would be the only alternative. The degree of detail in modelling generic environmental impacts is always a compromise between making the model feasible and sufficiently precise, and the simplifications suggested in the following is not an exception. However, nothing prevents that more details are taken into account in the future or in specific cases where more accurate results are required. Readers who are not familiar with measurements and calculations of noise are encouraged to consult Annex 9.1 in order to ease the understanding of the following sections. 9.2 General approachThe noise nuisance NNd at a specific distance d from a point source can be quantified in terms of "person hour" by the following equation: NNd = Pd• Tproc •NNFLp (9.1) where Pd is the number of persons in the distance d from the source [dim. less] Tproc is the duration of the noisy process [h] and NNFLp is a noise nuisance factor specific for the actual noise level, Lp relative to the background noise level. [dim. less]. The number of people in the distance d from the noise source can be determined by counting or by average estimation. The duration of the noisy operation is the time that is used to produce one unit of the product or service according to the functional unit, and can also be determined by measurements or by average calculations. The noise nuisance factor NNFLp, represents the inconvenience caused by the noise to humans. The noise nuisance factor is a subjective parameter, which is determined by several aspects such as
Formula 9.2 shows the relation between the noise pressure and the nuisance factor as determined by interviews for traffic noise (see Anonymous, 1989) and Annex 9.1). NNFLp = 0.01 •4.22 0,1 (Lp - K) (9.2) Where Lp is the noise level and K is the background noise level in dB relative to 20 µPa. The exponent factor (Lp – K) expresses the part of the noise that exceeds the background noise.
Figure 9.1. Relation between noise pressure level, Lp [dB(A)] and nuisance factor NNF Lp for traffic noise, (Anonymous, 1989). Figure 9.1 illustrates the relation between outdoor noise and the nuisance factor graphically. The magnitude of the noise, Lp in various distances from the source can be determined either by measurements or by calculations.
Figure 9.2. Distribution of population (x) within 10 meter wide circular rings (isobars) in different distances, d (0 - 85 m) around a point noise source (o). Figure 9.2 illustrates the noise isobars around a point noise source as well as the number of people in each isobar. The noise level is high close to the source and decreases with increasing distance from the source due to attenuation caused by divergence of the sound waves and by absorption. The reduction of the noise is determined by several factors, such as
Circular noise isobars as shown in Figure 9.2 appear only in open, flat landscapes when the atmosphere is still and homogenous. Thus, in most situations, the isobars are non-circular as they are shaped according to the actual conditions. As a simplified average consideration, however, the noise isobars are assumed in this model to be circular, and the noise level in various distances from the point source can be calculated according to the mathematics in Annex 9.1. As an example, the noise levels in various distances from the source are shown in Figure 9.3. The strength of the noise source and other input parameters for the calculations are listed in Table 9.1.
Figure 9.3. Example of calculated noise levels in various distances from the source. Table 9.1. Input parameters for calculation of noise levels in Figure 9.3.
Figure 9.3 shows that the noise level is quite high close to the source but decreases significantly within the first 100 m. At the distance of 300 m from the source the noise level is 38 dB(A) (near to the background noise level of the area). The number of people annoyed by the noise from a specific source may vary as people move between the noise isobars as well as in and out of the area influenced by the noise (see Figure 9.2). However, average considerations taking the general population density ( pop) into account provide a useful estimate of the number of people, Pn in each isobar around a noise source and are used in the present model. Hence the number of people in the n'th isobar is
Assuming as an example that the population density is 55 people pr. km2 around a noise source with no one situated closer to the source than 13 m, Figure 9.4 shows the average number of people within 10 meter circular rings (isobars) at various distances from the noise source.
Figure 9.4. Example of average number of people in 10 meter wide circular rings at various distances from a noise source. ( pop = 55 pr. km-2) The total noise nuisance caused by a specific process NNproc can be determined by summarising the nuisance on all persons within each isobar sector as shown in Formula 9.4.
Where Lp(d) is the noise level at the distance d from the noise source. The total noise nuisance NNprod from a product or a service can be determined by adding all nuisance contributions from all processes in the entire lifecycle of the products or the services.
Although some people located in the area influenced by the noise are actually situated in buildings, it is assumed in the present model that all noise perceived by humans is perceived outside. Thus, the noise level in various distances from the source is calculated as "outdoor-levels" and the noise nuisance is presented as a "noise nuisance impact potential" (NNIP). 9.3 TransportationTransportation by truck and train are generally quite noisy and the noise nuisance from these processes must be quantified. However, two aspects of the transportation processes complicate the quantification of the noise nuisance.
9.3.1 General approach to noise from transportationBecause the noise source is moving during the transportation process, the number of people who are hearing the noise changes continuously and the noise level experienced by individual persons changes as shown in Figure 9.5.
Figure 9.5. The noise level experienced by a person who is passed by the transportation unit. A person's experience of the noise level is low when the transportation unit is far from the perceiving person and increases up to a maximum when the person is passed. After that the noise level decreases to the level of the background noise as the transportation unit moves out of hearing. In the present model it is assumed that
Based on these assumptions, the noise nuisance impact potential from a moving transportation unit can be determined by Formula (9.4) as if the noise source was standing still. Figure 9.6 shows the noise isobars around a moving truck on the motorway. The figure illustrate that even though the truck is moving, the same average number of people are influenced by the same amount of noise as long as the landscape, the population density and the noise remain the same.
Figure 9.6. Example of noise isobars around a truck (o) on the motorway (dark grey). Light grey colour illustrates the road border, (x) represents the population density and rings represent noise isobars. Since the noise nuisance impact potential may vary significantly during a transportation process from A to B it can be useful to compose a transportation scenario with different transportation types, different population densities and different acoustic conditions as illustrated in Table 9.2. Table 9.2. Examples of transportation types during transportation from A to B by a truck.
Table 9.2 suggests that the transportation type can vary significantly even during a simple transportation with the same transportation unit and that the most important transportation types must be included to give a true impression of the noise nuisance impact potential. However, the degree of detail must always be determined according to the scope definition of the actual LCA. 9.3.2 Truck transportationThe noise emitted from trucks depends on the size of the truck, the transportation speed, the road surface and the general condition of vehicle. The noise-LCA model is implemented in a spreadsheet model (LCA-noise.xls) which as input parameters takes three different truck weight categories, five different types of residential areas and three different road types. The model can be downloaded from the homepage of the Danish EPA (www.mst.dk). Different noise propagation set-ups are used for calculations of noise levels in each type of area. Each set-up contains typical average parameters like minimum distances to the nearest houses, distances between houses and height of houses. The traffic noise propagation model then calculates the shielding effect and the damping due to terrain hardness in various distances from the road. Table 9.3 provides three examples of the input parameters from the transportation of 5 kg product in three different types of trucks each one driving on different types of road through different types of areas. Table 9.3. Examples of input parameters for model calculation of noise nuisance from three different trucks carrying cargo through different areas. Parameters marked with * are default in the model.
1) See (Vejdirektoratet, 1996). 2) See Annex 9.2. 3) See (Vejdirektoratet, 1998), 4) See Anonymous, 1989, Lydteknisk Institut, 1984 and sheet about background noise. Output from the noise model will be presented and discussed in Section 9.3.4. 9.3.3 Railway transportationThe sound emission from train transportation comes partly from the interaction between rail and wheels and partly from the locomotive. At high speed (>80 km/h) the noise from the rail and wheels contribute mostly to the noise emission while at low speed (<30 km/h) the noise from the locomotive is dominant. Table 9.4 shows the input parameters for noise nuisance calculations in the model at the transportation speed of 100 respectively 60 km/h. Table 9.4. Example from a case where a train passes through different landscapes. Parameters marked with * are default in the model.
1) Nielsen, 1999. 2) See Annex 9.2. 3) See Anonymous, 1989, Lydteknisk Institut, 1984 and sheet about background noise. 9.3.4 Output from spreadsheet modelThe output generated by the LCA-noise spreadsheet model is the noise nuisance impact potential, NNIP caused by the transportation of the product. Table 5 shows the result from the above-mentioned cases given the input parameters from table 3. The greatest contribution comes from the van driving on highway through countryside where the background noise level is low. Although the large truck is the noisiest and although it passes through the most densely populated area its contribution to the noise annoyance is small. This is partly due to the shielding effect of the houses close to the road and partly because of the higher background noise in inner city areas and the greater load capacity of the truck. Table 9.5. Example of outputs generated by LCA-noise model. Input parameters to the model are listed in Table 3.
The noise nuisance factor (NNFLp) in formula (1) is a unit less parameter, and hence the noise nuisance impact potential calculated in the model comes out in the unit "person seconds", which can be interpreted as a "number of persons annoyed by the noise in a certain time". It may surprise that the calculated noise annoyance is relatively small for all the three types of road transportation. Hence, it should be noted that the calculated noise nuisance impact potential (NNIP) is only calculated for transportation of 5 kg cargo. The noise nuisance impact potential for the whole transportation unit is 300 times higher for the small van (3 t) and 2000 times higher for the big van (20 t) with the actual payload of 50%. 9.4 DiscussionThe present report presents a method to calculate noise nuisance from transportation of cargo by road and railway. With a number of modifications the same principles can be used for noise nuisance calculations for other noise sources such as industry, cargo loading, construction work and transportation with ship or aeroplane. The sound propagation model for road traffic noise is in this paper applied on train noise as well. At low speed the sound propagation does not differ significantly from that of a moving truck but at higher speed this method needs to be modified with respect to calculation of noise propagation zones and data for the noise emission relative to the train length. In the present model, daytime levels of background noise are used as standard parameters when calculating the noise nuisance. Thus, if a certain transportation process actually takes place in the night-time, the actual noise nuisance is slightly higher than calculated in the standard mode of the model. However, it is possible in the present model to enter another background noise level and the calculations can thus be adapted to the actual time of the transportation. Since much transportation of goods actually takes place during the night time (especially in the countryside), it would be desirable if standard night transportation scenarios would be addressed in future studies. 9.5 References:Anonymous: Noise considerations in construction of new roads (in Danish) Vejregel 2.30.02, June 1989. DS/ISO: Acoustics - Description and measurement of environmental noise - Part 1: Basic quantities and procedures. DS/ISO 1996/1. March 1983. Lydteknisk Institut: Acoustic warning (in Danish). Report no. 116, 1984. Miljøstyrelsen: External noise from companies (in Danish). Miljøstyrelsens vejledning nr. 5/1984. Miljøstyrelsen: Calculation of external noise from companies (in Danish). Miljøstyrelsens vejledning nr. 5/1993. Nielsen, J.S.: Correspondence with John Skafte Nielsen, DSB. 07. Sept.1999. Nordic Council of Ministers: Road Traffic Noise – Nordic Prediction Method. TemaNord 1996. Vejdirektoratet : Routes to less noise (in Danish). Report no. 80, 1999. Vejdirektoratet: Road traffic and noise – basic concepts (in Danish) Report no. 146, 1998. Vejdirektoratet: Velocity report – velocities on the main roads 1992-1994 (in Danish). Report no. 49, 1996. Wenzel, H., Hauschild M.Z. and Alting, L.: Environmental assessment of products. Vol. 1 - Methodology, tools, techniques and case studies, 544 pp. Chapman & Hall, United Kingdom, ISBN 0 412 80800 5, Kluwer Academic Publishers, Hingham, MA. USA., 1997. Annex 9.1 Assessment and regulation of noiseSound pressure. The central parameter to measure sound is the sound pressure p (in Pascal) at a given point. The sound pressure can vary in time, in strength and in frequency composition. It is measured with a microphone and a sound level meter. For this purpose several measuring methods and -equipment can be used. The sound pressure level is defined as Lp = 20log (p/p0) where p0 is a reference sound pressure 20 µPa, which is the smallest detectable sound pressure for humans. The unit is deciBel or dB relative to the reference p0. When measuring noise a spectral filter - an A-weighting filter - is commonly used. A-weighting. The A-filtering assures that only the sound level frequencies where the human hearing is good is dealt with in the evaluation of the measurement. The shape of the filter is similar to the inverse of the human hearing threshold. Thus noise signals having the same A-weighted sound pressure level will also have approximately the same subjective hearing level - irrespective on their frequency composition. If the assessment of noise nuisance to animals should ever come in question, another frequency filter shape should be used since animals hearing threshold can look very much different from ours. Sound power level is a source specific acoustic parameter, which is commonly used as an objective way of describing noise sources since it is independent of time and place. The reference quantity is 1 picoWatt or 1 pW (10-12 W). Sound power level is derived from sound pressure level as LW = Lp + 10log (S) where Lp is the sound pressure level at a measurement surface surrounding the source and S is the surface area in m2. The sound power level is measured in dB relative to 1 pW. Example: A point sound source is emitting sound equally in all directions and is placed freely on the ground. The wave front surface is then a hemisphere in which the sound pressure is the same everywhere along the surface. The sound power for the source is then calculated as
The corresponding A-weighted levels are called LWA and LpA. Also see Miljøstyrelsen, (1993) and DS/ISO (1983). Predicting sound pressure level from several sound sources. When predicting sound from one or more sound sources - each with a given sound power Lw - in a noise sensitive spot e.g. at the neighbour of a industrial plant, the sound reduction (due to distance, shielding from buildings etc.) from each source is calculated to this receiving point (see Miljøstyrelsen, 1984). Each noise source contributes to the total sound pressure level with L p = L W - 10•log (S) - F, where 10•log (S) is the divergence correction factor and F is a factor depending on the obstacles on the sound's path to the receiving point. S is the surface area of hemisphere with a radius equal to the distance from each source centre to the receiving point. The total sound pressure level at the receiving point is finally calculated by logarithmically adding each contribution from n sources:
Mapping of traffic noise. Normally the method for calculating noise from road traffic is to regard the traffic noise as coming from an indefinitely long straight line, a so-called line source. The sound emission from a line source consists of a mean average of sound from several sources passing a given point over a time period. However when dealing with noise from one single source emitting to a whole area, we have to use a very different approach since
In order to consider the impact of the single moving vehicle on the environment we have introduced a method based upon a co-ordinate system that follows the moving vehicle. The vehicle is then assumed to be a point source in the centre of this co-ordinate system emitting spherical sound waves in all directions. For simplicity the co-ordinate system is considered to pass through different types of uniform landscapes with uniform distribution of inhabitants. The sound propagation is thereby the same within each type of landscape. The sound field from the vehicle is regarded as that of a point source. Area of influence. Point source. The influence area from a point source is a circle with a radius of d, which is the distance from the road centre of the vehicle to the furthest sound zone.4 The number of influenced people depends upon the population density in the areas that the vehicle passes through. These areas are chosen so that the population density and the shape of the landscape are reasonably constant within the area. For a point source the number of people in the n'th noise zone within an area is
where d i is the distance from the road centre to the n'th noise zone and σpopulation is the population density of the area (P of peoples per km2). The influence area is local in the sense that it is restricted to the point source circular surroundings. It does not cover the whole geographical area of the route of the vehicle. The total area of influence is then found by time weighing and adding the number of noise influenced persons for each type of area according to the duration of the vehicle passing through an area. Line source. The sound field along a road, where many vehicles pass, has the shape of a cylinder. The influence area from line sources is a rectangular area covering both sides of the road, i.e. 2 d multiplied with the transport distance, where d is the distance from the road centre line to the furthest sound zone. For 1 km of road in a population area the number of influenced people is
The total area of influence for the whole route is found by weighing and adding the number of noise influenced peoples for each type of population area according to the length of the area. Hence
where P j is the number of influenced people in the j'th area and D j is the length of the corresponding area. Sound propagation model. In this paper the sound propagation model "Road Traffic Noise - Nordic Prediction Method" (Nordic Council of Ministers, 1996) is used for calculating the equivalent sound pressure level LAeq in various distances form the road centre line. In (Nordic Council of Ministers, 1996) the road traffic is considered to be an infinitely long, straight-line source emitting constant noise in cylindrical sound waves. The passages of individual vehicles are not dealt with directly in (Nordic Council of Ministers, 1996). The formulas for maximum sound pressure level is comparable to the sound propagation from a point source and is in this paper used to describe noise from the passing a single vehicle. The validity of the method is restricted to distances up to 300 meter. LAeq is calculated from the following input parameters:
Noise nuisance. People living in the above described noise influenced areas do not all experience the same amount of noise since the sound pressure decreases with the distance from the road, shielding objects, height above terrain etc. Secondly the human ear does not perceive noise levels linearly in frequency but "A-filtered" and thirdly the nuisance experienced from a given noise level does not depend linearly of the level of the noise (Anonymous, 1989). A common used method of calculating noise annoyance in areas near to road (or railway) traffic lines is to estimate the number of inhabitants in a certain area and calculating how many of these that is exposed to certain noise levels. In Denmark the concept SBT (noise load figure, which in this paper is referred to as noise nuisance, NN) is widely used in traffic planning. SBT incorporates noise nuisance i.e. the fact that noise in different distances from the road causes different levels of nuisance for the people living close to and further away from the road. SBT contains an empirically found nuisance factor that weighs the calculated noise level according to the human hearing perception. The noise nuisance factor is
where LAeq, i is the A-weighted, energy equivalent sound pressure level in decibel (dB) in the i' th noise zone. K is 41 dB for outdoor noise in all-year residential areas. For summerhouses K = 36 dB because of the lower background noise in summerhouse areas. K = 16 dB for indoor noise. The noise nuisance factor is dimensionless. It is valid down to LAeq = 45 dB. Normally SBT is based upon 24-hour energy mean level of traffic noise but since we consider noise impact from single vehicles we will in this paper consider the nuisance factor to be applicable when using maximum sound pressure level of a single vehicle passing. The sound pressure LAF,max, 10m 10 meters from the road centre line is therefore used as a basic input parameter. The total noise nuisance caused by the road traffic in a certain area is calculated by multiplying the noise nuisance factor with the number of people in each noise zone and summarising all the noise zones
where P i is the number of people living in the i' th noise zone. The lower limit for summarisation of noise zones is set to 45 dB. Finally the noise nuisance for the whole route is found by summarising NN for all the different areas that the vehicle passes through. Hence
Annex 9.2 Estimation of average population densities in three different area categories in Denmark
Reference: Danmarks Statistik, kommuner i tal (http://www.dst.dk/siab.asp?o_id=11). Approximate average population density, pers./km2
Footnotes[1] Institute of Product Development (IPU) in Denmark until 2000, presently at the Center for Energy and Environmental Studies IVEM, University of Groningen [2] Institute of Product Development (IPU) in Denmark [3] Evaluation of threshold exceedance involves the comparison of a predicted environmental concentration (PEC) with a predicted no-effect-concentration (PNEC). There is anticipated risk of an effect when PEC/PNEC1, but absence of risk if PEC/PNEC<1. A typical LCA does not predict environmental concentrations, but increases of the environmental concentration (PEC). The comparison of this increase with a no-effect-concentration (PEC/PNEC) in LCA is performed only to allow aggregation of the contribution from different substances to toxicity (see also Footnote 2), and not with the aim to evaluate threshold exceedance. [4] The comparison in LCA of a concentration increase with a no-effect-concentration (PEC/PNEC) is performed only to allow aggregation of the contribution from different substances to toxicity (see also Footnote 1). The underlying assumption is that the toxicity impact from a quantity at the no-effect-concentration of a substance has the same importance as the toxicity impact from a quantity of another substance at the no-effect-concentration. In other words: If the quantities of both substances are at their no-effect-concentration, the impacts from a neuro-toxic substance and a skin irritating substance are regarded as equally important. Adding together completely different effects is one of the more serious problems in LCA, but not further addressed in this technical report. [5] This does not apply for categories as ozone depletion and the increased radiative forcing in global warming where substances distribute globally and therewith exert their impact globally. [6] Institute of Product Development (IPU) in Denmark until 2000, presently at the Center for Energy and Environmental Studies IVEM, University of Groningen [7] Institute of Product Development (IPU) in Denmark [8] This can also be deduced from Chapter 5. While benzene has less than 10% of its impact within the first 10km from the source, a relatively short-lived substance as hydrogen chloride has about 40% within the first 10km and almost 80% within the first 75km. However, at a moderate release height of 25m this short-lived substance will still have less than 10% of its impact within the first hundred metres from the source where the peak concentration occurs. Also a short-lived substance thus has most of its impact at longer than very local distance. This makes a site-dependent approach achievable also for these substances, though a higher spatial resolution is needed and the uncertainties will be larger (as also appears from Chapter 5). [9] The term "exposure" covers the same as the term "target" in Chapter 4, but has been preferred here to draw the connection with publications of Heijungs and Wegener Sleeswijk (1999) and Udo de Haes et al. (1999) that follow a similar line of reasoning. [10] The exposure increase in any receptor resulting from an emission is usually small or marginal compared to the background exposure in that receptor. The background exposure therefore determines whether a threshold value will be exceeded by an exposure increase. It is an ongoing discussion whether threshold exceedance should be accounted for in LCA. [11] No-effect-levels are based on experiments on test-species under laboratory conditions and therefore say something about the intrinsic substance characteristic to cause toxic effect (rather than something about the sensitivity of a species in real-life for this toxic substance). [12] The underlying assumption is that the toxicity impact from a quantity at the no-effect-level of one substance has the same importance as the toxicity impact from a quantity at the no-effect-level of another substance. To put it more clearly: If the quantities of both substances are at their no-effect-level, the impacts from a neuro-toxic substance and a skin irritating substance are regarded as equally important. The adding together of very different effect types is one of the more serious problems in LCA, but has not been further addressed in this dissertation. [13] "PAL" stands for predicted acidifying load (similar as to predicted environmental concentration in toxicity assessment). [14] Hence the impact area and the territory of the selected population are not the same. This is the logic consequence of Danish emissions having transboundary impact. [15] Actually, the definition of such impact area is not without problems. It is not really possible to isolate an area over which an impact extends, from an area in which this impact is not at stake. This creates arbitrariness in the normalisation factor. [16] Institute of Product Development (IPU) in Denmark until 2000, presently at the Center for Energy and Environmental Studies IVEM, University of Groningen [17] International institute for Applied Systems Analysis (IIASA) in Austria [18] University of Utrecht, Department of Science, Technology and Society, the Netherlands [19] Institute of Product Development (IPU) in Denmark [20] The most extreme exception is Bulgaria, where two power plants determine 45% of total sulphur dioxide emission (EEA 1996), while the contribution from Bulgaria to sulphur deposition on its own area is 55% (Barret and Berge 1996). This means that these two power plants contribute almost 25% to total deposition on Bulgaria. The first assumption holds reasonable well even for this extreme case. [21] Institute of Product Development (IPU) in Denmark until 2000, presently at the Center for Energy and Environmental Studies IVEM, University of Groningen [22] International institute for Applied Systems Analysis (IIASA) in Austria [23] Institute of Product Development (IPU) in Denmark [24] Finnveden et al. (1992) do not mention airborne emissions of phosphor. [25] All together, Finnveden et al. (1992) distinguishes five subcategories: Aggregation of airborne nitrogen emissions (terrestrial eutrophication), aggregation of waterborne phosphor emissions (and organic material), aggregation of waterborne nitrogen emissions (and organic material), aggregation of nitrogen emissions (and organic material) to water and airborne nitrogen emissions, aggregation of all phosphor and nitrogen emissions (and organic material). [26] For a comparison: Emissions of phosphor to air are approximately 130 kton per year, while phosphor emissions to water exceed 5,000 kton per year in the Netherlands (Berdowski and Jonker 1994). The atmospheric emission is thus less than 3 percent of the waterborne emission, whereas only a minor part of that airborne phosphor will reach surface water through first deposition and next topsoil erosion. [27] Each European country determines self which species have to be protected. Some countries might be more protective than others, and seem to have set stricter criteria in the critical load database more recent than the one used here. The influence by this new set of critical loads on the calculated factors is not clear. [28] For instance, a product designer should have knowledge about so many things that it may go too far to ask such additional data gathering from him. [29] Institute of Product Development (IPU) in Denmark until 2000, presently at the Center for Energy and Environmental Studies IVEM, University of Groningen [30] The Dutch National Institute of Public Health and Environmental Protection (RIVM) [31] The Danish Technological Institute (DTI) [32] The Dutch National Institute of Public Health and Environmental Protection (RIVM) [33] Institute of Product Development (IPU) in Denmark [34] Main uses being for public water supply, irrigation, industrial processes and cooling, hydroelectric power generation (inland freshwaters), and transport and waste disposal (both coastal and inland freshwaters). [35] Samuelsson lists also other ratios for carbon, nitrogen and phosphor reported in literature. This list shows considerable differences in nitrogen and phosphor ratios, the lowest being 5:1 and the highest being 19:1. However, those ratios are based on samples also containing organic matter originating from other organisms than phytoplankton. Therefore, the Redfield ratio is followed here since it is suggested to be valid for phytoplankton. An alternative might be, as recently suggested by Seppälä (2000), to employ a range of 13 to 19 for nitrogen versus 1 for phosphor around the Redfield ratio. This is not further elaborated here. [36] Terrestrial ecosystems are usually not limited by phosphor, and airborne phosphor has no role worth mentioning in eutrophication of surface waters. For a comparison: Emissions of phosphor to air are approximately 130 kton per year, while phosphor emissions to water exceed 5,000 kton per year in the Netherlands (Berdowski and Jonker 1994). The atmospheric emission is thus less than 3 percent of the waterborne emission, while only a minor part of that atmospheric phosphor will through topsoil erosion reach surface water. [37] Similar to Wenzel et al. (1997), Finnveden et al. (1992) also propose to separately aggregate nitrogen and phosphor. They suggest in addition to report airborne and waterborne nitrogen both separately (maximum scenario for terrestrial eutrophication) as well as summed together (representing a maximum scenario for aquatic eutrophication). All together, Finnveden et al. (1992) distinguish five subcategories: Aggregation of airborne nitrogen emissions (terrestrial eutrophication), aggregation of waterborne phosphor emissions (and organic material), aggregation of waterborne nitrogen emissions (and organic material), aggregation of nitrogen emissions (and organic material) to water and airborne nitrogen emissions, aggregation of all phosphor and nitrogen emissions (and organic material). [38] Eutrophication and nutrient enrichment may thus be taken as synonyms. Other terms sometimes used are "nutrifcation" and "oxygen depletion" and refer to the same group of impacts as covered by eutrophication. The term eutrophication is chosen here and refers in this chapter to aquatic eutrophication only (see Chapter 4 for terrestrial eutrophication). [39] Whereas nature is the predominant source for other nutrients, supply of nitrogen and phosphor largely originates from anthropocentric sources. Therefore, these nutrients are the only ones being amenable to control. This is an additional reason for those nutrients to be important for "chronic" nutrient limitation. [40] CARMEN does not explicitly address eutrophication of lakes, but lakes will usually be part of a river catchment and are thus implicitly covered by CARMEN. [41] The eutrophication factors calculated for rivers are therefore assumed to cover all inland waters (both rivers and lakes thus). This is defensible because the factors represent maximum potentially biomass growth. The maximum potential biomass growth will be larger than the actual expected occurrence of biomass growth both in rivers and lakes. [42] Similar differences probably exist for phosphor, but this is hardly visible because of the small shares. [43] If the emission is known for actually being released to either inland waters or to a coastal sea, this information should of course be used in stead of the factors from Table 5.5. Hence the emission of nitrogen should be multiplied with a factor 0.7 to account for nitrogen leaving the system after denitification. [44] Institute of Product Development (IPU) in Denmark [45] National Environmental Research Institute (NERI) in Denmark [46] International institute for Applied Systems Analysis (IIASA) in Austria [47] Institute of Product Development (IPU) in Denmark until 2000, presently at the Center for Energy and Environmental Studies IVEM, University of Groningen [48] Stuttgart University, Institute of Energy Economics and the Rational Use of Energy, Germany [49] Danish Toxicology Institute (DTC) [50] National Institute of Public Health and the Environment (RIVM), the Netherlands [51] Institute of Product Development (IPU) in Denmark [52] The framework applies to human toxicity assessment from emissions to outdoor air since toxicity assessment from indoor emissions needs a different kind of modelling and is not elaborated here. [53] Section 7.2.3. through Section 7.2.7 are, almost without changes, taken from Potting et al. 2000) [54] Guinée et al. (1996) and Huijbregts (1999) do unfortunately not provide the possibility to check above estimates since they report only factors where all types of exposure are aggregated per emission type. [55] Section 7.2.3. up to an including Section 7.2.7 are, almost without changes, taken from Potting et al. 2000) [56] The data have been received in 3 separate files that due to the structure of each file unfortunately are impossible to merge in an unique way into one data file. This has complicated the analyses because the results become somewhat arbitrary, especially when the number of observations is very small and some emission points relate to more than one type of process, while others relate more than once to the same process type. The covariance existing between the data about process type and the height of release could have been avoided by doing the analysis on the aggregate level of industrial sectors (ignoring a further breakdown in processes). The loss of information for the interpretation of the results was in our opinion, however, not in balance with the moderate gain of precision in the results. [57] The standard deviation and range unfortunately got lost. Given the results for individual sectors, however, they are expected to be rather large. [58] Actually, regulatory standards for evaluation of occupational situations were often used at this stage. Though also influenced by other aspects, those regulatory standards take often their basis in no-effect-concentrations. No-effect-concentrations are based on experiments on test-species under laboratory conditions and therefore say something about the intrinsic potential of a substance to cause toxic effect (rather than something about the sensitivity of a species in real-life to this toxic substance). [59] The adding of completely different human toxic effects is one of the more serious problems in life cycle assessment. Since this was no part of the methodology and consensus research project, the issue is not further addressed in this chapter. [60] This section is to a large extent taken over from Potting et al. 2000) [61] The urban exposure levels are declining in Europe (EEA, 1997). It is assumed that the air concentration levels of NO2, CO, benzene and to a minor extent particles will drop even further after introduction of new and more efficient three-way catalysts in gasoline-powered vehicles (EEA, 1997). [62] Point sources are usually regulated in most developed countries. Exposure levels and thereby health hazards in the vicinity of regulated point sources are therefore in most situations eliminated or considerably reduced (Potting and Hauschild, 1997). However, as also illustrated later on in the paper, significant exposures may still be encountered in some situations. [63] Co-operative Program for Monitoring and Evaluation of the long range transmission of air pollutants in Europe. [64] The urban exposure levels are declining in Europe (EEA, 1997). It is assumed that the air concentration levels of NO2, CO, benzene and to a minor extent particles will drop even further after introduction of new and more efficient three-way catalysts in gasoline-powered vehicles (EEA, 1997). [65] Point sources are usually regulated in most developed countries. Exposure levels and thereby health hazards in the vicinity of regulated point sources are therefore in most situations eliminated or considerably reduced (Potting and Hauschild, 1997). However, as also illustrated later on in the paper, significant exposures may still be encountered in some situations. [66] Corinair94 operates with 10 sector sources: 1. Combustion in energy and transformation industries, 2. Non-industrial combustion plants, 3. Combustion in manufacturing industry, 4. Production processes, 5. Extraction and distribution of fossil fuels/geothermal energy, 6. Solvent and other product use, Road transport, 8. Other mobile sources and machinery, 9. Waste treatment and disposal, 10. Agriculture and forestry, land use and wood stock change. [67] WHO used to operate with guidance values (NB! for combined exposure to particles and SO2) (WHO/UNEP, 1992):
[68] I.e. guidelines exceeded by more than a factor two. [69] I.e. guidelines exceeded by up to a factor two and short term guidelines regularly exceeded. [70] I.e. short term guidelines exceeded occasionally. [71] NMVOCs: non-methane volatile organic compounds [72] In Europe VOC's are to be further regulated by a new VOC -directive under way in the EU (EC, 1998). [73] The labelling scheme has been criticised for not including carcinogenic effects, which, however, may be introduced in connection with a revision (Wolkoff et al., 1998). [74] It is not stated whether or not these measurements have been conducted before or after the introduction of vapour collecting devices. [75] No air guidance value has been encountered, but an estimate of 23 g/m³ can be derived from the TDI assuming: 64 kg body weight, inspiration of 22 m³ air per day and the same bioavailability/uptake via oral and inhalation exposure routes. Especially the latter assumption may be questioned. The value should therefore be used with caution! [76] No air guidance value has been encountered, but an estimate of 0.47 g/m³ can be derived from the TDI assuming: 64 kg body weight, inspiration of 22 m³ air per day and the same bioavailability/uptake via oral and inhalation exposure routes. Especially the latter assumption may be questioned. The value should therefore be used with caution! [77] Danish Hydraulic Institute, DHI [78] Institute of Product Development (IPU) in Denmark [79] Institute of Product Development (IPU) in Denmark [80] dk-TEKNIK, ENERGY & ENVIRONMENT
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
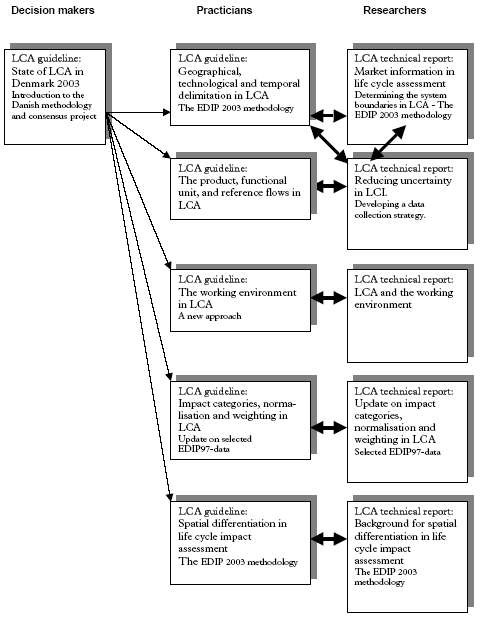
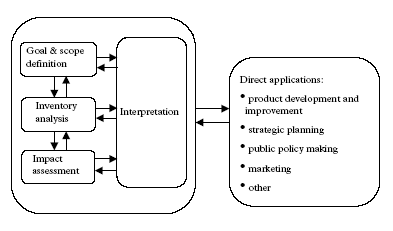




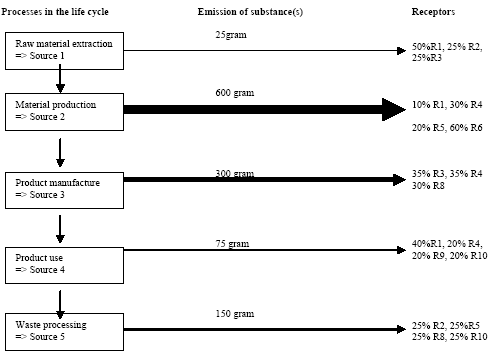




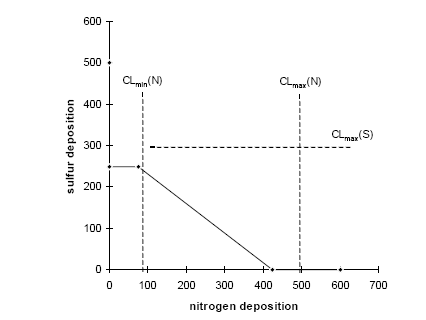
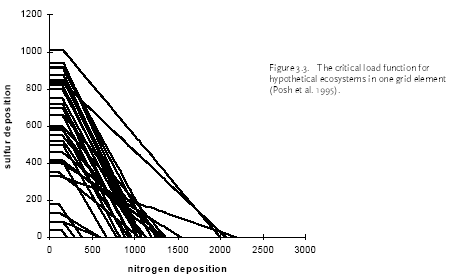
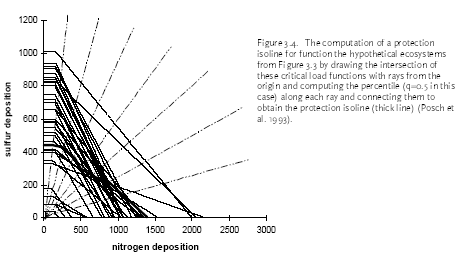
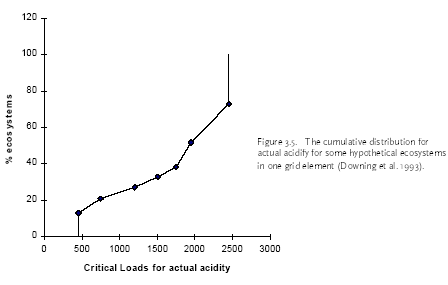


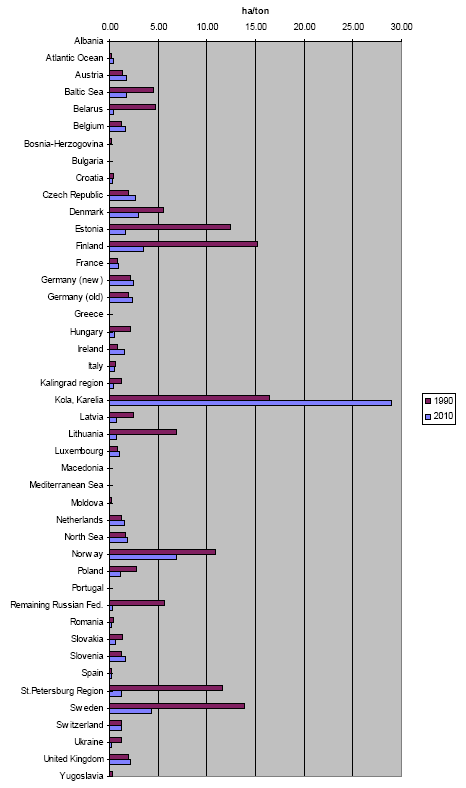
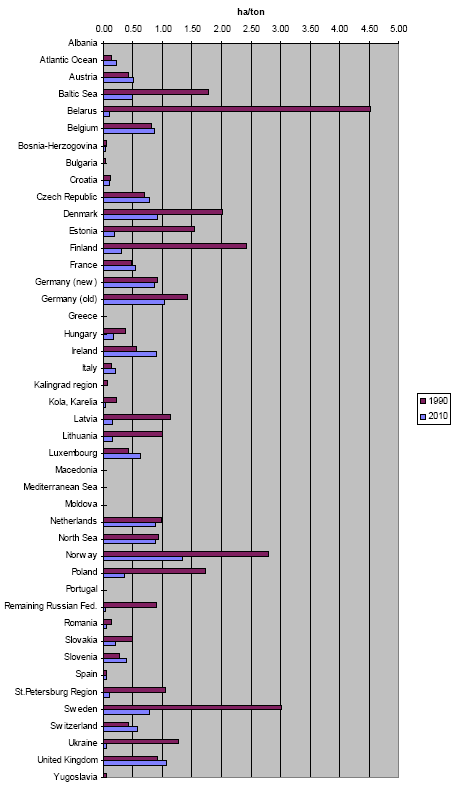
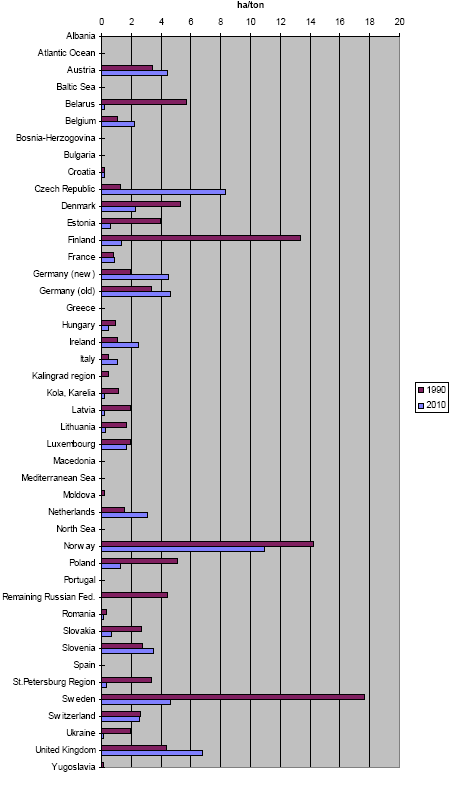
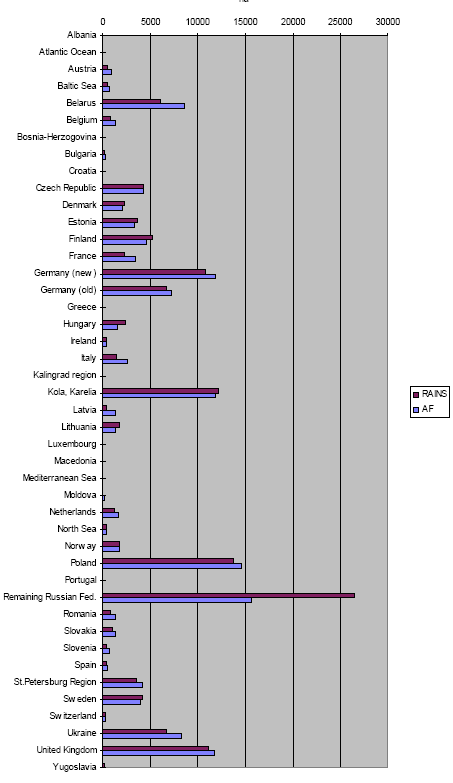
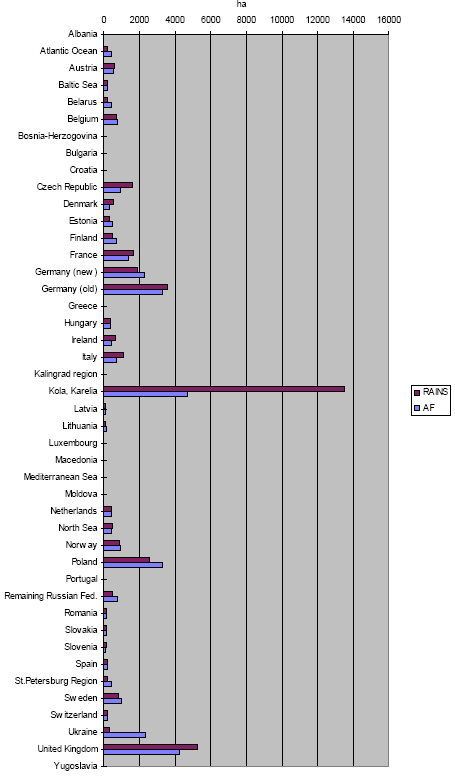
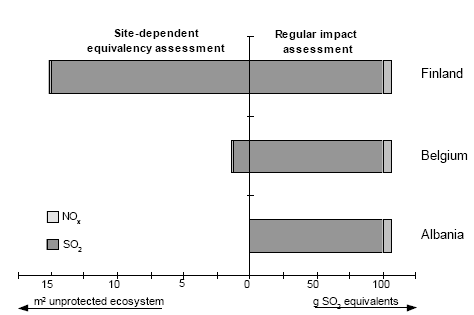
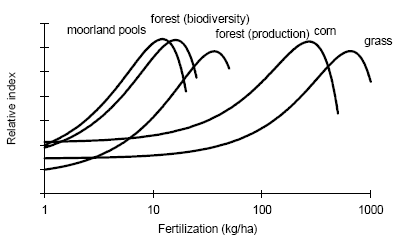
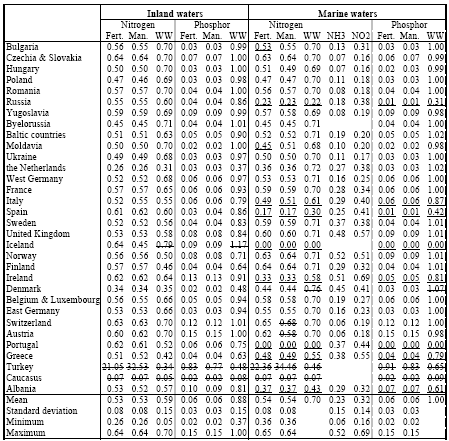


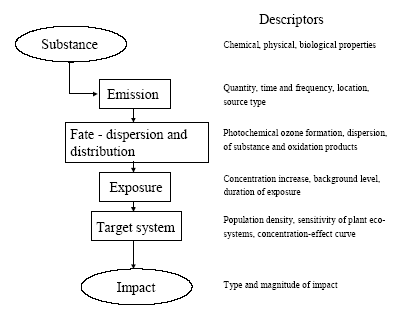
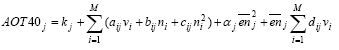
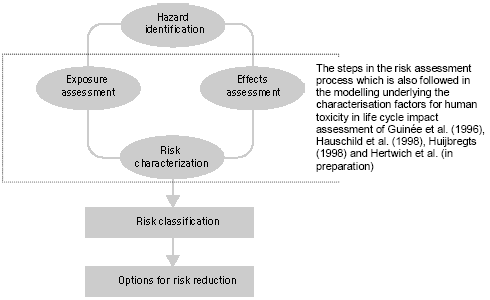
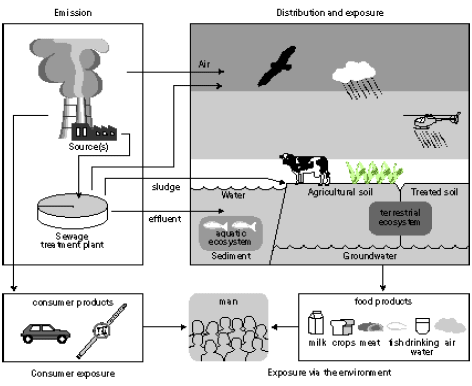
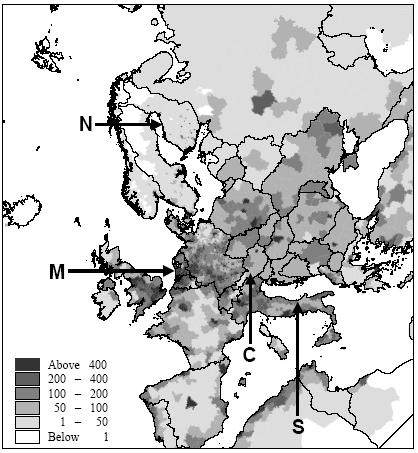
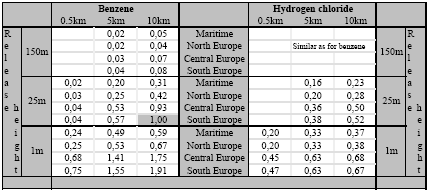
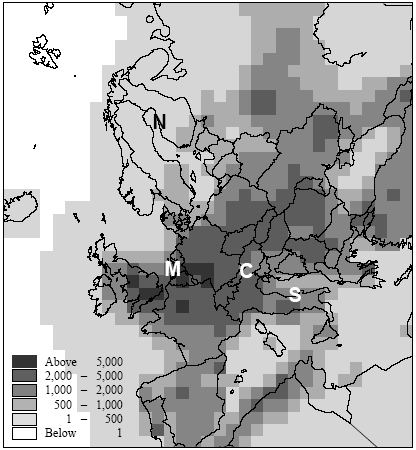
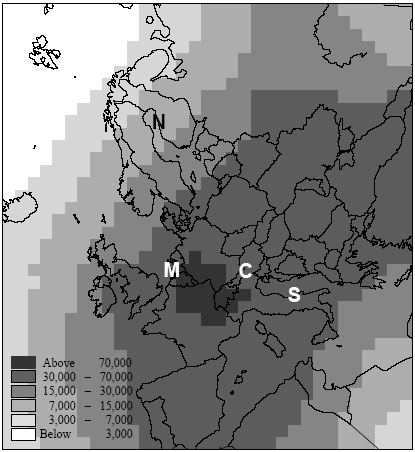

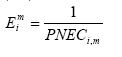
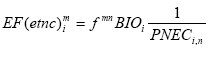
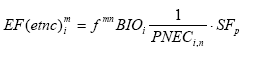
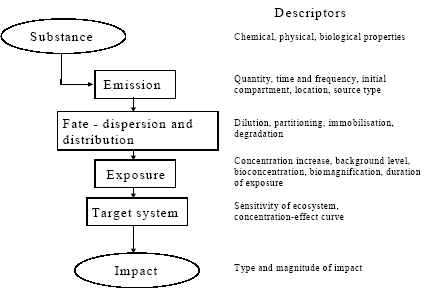
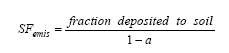

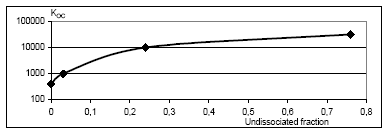
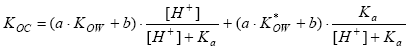
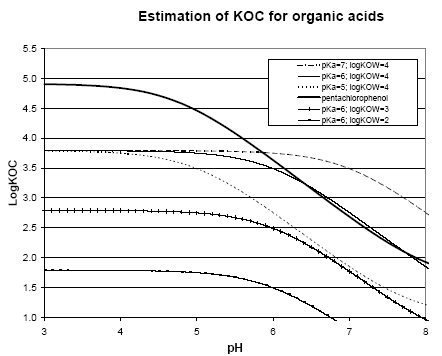
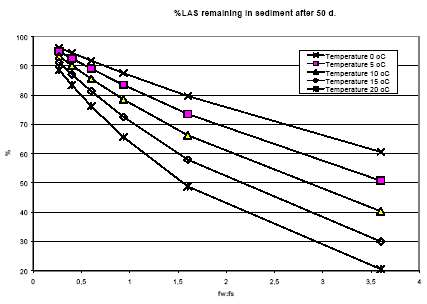
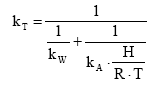
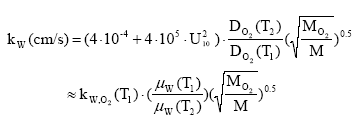
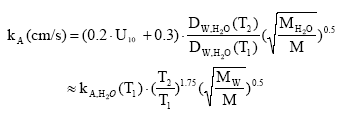
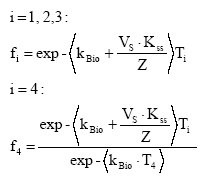
![Figure 9.1. Relation between noise pressure level, Lp [dB(A)] and nuisance factor NNF Lp for traffic noise, (Anonymous, 1989)](images/280.gif)