Miljøprojekt, 579; Teknologiudviklingsprogrammet for jord- og grundvandsforurening
Udvikling af metode til testning af udvaskning af organiske stoffer fra jord og restprodukter
Indholdsfortegnelse
Forord
Som led i jordlovsarbejdet og i forlængelse af udarbejdelsen af en ny bekendtgørelse om genanvendelse af restprodukter og jord til bygge- og anlægsarbejder har Miljøstyrelsen under Teknologiudviklingsprogrammet i 1998 og 1999 iværksat et projekt om udvikling af metode til testning af udvaskning af organiske stoffer fra jord og restprodukter. Projektet er udført af DHI – Institut for Vand og Miljø i samarbejde med Peter Kjeldsen, Institut for Miljøteknologi, med Ole Hjelmar, DHI, som projektleder.
Projektarbejdet har været styret af en arbejdsgruppe bestående af Povl Rasmussen (formand) og Lone Kielberg, Miljøstyrelsens Erhvervsaffaldskontor, Kim Dahlstrøm fra Miljøstyrelsens Jordforureningskontor, Peter Kjeldsen fra Institut for Miljøteknologi, DTU, Ole Hjelmar og Peter E. Holm fra Afdelingen for Grundvand, Affald og Mikrobiologi, DHI.
Rapporten er udarbejdet af Peter E. Holm, Kim Broholm, Peter Kjeldsen, Karen G. Villholth, Ole Hjelmar og Olaf W. Asmussen.
Sammenfatning og konklusioner
Som et led i det omfattende jordlovsarbejde er der udsendt en ny bekendtgørelse om genanvendelse af restprodukter og jord til bygge- og anlægsarbejder. Princippet for reguleringen/kravværdierne er som noget nyt baseret på en regulering af jordens og restprodukternes udvaskelige mængde af spormetaller og salte bestemt under nærmere specificerede omstændigheder. Erfaringerne med testning af udvaskningen af organiske stoffer fra forurenet jord og restprodukter er små, og der eksisterer kun få veletablerede udvaskningstests til undersøgelse af udvaskningen af organiske stoffer. Med henblik på at tilvejebringe det fornødne grundlag er der gennemført et projekt, som har haft til formål at vurdere primært de tekniske muligheder for at anvende udvaskningstests til karakterisering og klassificering af udvaskningen af organiske stoffer fra forurenet jord og restprodukter.
Projektet har taget udgangspunkt i det arbejde, der blandt andet er udført i Miljøstyrelsens regi vedr. organiske stoffers opførsel i jord og grundvand. Der er etableret en oversigt over "state of the art" med hensyn til de processer og faktorer, som er styrende for udvaskningen/desorptionen af organiske stoffer fra forurenet jord og restprodukter. Ligeledes baseret på sammenstilling af viden og eksperimentelle erfaringer er der foretaget en vurdering af anvendelsesbetingelser og begrænsninger for forskellige testmetoder og testprincipper i relation til udvaskning af organiske stoffer fra jord og restprodukter.
Gennem opdeling af relevante organiske stoffer i grupper efter deres fysisk/kemiske egenskaber i relation til stoffernes fordeling i et kombineret luft-vand-jord system er der foretaget en klassifikation af stofferne i forhold til mulighederne og relevansen af at teste deres udvaskning fra jord og restprodukter.
Endelig er der på baggrund af den indsamlede og behandlede viden og den gennemførte klassifikation af de enkelte stofgrupper gennemført eksperimentelle afprøvninger af testmetodikker på 5 udvalgte forskellige jordprøver.
Afslutningsvis er der foretaget en samlet vurdering af den indsamlede viden, de gennemførte beregninger og resultaterne fra de eksperimentelle undersøgelser samt opstillet anbefalinger for den eventuelle videre udvikling af en testmetodik.
Gennem sammenstilling af erfaringer fra litteraturen er der identificeret en række forhold, der er centrale for den videre udvikling af udvaskningstests til forurenet jord:
| I de situationer, hvor forurenet jord og restprodukter indeholder fri fase af organiske stoffer, vil opløsningen af de organiske stoffer være styrende for udvaskningen af stoffer. I tilfælde af, at forureningen ikke indeholder fri fase, eller den frie fase er opløst, vil udvaskningen af organiske stoffer være styret af desorption fra de faste partikler i jorden/rest-produktet. |
| Opløsning fra frie og residuale faser af organiske stoffer i jord er en relativt hurtig proces (minutter/timer). Udvaskningsforløbet ved tilstedeværelse af en fri fase kan forudsiges ud fra en kendt sammensætning af den frie fase. Dette kræver imidlertid kendskab til stofferne og jorden, og sådanne oplysninger er det vanskeligt at fremskaffe i praksis. |
| Desorptionen af organiske stoffer fra jord er i mange tilfælde langsom (dage/uger). Desuden er den vanskelig at forudsige, hvilket antagelig skyldes, at den er afhængig af såvel stof- som jordtype. Specielt ses meget lave desorptionsrater for hydrofobe stoffer. Med hensyn til jordtypernes betydning er der indikationer af, at unge jordtyper er karakteriseret ved relativt høje desorptionsrater og små hystereseeffekter, mens det omvendte er tilfældet for geologisk gamle jordtyper. Ud over bestemmelser af O/C-forholdet, som stiger med jordens alder, findes der ingen umiddelbare eller veletablerede metoder til karakterisering af jordens alder. Endvidere skal det bemærkes, at topjord godt kan bestå af eller indeholde geologisk set gamle materialer. |
| Bestemmelse af jordmaterialets indhold af naturligt organisk kulstof kan benyttes til at få et generelt indtryk af prøvens forventede sorptionskapacitet. Herudover menes forholdet mellem ilt og kulstof, det såkaldte O/C-forhold at være indikator for alderen af det faste organiske kulstof i prøven. Lave værdier af O/C-forholdet indikerer, at det organiske stof er gammelt, og at der derfor kan forventes lave desorptionsrater. |
| I forhold til testning af stofudvaskningen er det centralt, at udvaskningen af organiske stoffer fra jord i mange tilfælde vil være styret af desorptionskinetik. Dette stiller direkte krav til kontakttiden mellem materialet og udvaskningsmediet og således til den endelige testprotokol for testningen. I den forbindelse bør det også overvejes, hvad der kan betragtes som feltrealistiske kontakttider i de relevante fysiske scenarier. Umiddelbart er det oplagt at udvikle testsystemer, hvor der opnås ligevægtslignende forhold mellem de organiske stoffer i faststofprøven og udvaskningsmediet. I de tilfælde, hvor tidshorisonten for at opnå ligevægtslignende forhold er lang, vil denne ikke nødvendigvis være et hensigtsmæssigt kriterium for fastlæggelse af testens kontakttid. Dette skyldes dels praktiske forhold, dels hensyn til, at de samme ligevægtslignende forhold ikke altid opnås i felten. |
| De fleste af ovennævnte forhold, som påvirker og er af betydning for udvaskningen af organiske stoffer fra jord/forurenet jord, kan forventes at have en tilsvarende betydning for udvaskningen af organiske stoffer fra restprodukter. For mange restprodukter vil det dog gælde, at indholdet af organiske forureningskomponenter er en mere eller mindre integreret del af materialet, mens de for jords vedkommende normalt er associeret med overfladen af jordpartiklerne eller porerne i disse. I modsætning til jord vil en del restprodukter reagere kemisk med vand, når de befugtes eller gennemstrømmes, hvilket f.eks. kan give anledning til mere ekstreme pH-værdier i vandfasen, end man normalt ser for jord. |
På grundlag af de indsamlede erfaringer om testtekniske forhold samt de gennemførte eksperimentelle afprøvninger af testsystemer kan der fremhæves en række forhold, som er af betydning for udvikling af en udvasknings-test, der i princip og udformning ligner den, som Miljøstyrelsen har foreslået anvendt til sporelementer og salte:
| En vis forbehandling af prøven er nødvendig som minimum af hensyn til homogenisering af prøven. En sådan forbehandling må være skånsom og bør i mange tilfælde foregå i et lukket system for at mindske fordamp-ningstabet af organiske stoffer. Dette bevirker enten, at sigtning og evt. nedknusning af prøven i praksis er umulig, eller at fordampningstabet må accepteres. |
| Væske-/faststofforholdet er helt centralt for gennemførelsen og tolkningen af resultaterne af en udvaskningstest. De eksperimentelle afprøvninger er gennemført med et væske-/faststofforhold på 2 l/kg, hvilket i mange tilfælde er nær det laveste forhold, som en simpel batchtest i praksis kan udføres ved. De praktiske vanskeligheder med partikler i systemet kombineret med behovet for store volumener af eluat til videre analysering betyder, at det lave L/S-forhold, som er benyttet, ikke har været optimalt. I de gennemførte tests har volumenet af luft (headspace) i testbeholderen været minimeret for at mindske risikoen for afdampningstab af stoffer samt for at reducere tilførslen af ilt til testsystemet under testningen. |
| Separationen af faststoffasen fra eluatet har vist sig at være et særdeles kritisk punkt i forhold til udviklingen af en metode. Til sammenligning med arbejdet vedrørende testning af udvaskningen af salte og sporelementer fra jord og restprodukter vanskeliggøres disse forhold af, at organiske stoffer i langt højere grad end de uorganiske salte og sporelementer kan være flygtige, og det er tilsyneladende vanskeligere at undgå tab af stofferne til filtermaterialer m.v. Endvidere er der generelt behov for langt større prøvevolumener til analyse for organiske stoffer sammenlignet med de uorganiske. |
| Til de gennemførte forsøg er der for at mindske udvekslingen af organisk stof med de anvendte materialer primært anvendt glas og rustfrit stål samt i mindre omfang teflon. Filtermaterialer af glas har resulteret i stoftab, men herudover har testsystemet virket efter hensigten. Det ser endvidere ud til, at sammensætningen af udvaskningsmediet, temperaturen og agitationsformen ikke har givet anledning til problemer i forbindelse med afprøvningen på de udvalgte jordprøver. |
For mange af de relevante stofgrupper findes der et godt fagligt grundlag for at arbejde videre med en testmetodik for stofudvaskningen. Dette gælder for oliestoffer, klorerede opløsningsmidler og polycykliske aromatiske hydrokarboner (PAH), mens stoffer som MTBE og DEHP i mindre grad er undersøgt. Forholdene omkring partikler i prøverne og det resulterende eluat fra testen er undersøgt (og undersøgelser er i gang), for så vidt angår PAH-forbindelser. På grund af PAH'ernes affinitet for partikler vurderes denne stofgruppe at være den væsentlige for denne problemstilling, men når den endelige form for partikelafskæring er fastlagt, bør det afklares, hvorvidt tilstedeværelsen af partikler har betydning for testningen af andre prioriterede organiske stofgrupper. I denne undersøgelse er der af praktiske årsager bl.a. brugt en partikelafskæring på 0,7 m m i stedet for de 0.45 m m, som normalt anvendes i forbindelse med udvaskning af sporelementer og uorganiske salte. Ved at anvende den større partikelafskæring reduceres problemerne med håndtering af de forholdsvis store eluatmængder, som nogle af analyserne af organiske stoffer kræver, noget.
Baseret på forenklede beregninger af fordelingen mellem faserne i et kombineret luft-/vand-/jordsystem foreslås det at klassificere de organiske stoffer, som der er fastsat toksikologiske kvalitetskriterier for i jord og grundvand, i forhold til mulighederne for og relevansen af at teste deres udvaskning fra jord og restprodukter. De foreslås inddelt i tre grupper:
| Første gruppe indeholder stoffer, der sorberer kraftigt til jorden, har en lille opløselighed i vand og er ikke-flygtige. Til denne gruppe hører de fleste PAH'er og dieselstoffer. Det er oplagt, at en metodik for testning af udvaskningen af organiske stoffer fra jord og restprodukter skal omfatte disse stoffer, der vil optræde og flyttes sammen med jorden, men i et vist omfang samtidigt udvaskes. Det vil specielt være separationen af fast fase fra eluatet, herunder hensyntagen til kolloidbundet stof og forsøgstiden, der vil være kritisk for testningen. Der er udarbejdet et forslag til metode til testning af udvaskning af ikke-flygtige organiske stoffer fra forurenet jord, hvori der ikke er foreslået nogen endelig kontakttid og ikke taget stillling til problematikken omkring kolloider. |
| Den anden gruppe omfatter flygtige stoffer med en forholdsvis høj opløselighed i vand. Stofferne i denne gruppe er bl.a. de klorerede opløsningsmidler og BTEX. Mange af disse stoffer vil det være hensigtsmæssigt at kunne teste for, da de har lave kriterieværdier i vand. En del af dem har langt højere diffusionskoefficienter i luft end i vand og vil derfor spredes hurtigere i luft. I forhold til det konkrete arbejde, hvor den potentielle anvendelse af jord og restprodukter primært vil bestå i bygge- og anlægsarbejder, vil stofferne dog sjældent udgøre et luft(indeklima) problem, da der er krav om overdækning, så kontakten afskæres. Det kritiske forhold ved testning af stofudvaskningen vil for denne gruppe af stoffer derfor være at forhindre tab af stof til luften.Ved at minimere luftvolumenet i testsystemet kan dette håndteres i en batch, da kun en mindre del af den totale stofmængde vil findes på luftform, men i forhold til håndtering af prøven før og efter testningen vil flygtigheden være vanskelig at kontrollere. Specielt separationen af faststoffasen og eluatet vil være kritisk for de flygtige stoffer og dermed testens anvendelighed. |
| Den tredje gruppe af stoffer med kriterieværdier indeholder stoffer med meget høj opløselighed eller blandbarhed i vand, og de sorberer meget lidt til jord. Til denne gruppe hører bl.a. methyltertiærbutylether (MTBE). Phenolerne tilhører også denne gruppe, idet de har en høj opløselighed i vand, men de sorberer kraftigere end f.eks. MTBE. Andre stoffer i gruppen vil kun i meget begrænset omfang kunne findes associeret med de faste faser, og det vil derfor være vanskeligt eller irrelevant at søge at beskrive deres opførsel på grundlag af udvaskningstests. Stofferne i tredje gruppe kan, i det omfang det er relevant, også testes med den ovenfor foreslåede metode. For disse stoffer vil kolloider sandsynligvis ikke være af stor betydning for transporten, da de ikke sorberer så kraftigt som stofferne i første gruppe. |
For alle prioriterede stoffer vil det være påkrævet at foretage en videre udvikling af en testmetodik, også af den i dette projekt foreslåede testprotokol. Dette vil kræve tiltag på en række af de tidligere nævnte kritiske faktorer. Af høj prioritet er udvikling eller tilpasning af metoder til analyse af stofferne med opretholdelse af de detektionsgrænser, som er gældende for analyse af grundvand. Ved anvendelse af de eksisterende metoder bevirker kravene til prøvevolumener, at testsystemerne bliver uhensigtsmæssigt store (eller mange), og at separationen af eluatet fra faststofprøverne bliver vanskelig og tidskrævende.
Det er sandsynligvis mest oplagt at gå videre med udformningen af batchforsøg, men det er ikke uden problemer, og en eksperimentel sammenligning med små kolonneforsøg vil være hensigtsmæssig. En nærmere afklaring kan nås i forbindelse med fastlæggelse af, hvordan resultaterne fra en test forventes anvendt. Som nævnt ovenfor er problematikken vedrørende separation af partikler fra eluatet central. For nogle af de mindre hydrofobe stoffer er betydningen af partikelafskæringen antagelig minimal, mens den for mere hydrofobe stoffer kan have større betydning. Det anbefales at satse på sådanne teknikker til separation af eluat fra faststoffasen, hvor anvendelse af filtre ikke er nødvendig (f.eks. centrifugering). Disse teknikker bør udvikles/afprøves sammen med udviklingen af de omtalte analyseteknikker. Det er antagelig for optimistisk at designe testsystemerne, således at de kan tage hensyn til alle potentielle stofgrupper, da de så vil blive uhensigtsmæssigt komplicerede. Testsystemet bør kunne tage hensyn til de typisk forekommende blandingsforureninger, f.eks. BTEX og tungere oliestoffer, mens mere sjældent forekommende blandingsforureninger, f.eks. klorerede opløsningsmidler og benzinstoffer, om fornødent må testes i to parallelle tests. Muligheden for at gennemføre testningen ved et højere væske-/faststof-forhold end 2 l/kg, f.eks. 5 eller 10 l/kg bør også afprøves. Denne mulighed vil alt andet lige øge volumnet af eluat og muligvis reducere problemerne med separation af eluat fra faststof. Ulemperne kan være, at koncentrationerne af de organiske stoffer i eluatet reduceres, hvilket kræver mere følsom analyseteknik, samt at det hidtidigt valgte væske-/fast-stofforhold fraviges.. Dette vil i givet fald medføre, at de forudsætninger og antagelser, der ligger til grund for det hidtidige valg af væske-/faststofforholdet, bør revurderes. Et alternativ vil være at bruge samme L/S-forhold, men benytte større beholdere. Håndtering af større eluatvolumener kan også give betydelige problemer med hensyn til filtrering og/eller centrifugering.
Agitationen kan også vise sig at have væsentlig betydning, idet den påvirker jordpartiklerne fysisk. Aggregater og belægninger kan ødelægges, således at sorptionskapaciteten ændres. Hvis f.eks. den fysiske påvirkning resulterer i, at fast organisk stof bliver mere tilgængeligt, vil desorptionen underestimeres. Omvendt vil frigivelse af kolloider kunne resultere i overestimering af udvaskningen af sorberede stoffer. Det sidste er blevet vist eksperimentelt, hvor partikler i størrelsen 0,45 – 10 m m fandtes i eluatet efter agitation, og der væsentlige mængder PAH var associeret med disse partikler. I eluatet fra søjleforsøg (som i højere grad end batchforsøg må antages at simulere virkelige forhold) blev der ikke fundet samme mængde partikler, som i batchforsøgene.
De tidligere omtalte konstaterede forskelle i nødvendig kontakttid bør opfølges med videre forsøg kombineret med en vurdering i de konkrete tilfælde. Som en mulig strategi for opsætning af forsøg, der tager hensyn til kinetikken i udvaskningen, kan der foreslås opstillet parallelle batchtest, der prøvetages efter forskellige tidsperioder. De første to prøvetages f.eks. efter henholdsvis 1 og 3 timer, og hvis der ikke ses en væsentlig forskel mellem koncentrationerne af stofferne i disse to systemer, slutter testningen her. Hvis der ses en betydelig forskel, fortsætter testningen og analyseringen til f.eks. 24 timer og 5 døgn. I sidstnævnte tilfælde indlægges resultaterne i en model, som tager hensyn til feltrealistiske forhold.
Temperaturen er ikke af væsentlig betydning for sorptions- og desorptionsprocesser, mens den naturligvis har betydning for flygtigheden og nedbrydeligheden af visse stoffer. I det omfang, nedbrydeligheden og flygtigheden kan kontrolleres, anbefales det at udføre forsøgene ved stuetemperatur.
Det vil være rimeligt at antage, at metoder til testning af udvaskningen af organiske stoffer fra granulære, mineralske restprodukter i vidt omfang kan baseres på de samme principper og hensyn, som metoder til udvaskning af organiske stoffer fra jord. Dog vil der for restprodukternes vedkommende i mange tilfælde skulle tages specielle forholdsregler med hensyn til forbehandling af prøverne (f.eks. tørring og nedknusning), ligesom restprodukternes potentielle reaktivitet med vand og de i nogle tilfælde ret ekstreme pH- og redoxforhold, som reaktionerne med vand kan medføre, må tages i betragtning. Det bør eventuelt foretages en vurdering af værdien af de resultater man opnår, når man, som det i praksis ofte er tilfældet, udvider analyseprogrammet for udvaskningstests til bestemmelse af udvaskningen af uorganiske komponenter fra restprodukter til også at omfatte indholdet af opløst organisk stof.
Sammenfattende må det anføres, at den gennemførte undersøgelse har tilvejebragt et fagligt grundlag for at opstille forslag til en udvaskningstest til organiske stoffer. Det er lykkedes at identificere en række forhold, der er centrale for udviklingen af udvaskningstests til forurenet jord. Der er foretaget en klassifikation af de vigtigste organiske stoffer, som kan benyttes til at målrette eventuel videre udvikling. Det er imidlertid klart, at den præcise viden om de organiske stoffers egenskaber og forhold, der er kritiske for opstilling af en testprotokol til organiske stoffer, er mangelfuld. Det betyder, at på trods af, at eksperimentelle afprøvninger har bidraget med yderligere specifik information i relation til udvalgte jordtyper og stofgrupper, er det blevet klart, at flere forhold behøver videre afklaring og nærmere undersøgelse, før der kan opstilles en egentlig protokol for udvaskningstests til klassificering af udvaskningen af organiske stoffer fra jord og restprodukter i Danmark.
Der er udarbedet et indledende forslag til en testmetode til bestemmelse af udvaskningen af ikke-flygtige organiske stoffer som olieprodukter (benzin undtaget) og PAH’er fra jord. Testen foreslås udført som en enkelt batchtest ved L/S = 2 l/kg, og for at simulere jordvæske foreslås en 0,001 M CaCl2-opløsning i demineraliseret vand anvendt som udvaskningsmedium. Der anvendes en kontakttid på 1 eller 5 døgn, afhængigt af den aktuelle situation og formålet med testen. For at forebygge biologisk nedbrydning af de organiske stoffer fjernes ilten fra testsystemet med kvælstof og NaSO3.Det foreslås, at separationen af eluatet fra jordfasen sker ved centrifugering i stedet for filtrering. Der er efterfølgende givet et eksempel på, hvorledes resultatet af en udvaskningstest på en forurenet jord kan anvendes til at estimere indholdet af forureningskomponenter i porevandet i jorden lige under et lag af den samme forurenede jord, som gennemsives af infiltrerende nedbør.
Executive Summary
Summary
As part of a comprehensive legislative effort in the area of soil protection and soil remediation, a new statutory order on the utilisation of soil and certain inorganic waste materials for building and construction purposes has been issued (Miljøstyrelsen, 2000). In previous regulation, requirements on soil and waste materials to be utilised only concerned total or partial contents of regulated substances, determined by chemical analysis of the solid materials. The new statutory order also prescribes testing of the leaching of trace elements and salts under specified conditions and sets leaching limit values, which must be met by soil and inorganic waste materials in order to allow their use in certain applications.
Practical experiences with testing of the leaching of organics from contaminated soil and (largely) inorganic waste materials are limited, and very few well-established test procedures exist for the examination of the leaching behaviour of organic contaminants. In order to improve this situation a study has been carried out with the objective to evaluate primarily the technical feasibility of using leaching tests for the characterisation and classification of the leaching of organic components from contaminated soil and inorganic waste materials.
The project has benefited from previous work on the behaviour of organic components in soil and groundwater carried out under the auspices of the Danish Environmental Protection Agency (DEPA). A state-of-the-art overview of the processes and factors controlling the leaching/desorption of organic compounds from soil and waste materials has been established. Based on this information and experience from experimental work, the preconditions and limitations of various test methods and test principles related to leaching of organic compounds from soil and (largely) inorganic waste materials have been evaluated.
By grouping the relevant organic compounds according to their physical/chemical properties in relation to their distribution in a combined air-water-soil system, they have been classified in relation to the feasibility and relevance of testing their leaching from soul and waste materials.
Based on the collected information and the classification of the organic compounds, some of the leaching procedures have been tested experimentally on 5 selected samples of contaminated soil. Some of the results have been compared to results of fugacity/total analysis based distribution calculations performed on the same samples.
Finally, an overall evaluation of the compiled information has been performed, and a number of recommendations concerning the development of methods for the determination of the leaching of certain organic compounds from contaminated soil is given. A first preliminary draft version of a test protocol is presented.
Conclusions and recommendations
Through the collection and compilation of experience and data from literature, a number of factors of importance to further developments of test procedures for examination of the leaching of organic compounds from soil and waste materials have been identified:
| When organic compounds are present in contaminated soil and waste materials as free phases, the leachability of these organic compounds will be controlled by their water solubility. If no free phase of an organic compound is present, or the free phase is totally dissolved, the leaching will be controlled by desorption from the solid particles of the soil/waste material. |
| Dissolution from free and residual phases of organic compounds in soil is a relative fast process (minutes to hours). The course of the release of organic compounds from a free phase may be predicted if the composition of the free phase is known. This would, however, require additional information on the properties of the compounds and the soil. Such information is often difficult to procure. |
| Desorption of organic compounds from soil is often a slow process (days to weeks). Desorption rates are difficult to predict, probably because they depend both on the organic compounds and the soils in question. Slow desorption rates are seen in particular for hydrophobic compounds. There are indications that "young" soil types in general are characterised by exhibiting relatively fast desorption rates and small hysteresis effects, whereas the opposite is true for geologically older soil types. Except for determinations of the O/C ratio, which increases with the age of the soil, no easy or well-established methods for the determination of the age of soils exist. It should be noted that top -soils in some cases may consist of or contain geologically old materials. |
| A general impression of the expected sorption capacity of a soil may be based on a determination of its content of natural organic carbon. Furthermore, the above mentioned ratio between oxygen and carbon (O/C) is considered an indicator of the age of the solid organic carbon present in the soil. Low values of O/C indicate that the organic material is old and that low desorption rates may consequently be expected. |
| In relation to testing of the leaching of organic compounds it is of key importance that their release from soils in many cases will be controlled by desorption kinetics. This has an impact on the required contact time between the soil and the leaching agent in the test. Considerations should be given to the determination of realistic contact times under field conditions for relevant physical scenarios. It seems obvious to attempt to develop test systems, which ensure that equilibrium-like conditions between the organic compounds in the solid phase and the leaching agent, respectively, are achieved. In those cases where long time periods are required to achieve equilibrium-like conditions, this would not necessarily be a suitable criterion for establishing the contact time. The reason for this is partly the impracticability of long contact times, partly the fact that equilibrium-like conditions are not always attained in the field. |
| Most of the above mentioned factors, which influence and are of importance to the leaching of organic compounds from soil/contaminated soil, may be expected to be of similar importance to the leaching of organic compounds from (largely) inorganic waste materials. However, for many such waste materials the organic contaminants constitute a more or less integral part of the materials themselves, whereas for soils they normally are associated only with the surface or the pores of the particles. In contrast to soil, a number of waste materials may react chemically with water when they are moistened or percolated, which e.g. may give rise to more extreme pH values in the water phase than usually seen for soils. |
Based on the in formation collected on test methodology and the experimental work conducted in this study, a number of factors of importance to the development of a batch leaching test for organic compounds have been identified:
| A certain minimum of pre-treatment of a soil or waste sample is necessary to ensure sufficient homogenisation of the sample. Such pretreatment procedures should be gentle and should in many cases be carried out within a closed system to minimise loss by evaporation of volatile organic components. This means either that sieving and crushing of the sample is not possible, or that a certain loss by evaporation must be accepted. |
| The liquid to solid ratio (L/S) is of key importance to the performance of a leaching test and the interpretation of the results. The leaching experiments performed in this study were carried out at L/S = 2 l/kg, which in many cases approaches the lowest practicable L/S ratio at which a batch leaching test can be executed. The practical difficulties with suspended particles in the system combined with the need for relatively large volumes of eluate for chemical analysis have meant that the use of the low L/S value has not been optimal from all perspectives. During the performance of the batch tests, the headspace (the volume of air) in the test bottles has been minimised in order to reduce the loss of volatile organic compounds and to reduce the amount of oxygen in the test systems |
| The separation of the eluate from the solid phase has turned out to a very critical part of the leaching test procedure. Compared to the application of a similar procedure to the testing of the leaching of trace elements and inorganic salts, the separation procedure for organic components is made much more difficult by the fact that organic compounds are far more prone to evaporation and to sorption to filter materials etc. than trace elements and salts. In addition, there is a general requirement for larger volumes of eluate for chemical analysis of organic compounds than for analysis of trace elements and salts. |
| In order to minimise the losses (or gains) of the organic compounds being studied, equipment primarily made of glass and stainless steel, and to a lesser extent teflon, has been used in the leaching experiments carried out in this study. Filter materials made of glass resulted in losses, but apart from this, the test system performed as intended. The composition of the leaching agent, the temperature and the mode of agitation appear to have be non-critical in the leaching tests carried out on the selected leaching tests. |
For some of the relevant groups of organic compounds a reasonable amount of background information exists on which further development of procedures for the testing of the leaching properties may be based. This is true for oil components, chlorinated solvents and polycyclic aromatic hydrocarbons (PAH), whereas less information is available on components such as MTBE and DEHP. The potential problems associated with small particles in soil samples and the eluate resulting from the leaching test has been (and is being) examined in relation to the leaching of PAHs. Due to the high affinity of PAHs for solid particles, the presence of small particles/colloids in the test system is expected to be potentially important primarily to the leaching of PAHs. However, once the final particle cut-off has been decided, it should be examined whether the presence of particles influences the test results for other organic compounds of high priority. For practical reasons, i.e. to save time and reduce the difficulties involved in the handling of larger volumes of eluate, particle cut-offs corresponding e.g. to 0,7 m m have been used in this study instead of the 0,45 m m cut-off, which is most commonly used for trace elements and inorganic salts.
Based on simplified calculations of the distribution between the phases in a combined air/water/soil system it is proposes to classify those organic compounds for which ecotoxicological criteria in soil and water exist according to the feasibility and relevance of testing their leaching from soil and waste materials. It is proposed that they should be divided into three groups:
| The first group consists of consists of non-volatile compounds which sorb strongly on soil and have low solubilities in water. Most PAHs and diesel oil components belong to this group. It seems obvious that a methodology for determination of the leaching of organic compounds from soil and waste materials should include these compounds, which on the one hand will be present in and move with the soil and on the other hand to a certain extent will be leachable. The most critical factors that may influence the leaching procedure and results and need consideration are likely to be the separation of the eluate from the solid phase, including the material which may be sorbed on colloids, and the contact time. A preliminary proposal for a protocol for testing of the leaching of non-volatile organic compounds from contaminated soil has been produced. The proposed protocol does not specify a definite contact time (1 to 5 days are suggested) and it does not address the question of colloidal transport. |
| The second group includes volatile compounds with relatively high water solubilities. The chlorinated solvents and BTEX are among the compounds in this group. Many of these compounds have low criteria values in groundwater and it would therefore be advantageous to be able to test their leachabilities. Some of them have far higher diffusion coefficients in air than in water and will therefore migrate faster through air that through water. In the context of utilisation of soil and waste materials for building and construction purposes, these compounds are not likely to cause a general (indoor) air problem, since covering is required and may be assumed hinder any substantial transport by air. In relation to testing of the leaching of these compounds, the potential loss of material through volatilisation is likely to pose the major problem. By minimising the headspace in the test system this may be taken care of in a batch test since only a minor part of the total amount be present as a gas, but the volatility will be difficult to control when handling the solid sample and the eluate before and after the test itself. Especially the separation of the solid phase and the eluate will be critical in relation to loss of material and hence the feasibility of the test. |
| The third group consists of compounds, which have very high water solubilities or are water miscible, and which do not sorb very strongly on soil. This group includes e.g. MTBE. The phenols have high water solubilities and also belong to this group, although they sorb stronger on soil than e.g. MTBE. Other compounds in this group will only to a very limited extent be associated with the solid phases, and it will therefore in general be difficult or irrelevant to attempt to use leaching tests to describe their behaviour. To the extent it seems relevant, the leachability of compounds belonging to this third group may also be tested by the method mentioned above. Colloids are not likely to play any significant role in the transport of these compounds due to their weak sorbing properties. |
It will be necessary to develop appropriate methodologies for testing the leaching properties of all the organic priority compounds. This includes further develop the proposed test protocol. A number of the critical factors identified in this study and their influence on the test results must be coped with and accounted for. High priority should be given to the development or adjustment of methods for chemical analysis of the organic compounds with detection limits comparable to those applicable to analysis of groundwater. Application of most of the existing analytical methods leads to requirements for relatively large volumes of eluate, which again leads to a need for inconveniently large test systems (or multiple systems) causing the separation of the eluate and the solid phase to become difficult and time consuming.
From a practical perspective, it will probably be most useful to continue the development of a batch leaching test for organic compounds. The choice of test conditions and the interpretation of the results do present some problems and a comparison of the results of batch test results with the results of column leaching tests would be appropriate. Further discussion of the exact objective and expected use of the result may help. One of the most important technical issues is, as already mentioned, the separation of the eluate from the solid phase. The importance of the particle cut-off is probably negligible for some of the less hydrophobic compounds, whereas it may be more significant for the more hydrophobic compounds. Based on the experiences from the experimental part of this project it is recommended to use leaching procedures, which do not require filtration. Centrifugation is recommended as an alternative to filtration. Such procedures should be developed and evaluated in parallel with the development and evaluation of the above mentioned analytical techniques which do not require large volumes of eluate. It will probably be too optimistic to attempt to design the test systems to be applicable to all the potentially interesting groups of organic compounds, since this would lead to inappropriately complicated procedures. The test system should be able to accommodate the most frequently occurring multiple compound contaminations (e.g. contaminations with BTEX and heavier oil components), whereas soil contaminated with combinations of organic compounds which occur less frequently, e.g. chlorinated solvents and gasoline components, could, if necessary, be subjected to two (different) parallel test procedures. The feasibility of testing at a higher L/S ratio than 2 l/kg, e.g. 5 or 10 l/kg, could also be examined. All other things equal, this would increase the volume of eluate produced and possibly reduce the problems involved in the separation of the eluate from the solid phase. Among the disadvantages of using higher L/S values would be the fact that the test conditions would move further away from simulating the L/S ratio corresponding to the pore volume of the soil, which in many cases is of primary interest to the investigator. Another disadvantage is that the concentration of the organic compounds in the eluate in general will be lower than it would be at L/S = 2 l/kg, thus possibly requiring a lower analytical detection level anyway. A deviation from the currently chosen L/S ratio would also require a re-evaluation of preconditions and assumptions upon which that choice has been based. Another alternative would be to apply the same L/S (= 2 l/kg) but use larger test bottles. The handling of larger volumes of eluate may, however, as already mentioned be both troublesome and time consuming.
The mode and degree of agitation may possibly have a significant influence on the performance and results of a batch leaching test because they have a physical impact on the soil particles. Agglomerates and surfaces may be destroyed in such a way that the sorption capacity of the soil changes. If the physical influence on the soil e.g. results in an increased availability of solid organic material, the test may underestimate the desorption of organic contaminants. In contrast, the release of colloids may result in an overestimation of sorbed contaminants. The latter has been shown experimentally, where particles in the 0,45 to 10 m m range were observed in the eluate after the agitation, and substantial amounts of PAH were shown to be associated with those particles. Eluate from corresponding column leaching tests, which may be assumed to simulate field conditions better than batch leaching tests, did not contain significant amounts of particles.
The previously discussed differences in the contact times needed to achieve equilibrium-like conditions for different types of organic compounds should be further examined and evaluated for specific test systems and materials. A possible test straetegy that would enable the kinetics of the leaching process in question to be taken into account, would be to run a number of identical batch leaching tests in parallel and analyse the eluates at various time intervals. The first two batch tests could be terminated e.g. after 1 and 3 hours, respectively, and if no significant differences between the concentrations of the organic contaminants are observed, the testing stops here. If a significant difference is observed, additional batch leaching tests are continued and eluates are analysed e.g. after 24 hours and 5 days, respectively. In this case, the results could be fed into a model that is able to account for the field conditions, particularly with respect to contact time and equilibrium conditions.
The temperature has no significant influence on sorption and desorption processes, but it does, of course, influence the volatility and biodegradability of certain compounds. In general, it is recommended to perform leaching tests for organic contaminants at room temperature, provided the volatility and biodegradability of the organic compounds in question is under control. Minimising the oxygen concentration in the test system may reduce the biodegradation.
It is reasonable to assume that test procedures for examining the leaching of organic compounds from granular, mineral waste products to a large extent may be based upon the same principles and considerations as test procedures for examining the leaching of organic compounds from soil. However, some specific measures will in many cases be necessary for waste products as regards pre-treatment of the samples to be tested (e.g. drying and crushing), and the potential reactivity of some waste materials with water as well as the changes in pH and redox conditions of the eluate these reactions may cause must be taken into consideration. An evaluation should be made of the results obtained when dissolved organic carbon (DOC ), as is often the case, is determined from analysis of eluates from leaching tests for examination of the leaching of inorganic components from waste products.
The study described in this report has provided a foundation which allows the presentation of a first draft proposal for a test procedure for the examination of the leaching of certain organic compounds from soil. A number of factors and issues which are essential to the development of leaching tests for soil contaminated with organic compounds have been identified, discussed and in some cases subjected to experimental investigations. A classification of the most important organic contaminants has been established and may used to focus further developments. It is, however, quite clear that the precise knowledge on the organic compounds, particularly as regards properties and factors which are critical in relation to the development of a more comprehensive test protocol for leaching procedures for organic compounds, is incomplete and insufficient. This means that even though some specific information was produced in this study from experimental work on selected soil types and selected organic compounds or groups of compounds, it will be necessary to further clarify and examine (experimentally) a number of important issues, before a more comprehensive protocol for testing of the leaching of organic compounds from soil and waste products in Denmark can be elaborated.
A preliminary draft protocol describing a single batch leaching test for the examination of the leaching of non-volatile organic compounds such as oil products (not gasoline) and PAH’s at L/S = 2 l/kg is presented presented. To simulate natural soil water and facilitate the separation of the eluate from the soil phase, the proposed leaching agent is 0.001 M CaCl2 solution in demineralised water. Centrifugation rather than filtration is recommended for the separation of the eluate from the soil. In some cases and with great caution, the results of the leaching test may be used to estimate the composition of the pore water in the soil just below a layer of contaminated soil that is being percolated by infiltrating rain. An example of such an estimation is presented.
1. Introduktion
| 1.1 | Baggrund |
| 1.2 | Organiske stoffer i jord og restprodukter |
| 1.3 | Formål |
| 1.4 | Rapportens opbygning |
1.1 Baggrund
Som et led i det omfattende jordlovsarbejde er der udsendt et forslag til bekendtgørelse om genanvendelse af restprodukter og jord til bygge- og anlægsarbejder. Princippet for reguleringen/kravværdierne er som noget nyt baseret på en regulering af jorden og restprodukternes udvaskelige stofmængde bestemt under nærmere specificerede omstændigheder. Under arbejdet med koncepter, metodik og tests til klassificering af stofudvaskningen fra jord og restprodukter (Hjelmar et al. 1998) blev det klart, at det faglige grundlag for vurdering af jord generelt samt flere typer af mineralske restprodukter er utilstrækkeligt. Før eller parallelt med, at retningslinierne effektueres, er det derfor nødvendigt, at der gennemføres undersøgelser, som tilvejebringer det fornødne grundlag, og som om nødvendigt kan benyttes til justering af metoder og kriterier.
Såvel for restprodukter som for jord gælder det, at testning for udvaskning af organiske forureningskomponenter ikke er velundersøgt, og eftersom de organiske forureningsstoffer udgør en meget omfattende og udbredt gruppe af stoffer med forskellige egenskaber, er det således af stor betydning, at der igangsættes udvikling af tests, der kan anvendes i forbindelse med undersøgelser af de organiske forureningsstoffer. På grund af de organiske stoffers forskellige egenskaber, f.eks. med hensyn til nedbrydelighed og flygtighed, er det vanskeligt og tidskrævende at udvikle selv enkle udvaskningstests til organiske stoffer. Det er således af stor betydning at udvikle testmetoder til organiske stoffer og at relatere erfaringerne herfra til de koncepter og testmetoder, der er udviklet for uorganiske stoffer. Muligvis vil nogle af de mest vandopløselige organiske stoffer med lav flygtighed kunne testes i systemer, der minder om dem, som anvendes til testning for uorganiske stoffer, hvilket i størst muligt omfang bør tilstræbes. I forskningssammenhænge har der gennem en årrække været fokus på sorption og desorption af organiske stoffer i jord og akvifermaterialer. Denne viden udgør et vigtigt udgangspunkt for forståelsen for de konkrete testrelaterede problemstillinger.
På denne baggrund har Miljøstyrelsen med midler fra Miljøstyrelsens Teknologiudviklingsprogram igangsat projektet "Udvaskning af organiske stoffer fra jord og restprodukter: Metodeudvikling". Projektet er udført i 1998 og 99 af DHI i samarbejde med Peter Kjeldsen, Institut for Miljøteknologi, DTU.
1.2 Organiske stoffer i jord og restprodukter
Jord fra forurenede grunde kan være blevet forurenet på grund af spild,
utætheder i tanke eller rørføringer samt deponering af kemikalieaffald på grundene. I
flere tilfælde sker kontamineringen i form af ikke-vandblandbare organiske væsker, også
kaldet NAPL (benzin, olie, opløsningsmidler) efter den engelske forkortelse af Non
Aqueous Phase Liquid. En forurenet jord kan således i nogle
tilfælde bestå af fire faser (fast fase i form af jordpartikler, vand, luft og NAPL i
form af små klumper tilbageholdt af jorden). I denne situation vil udvaskningen være
styret af opløsningen af organiske stoffer fra den frie NAPL-fase. I flere tilfælde har
forureningen ikke hidrørt fra nedsivning af NAPL, eller den frie NAPL-fase er forsvundet
- i disse tilfælde består jorden kun af tre faser. Her vil udvaskningen af organiske
stoffer været styret af desorption fra de faste jordpartikler.
Vurdering af udvaskningen af organiske stoffer fra jord kan blive aktuel under flere
scenarier, nemlig in-situ, samt som opgravet og behandlet. Oprensning af forurenede grunde
sker enten ved opgravning af jorden med efterfølgende behandling eller ved
in-situ-teknikker. Ved de fleste oprensningsprojekter vil man normalt ikke fjerne
forureningen til baggrundsniveau alle steder - der vil i mange tilfælde kunne accepteres
en vis mængde restforurening, som efterlades på lokaliteten. Udvaskningstests kunne
være metoden til at fastlægge acceptable restforureningsniveauer, idet en total analyse
af jorden siger meget lidt om udvaskningspotentialet fra jorden. Ved opgravningsprojekter
vil man stå med forurenet jord (evt. opdelt i forskellige forureningsklasser), som
ønskes behandlet - i mange tilfælde biologisk. Her vil udvaskningstests kunne benyttes
til at vurdere forureningspotentialet, som baggrund for at vælge behandlingsform/-pris.
Resultatet af en sådan test kunne være, at jorden kunne deponeres direkte, eller at den
skulle behandles først. Endelig kunne udvaskningstests benyttes efter en behandling til
dokumentation af ønsket behandlingsgrad, idet totalanalyser heller ikke i dette tilfælde
siger meget om udvaskningspotentialet.
Andre mineralske restprodukter som f.eks. slagge og flyveakse fra affaldsforbrænding kan også indeholde forskellige organiske stoffer. Der er meget lille erfaring med udvaskningen af organiske stoffer fra sådanne restprodukter, men mange af de processer og faktorer, der styrer udvaskningen af organiske stoffer fra jord, vil også være gældende for restprodukter. En væsentlig forskel er, at de potentielle forureningsstoffer i større grad findes i selve restproduktet, mens de i jord primært er tilknyttet overfladerne af jordpartikler eller de frie faser mellem partiklerne. Endvidere vil mange typer af restprodukter i den deponerede form bestå af "partikler" eller aggregater af væsentlig større dimensioner, end hvad der normalt findes i jord. Dette kan blandt andet stille krav til den skala, hvormed en udvaskningstest skal gennemføres.
1.3 Formål
Projektet har til formål at vurdere de tekniske muligheder for at anvende udvaskningstests til karakterisering og klassificering af udvaskningen af organiske stoffer fra forurenet jord og restprodukter. Projektet fokuserer specielt på forurenet jord, men i det omfang, der eksisterer viden om udvaskningen af organiske komponenter fra mineralske restprodukter, er dette medtaget i rapporten. Med udgangspunkt i Miljøstyrelsens forslag til bekendtgørelse om genanvendelse af restprodukter og jord til bygge- og anlægsarbejder og i forlængelse af arbejdet med de uorganiske stoffer har projektet desuden til formål at identificere de grupper af organiske stoffer, som bør kunne håndteres i et testsystem. Endelig skal der gennem videnindsamling, laboratorieforsøg og eksperimentelle afprøvninger så vidt muligt stilles forslag til en metode og udarbejdes en testprotokol for denne.
1.4 Rapportens opbygning
Rapporten indledes med tre kapitler som præsenterer "state-of-the-art" for de processer, som styrer udvaskningen af organiske stoffer. I kapitel 2 præsenteres opløselighedsstyret udvaskning, som er gældende i tilfælde af, at jorden indeholder fri fase. I kapitel 3 præsenteres de sidste ti års intensive forskning inden for desorptionsstyret udvaskning, som er gældende i de fleste tilfælde (jord uden fri fase). I kapitel 4 beskrives kort den nuværende viden om transport af organiske stoffer med kolloider og kolloiders betydning for tests af stofudvaskning. Kapitel 5 indeholder de seneste års erfaringer med udvikling af udvaskningstestmetodikker med særlig fokus på organiske stoffer og kritiske faktorer for udvikling af en testmetodik til organiske stoffer. I kapitel 6 er der med udgangspunkt i de stoffer, som der findes kriterieværdier for i jord og grundvand, foretaget forenklede fasefordelingberegninger af stoffernes fordeling i kombinerede luft-/vand-/jordsystemer. På baggrund heraf er der foretaget en klassificering af stofferne i tre grupper med forskellige karakteristika i relation til behovet for at udvikle en testmetodik for de enkelte grupper. Kapitel 7 beskriver de gennemførte eksperimentelle undersøgelser. Resultaterne præsenteres og diskuteres. Endelig indholder kapitel 8 en samlet evaluering af erfaringerne fra sammenstilling af litteraturen og de eksperimentelle undersøgelser. Kapitlet afsluttes med anbefalinger vedrørende testmetodik, og der opstilles forhold, som behøver nærmere undersøgelse. I kapitel 9 er der opstillet et indledende forslag til en testprocedure, og der er givet et eksempel på anvendelse af testresultater til estimering af sammensætningen af porevandet i den nederste del af et lag jord, der er forurenet med organiske stoffer. I kapitel 10 er der foretaget en opsummering af de indsamlede erfaringer, og der præsenteret en række anbefalinger
2. Opløselighedsstyret udvaskning fra frie faser
2.1 Konceptuel model
I et system med fri fase vil den frie fase altid kontrollere koncentrationen af organiske stoffer i vandfasen og derved også koncentrationen af stof sorberet til jorden samt i en eventuel luftfase. Når der er blevet udvasket så meget stof, at der ikke længere er fri fase tilbage i jorden, er koncentrationen af stof i vandfasen derefter kontrolleret af ligevægt (sorption/desorption) med jorden. Hvis den frie fase indeholder flere stoffer, vil sammensætningen af stofferne i den frie fase ændres i løbet af udvaskningsforløbet, da stofferne med den højeste opløselighed i vand udvaskes hurtigst. Når sammensætningen af den frie fase derfor ændres, vil koncentrationen i vandfasen ændres, hvilket medfører, at koncentrationen på jorden og i luften ligeledes ændres.
2.2 Styrende mekanismer
2.2.1 Opløselighed af enkeltstoffer
Koncentrationen af et stof i vand, som er i ligevægt med en fri fase, bestående udelukkende af dette ene stof, svarer til opløseligheden af stoffet. Opløseligheden af et stof afhænger af temperatur og vandets indhold af andre stoffer, som eksempelvis salte (Miljøstyrelsen 1996). Tabel 2.1 indeholder eksempler på opløseligheden af udvalgte organiske stoffer. Det fremgår tydeligt af tabel 2.1, at opløseligheden varierer meget mellem forskellige stoffer.
Tabel 2.1
Opløseligheden i vand ved 20-25°C af udvalgte stoffer (Miljøstyrelsen 1996).
| Stof | Opløselighed (mg/l) |
| Phenol | 84.000 |
| Benzen | 1760 |
| o-Xylen | 180 |
| Naphthalen | 31 |
| Phenanthren | 1,2 |
| Pyren | 0,14 |
| Benz(a)pyren | 0,0038 |
2.2.2 Opløselighed af stofblandinger
Det er sjældent, at en forurening kun består af ét stof. Når der er flere stoffer i den frie fase, beregnes ligevægtskoncentrationen ud fra Rauolt's lov (f.eks. Miljøstyrelsen 1996):
|
(2.1) |
hvor
Ci er ligevægtskoncentrationen af stof i vand
xi er molfraktionen af stof i den frie fase
Si er opløseligheden af stof i vand.
Molfraktionen er defineret i ligning 2.6.
Opløseligheden, Si, der benyttes i Raoult's lov, er den normale opløselighed for de stoffer, der er væsker ved den aktuelle temperatur. For stoffer, der er faste ved den aktuelle temperatur, men som normalt er en væske ved blanding med andre væsker, benyttes den såkaldte underafkølede opløselighed, der beregnes som (f.eks. Miljøstyrelsen 1996):
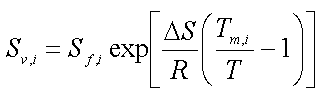 |
(2.2) |
hvor
Sv,i er den beregnede opløselighed for stof i
Sf,i er opløseligheden af det faste stof i i vand
D S er fusionsentropien
R er gaskonstanten
Tm,i er smeltepunktet for stof i (K)
T er den aktuelle temperatur (K).
Forholdet mellem D S og R kan, hvis det ikke kendes, sættes
til 6,8.
I tabel 2.2 er der angivet eksempler på faststofopløseligheder og de dertil svarende underafkølede opløseligheder. Det fremgår af tabellen, at der er stor forskel på de to typer af opløselighed (op til en faktor 40 for benz(a)pyren).
Tabel 2.2
Eksempler på udregning af den underafkølede opløselighed. Faststofopløseligheder
og smeltepunkter er fra Miljøstyrelsen (1996).
| Stof | Faststof- opløselighed (mg/l) |
Smeltepunkt (°C) |
Underafkølet opløselighed ved 20°C (mg/l) |
| Naphthalen | 31 | 81 | 128 |
| Phenanthren | 1,2 | 98 | 7,3 |
| Pyren | 0,14 | 156 | 3,3 |
| Benz(a)pyren | 0,004 | 178 | 0,16 |
Rauolt's lov udtrykt ved ligning 2.1 gælder for blandinger af stoffer af ensartet
karakter. Den simple udgave af Raoult's lov gælder bl.a. for blandinger af klorerede
opløsningsmidler (Broholm og Feenstra 1995) og blanding af octan og methylcyclohexan
(Burris og MacIntyre 1985), hvorimod eksempelvis opløselighederne i blandinger af octan
og naphthalen, methylnaph-thalener eller dimethylnaphthalener ikke kan beregnes eksakt ud
fra dette udtryk (Burris og MacIntyre 1985). Disse afvigelser er imidlertid i de fleste
tilfælde mindre end en faktor 2.
Hvis der er interaktioner mellem stofferne, ændres Rauolt's lov til:
|
(2.3) |
hvor g i er aktivitetskoefficienten for stof i.
Aktivitetskoefficienterne kan estimeres ved hjælp af f.eks. UNIFAC, der er en forkortelse for Universal Quasi Chemical Functional Group Activity Coefficients Method. UNIFAC er en semi-empirisk model baseret på et princip om forskellige kemiske gruppers bidrag til aktivitetskoefficienter i relation til deres struktur. Fordelen ved metoden er, at aktivitetskoefficienterne kan estimeres for et hvilket som helst kemisk stof, hvis blot stoffet består af nogle grupper, som UNIFAC kender konstanterne for. UNIFAC- metoden blev introduceret af Fredenslund (Fredenslund et al. 1977) og er siden blevet udvidet og forbedret (Skjold-Jørgensen et al. 1979; Gmehling et al. 1981). I dag findes UNIFAC som et PC-program.
2.3 Beregning af udvaskningen fra en fri fase
Til illustration af det dynamiske i udvaskningen beregnes udvaskningsforløbet fra 12,2 ml fri fase, der består af benzen (8,2 vol-%), phenol (82,0 vol-%), naphthalen (8,2 vol-%) og phenanthren (1,6 vol-%). Deres opløseligheder er henholdsvis 1760, 84000, 31 og 1,2 mg/l (Miljøstyrelsen 1996). Beregningen foregår trinvist. Det første grundvand, der kommer i ligevægt med den frie fase, indeholder de ved hjælp af Raoult's lov beregnede ligevægtskoncentrationer. Da koncentrationen af phenol er størst, fjernes der mere phenol, end der fjernes af de øvrige stoffer fra den frie fase. Dette medfører, at molfraktionen i den frie fase ændres med tiden. Dette problem kan matematisk ikke løses eksakt, men det er relativt enkelt at beregne koncentrationsændringerne numerisk i et regneark. Det nødvendige ligningssystem er følgende:
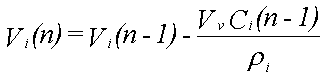 |
(2.4) |
|
(2.5) |
|
(2.6) |
|
(2.7) |
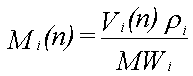
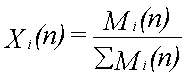
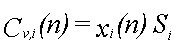
hvor
Vi(n) er volumenet af stof i til beregningstrin n
Ci(n) er væskekoncentrationen af stof i til beregningstrin n
Vv er volumenet af vand i ligevægt med den frie fase
Mi(n) er antallet af mol af stof i til beregningstrin n
xi(n) er molfraktionen af stof i til beregningstrin n
r i er massefylden af stof i
MWi er molvægten af stof i
Si er opløseligheden af stof i.
I ligning 2.4 beregnes de nye volumener af stoffer i den frie fase ud fra informationer om de gamle volumener og ligevægtskoncentrationer. Ud fra de nye volumener omregnes dernæst antallet af mol (ligning 2.5). Herefter beregnes de nye molfraktioner (ligning 2.6) og de nye ligevægtskoncentrationer (ligning 2.7). Til beregningstrin 1 benyttes begyndelsesvolumenet af de enkelte stoffer og begyndelsesligevægtskoncentrationerne, som er beregnet ved hjælp af Raoult's lov.
Volumnet af vand i ligevægt med den frie fase, Vv, er en beregningsstørrelse, hvis størrelse i forhold til volumnet af den frie fase bestemmer, hvor hurtigt beregningerne forløber, og hvor præcise de er. Hvis Vv er lille, bliver beregningerne mere præcise, men til gengæld kræver det flere beregningstrin. Vv svarer til en bestemt tid, som afhænger af grundvandshastigheden og fordelingen af den frie fase i forhold til strømningsretningen.
Figur 2.1 viser resultatet af beregningen for en blanding på i alt 12,2 ml fri fase, der består af benzen (8,2 vol-%), phenol (82,0 vol-%), naphthalen (8,2 vol-%) og phenanthren (1,6 vol-%). Figuren viser tydeligt det dynamiske opløsningsforløb, der er fra en kilde af fri fase, som indeholder flere stoffer. Phenol, som har den højeste opløselighed, opløses hurtigst fra kilden, hvorefter benzen, der er stoffet med den næsthøjeste opløselighed, fjernes næst hurtigst fra kilden. Koncentrationen af phenanthren, der har den laveste opløselighed blandt stofferne i dette eksempel, stiger i løbet af hele forløbet og falder først, når hele kilden af fri fase er opløst.
Figur 2.1
Beregnet udvaskningsforsøg for 12,2 ml fri fase bestående af benzen (8,2 vol-%),
phenol (82,0 vol-%), naphthalen (8,2 vol-%) og phenanthren (1,6 vol-%).
2.4 Kinetik
2.4.1 Batchforsøg
Det er centralt at kende den tidsperiode, der er nødvendig, før der opnås ligevægt i et batchudvaskningsforsøg. I litteraturen findes der afrapporteret to detaljerede undersøgelser, hvor koncentrationen i væskefasen er målt som funktion af tiden. Et af forsøgene omhandlede opløseligheden af BTEX fra benzin i vand med methanol eller MTBE og blev udført i en 60 ml glasflaske med ca. 55 ml vand og ca. 5 ml benzin. Flaskerne blev omrystet ved 10°C i forskellige tidsrum fra 30 min. til 50 timer. Forsøget viste, at 1 time næsten var tilstrækkelig til at opnå konstant koncentration i væskefasen, men kontakttiden blev valgt til 4 timer (Poulsen et al. 1991). Burris og MacIntyre (1985) undersøgte koncentrationen i vand som funktion af tiden i et system med en blanding af 1-methylnaphthalen og methylcyclohexan. Forsøget blev udført i en 500 ml glasflaske med 400 ml vand og 0,5-3 ml organisk fase. Flasken stod på en magnetomrører, der blev indstillet, således at overfladen mellem vandet og den organiske fase ikke blev væsentligt forstyrret. Ligevægt blev opnået i løbet af 24 timer, og mere end 90% af ligevægtskoncentrationerne blev opnået på 12 timer. En kontakttid på 12-24 timer benyttes ofte i opløselighedsforsøg med en eller anden form for omrøring (Eganhouse og Calder 1976; Leinonen og Mackay 1973). I et forsøg uden omrøring til bestemmelse af opløseligheden af trichlorethylen blev ligevægt opnået på mellem 5 og 19 dage i forsøg med 3 ml trichlorethylen og 14 ml vand (Broholm og Feenstra 1995).
Ovenstående kontakttider er anvendt til at opnå ligevægt i systemer uden jord eller andet faststof. Tilstedeværelsen af jord medfører ikke nødvendigvis længere kontakttider, hvis jorden omrøres så kraftigt, at den holdes i suspension. Indgår der derimod større klumper jord eller aggregater, som kan indeholde fri fase, vil dette forøge tiden til ligevægt, da sorptionsligevægt til jorden også skal indtræde.
2.4.2 Kolonneforsøg/naturlige systemer
I naturlige systemer er der sjældent ligevægt. At der ikke er ligevægt skyldes formentlig, at opholdstiden for vandet ikke er tilstrækkelig lang til, at en proces løber til ende. Dette skyldes enten hurtige strømningshastigheder, eller at fordelingen af de frie faser medfører en ringe kontakt mellem vand og den frie organiske fase. Den matematiske beskrivelse af opløsningskinetik er gennemgået i Miljøstyrelsen (1996) og vil derfor ikke blive gennemgået her. I grundvandet er det sandsynligt, at der er ligevægt ved grundvandshastigheder på mindre end 1 m/dag med zoner med frie faser, der har en udbredelse på mere end nogle få centimeter i strømningsretningen (Pankow og Cherry 1996).
2.5 Forudsigelser
Selve det dynamiske udvaskningsforløb ved tilstedeværelse af en fri fase kan let forudsiges ud fra en kendt sammensætning af den frie fase. Ligeledes kan det, hvis koncentrationen i jorden kendes, på basis af fasefordelingsberegninger forudsiges, om der er fri fase i prøven, og hvilken ligevægtskoncentration der vil forekomme i vandet. Det er imidlertid umuligt at forudsige væskekoncentrationen nedstrøms en forurening, fordi det ikke er alt vandet, der kommer i kontakt med den frie fase, og der er ikke nødvendigvis ligevægt. Fasefordelingsberegninger forudsætter et kendskab til egenskaberne af de organiske stoffer og jorden, og det er ikke altid let at fremskaffe sådanne oplysninger. Indholdet af naturligt organisk stof betyder meget for andelen af det stof, der sorberer til jorden. Dette kan medføre, at en jordprøve ud fra kendskab til totalt indhold af forureningsstoffer vurderes ukorrekt med hensyn til tilstedeværelsen af fri fase. Fasefordelingsberegninger og udvaskningsforløb er komplicerede at udføre for blandingsforureninger som diesel og tjære, fordi alle stofferne i blandingen ikke kendes.
2.6 Forhold af betydning for opløselighed/udvaskning
Opløseligheden af et stof afhænger af temperatur og vandets sammensætning. Det sidste har dog kun betydning i saltvand.
I stofblandinger kan ligevægtskoncentrationen i vand forudsiges ved at bruge Raoult's lov. Den simple udgave gælder for ideelle stofblandinger, men hvis den benyttes ved komplekse blandinger, vil fejlen ofte være mindre end en faktor 2.
Udvaskningen fra en kilde med blandingsforureninger kan let beregnes. Stoffet med den højeste opløselighed udvaskes hurtigst, hvorimod koncentrationen af stoffet med den mindste opløselighed vil stige i restfasen, indtil det hele er opløst.
Batchforsøg med forskellige typer af omrøring har vist, at det typisk tager mindre end 24 timer at opnå ligevægt i systemer, hvor der ikke er jord eller andet faststof til stede.
Ved udvaskningsforsøg med jord med fri fase vil koncentrationen i væsken være konstant, så længe der er fri fase til stede. Koncentrationen i væsken vil svare til opløseligheden. Dette vil som regel ikke være gældende for desorptionsstyret udvaskning.
Både batch- og kolonneudvaskningstests kan benyttes i situationer med fri fase. Det mest realistiske udvaskningsforsøg opnås med kolonneforsøg, hvorimod et batchforsøg vil give information om den maksimale udvaskelige mængde/koncentration. Hvis der skal opnås ligevægt i et batchudvaskningsforsøg med fri fase, kræves der formentlig en kontakttid på mindst 24 timer. Denne tid er dog svær at fastsætte præcist, da den afhænger af mængden af fri fase i forhold til vandmængden i udvaskningsforsøget og af agitationen.
Man kan eksperimentelt undersøge, om en jordprøve indeholder fri fase af et organisk stof, ved til en vandig suspension af prøven at tilsætte et farvestof, Sudan IV. Hvis der er en fri fase, vil farvestoffet fordele sig i og synliggøre denne. En indirekte metode er at vurdere koncentration i vandfasen og sammenligne denne med opløseligheden af stoffet eller stofferne.
3. Desorptionsstyret udvaskning
Dette afsnit vil beskrive de mekanismer og faktorer, der styrer desorptionen af organiske stoffer fra jord. I teksten vil der blive benyttet forskellige ord og begreber, som kort skal defineres her. Ordet sorption er en samlet betegnelse for flere fysisk-kemisk set forskellige processer, nemlig absorption (binding i en anden fase) og adsorption (binding til overflader). Hvis de organiske stoffer kan ioniseres, dvs. blive negativt eller positivt ladet (som organiske syrer og baser), kan denne overfladebinding helt eller delvist bestå af en ionbytning. Bindingen af de organiske stoffer kan være mere eller mindre reversibel, dvs. at sorptionsprocessen kan gå "baglæns", hvorved stoffet overføres fra jordpartiklerne til porevandet. Dette kaldes desorption. I den kemiske litteratur samt også i noget af sorptionslitteraturen benyttes ordet sorbent om det materiale, der kan binde forureningsstoffet (i samme litteratur kaldet sorbatet). Her benyttes ordet jord for det sorberende materiale. Begrebet dækker topjorde, akvifermaterialer, m.m. Frasen det organiske stof benyttes for det organiske forureningsstof, som sorberer/desorberer. Jordenes væsentligste fraktion for sorption af organiske stoffer benævnes her det naturlige organiske stof. Dette svarer til en anden ofte benyttet term, det faste organiske stof. Denne fraktion kan i nogle tilfælde omfatte menneskeskabt organisk stof i form af rester af ikke vandblandbare organiske væsker (NAPL). I gennemgangen vil der også blive benyttet termer som partikler og aggregater. I mange tilfælde udgøres mange af en jords korn af aggregater af grundpartikler, som er sammenkittet af oxider og andre stoffer.
3.1 Den traditionelle beskrivelse og dens forudsætninger
I en lang årrække har man anvendt en forenklet beskrivelse af organiske stoffers binding i jord. Beskrivelsen gjorde det muligt ud fra få parametre at kvantificere sorptionen. Den traditionelle beskrivelse er indgående beskrevet i (Kjeldsen,1996) og skal kort resumeres her.
Den traditionelle beskrivelse baserer sig på en række forenklede forudsætninger:
| Sorptionen for et aktuelt organisk forureningsstof kan beskrives ved en lineær sorptionsisoterm, hvilket betyder, at der altid er et fast forhold mellem stofkoncentrationen i væskefasen, Cw, og koncentrationen i den faste fase, Cs. Forholdet, som kaldes fordelingskoefficienten, Kd (opgivet i l/kg), er således uafhængigt af koncentrationsniveauet for det organiske stof. | |
| Sorptionen sker alene til jordens indhold af naturligt organisk stof, dvs. at sorption til jordens indhold af mineralske bestanddele (ler og diverse metaloxider/hydroxider) kan negligeres. Typen af naturligt organisk stof betragtes også som underordnet. | |
| Fordelingskoefficienten er proportional med jordens indhold af organisk kulstof, foc,
og fordelingskoefficienten, Koc, mellem organisk kulstof og vand: | |
| Der er fuld reversibilitet. Det vil sige, at sorptionsprocessen, hvor stof optages fra vandfasen til den faste fase, forløber fuldstændigt som desorptionsprocessen, hvor stof frigives fra den faste fase til vandfasen. Dette betyder, at man beskriver begge processer med den samme fordelingskoefficient, Kd. | |
| Momentan indstilling af sorptionsligevægt. Dette betyder, at den stof-overførsel, der skal ske fra vandfasen og over i den faste fase (eller omvendt) for at opnå ligevægt (beskrevet ved fordelingskoefficienten, Kd), sker øjeblikkeligt. Dette skal i praktisk henseende tolkes, således at stof-overførelseshastigheden er så høj, at den ingen begrænsende virkning har for procesbeskrivelsen under enhver praktisk situation. | |
| Negligibel indflydelse fra andre opløste eller kolloide stoffers tilstedeværelse. Dette betyder bl.a., at der kan ses bort fra konkurrenceeffekter, hvis flere organiske stoffer er til stede på samme tid. |
Hvis man, udover de ovennævnte forudsætninger, forudsætter, at det jordvolumen, som betragtes, kan forudsættes homogent med hensyn til rumvægt (r b), porøsitet, (e ), vandindhold, (e w) og indhold af organisk kulstof (foc), kan sorptions-/desorptionsprocessen beskrives ved retardationsfaktoren, R:
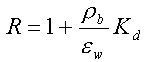
For et homogent jordlag med tykkelsen, L, som er styret af de forenklede forhold beskrevet ovenfor, vil man opnå et udvaskningsforløb som vist på figur 3.1. Man ville således opnå en konstant koncentration, Cw, i udløbet fra jordlaget. Koncentrationen ville holde sig konstant, indtil det organiske stof er blevet udvasket fra hele jordlaget. Tiden, der ville gå til koncentrationen, ville falde til nul, Tg, er:
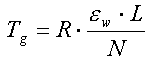
hvor N er nedsivningen gennem jordlaget (f.eks. i mm/år).
Da R er uafhængig af tid og koncentration (under de benyttede forudsætninger), vil R kunne bestemmes ud fra en Kd-måling, eventuelt via et simpelt batchudvaskningsforsøg, hvor et sammenhørende sæt af væske- og faststofkoncentrationer bestemmes, hvorfra Kd beregnes.
Figur 3.1
Ideelt udvaskningsforløb fra homogent jordlag, forudsat at den traditionelle
sorptionsbeskrivelse er gældende er angivet ved den stiplede linie, mens den fuldt
optrukne kurve illustrerer en mere realistisk udvaskning.
3.2 Begrænsninger i den traditionelle beskrivelse for desorptionsstyret udvaskning
Især inden for de seneste fem år har en meget omfattende forskningsindsats vist, at der er flere ting, som gør en udvaskningsbeskrivelse baseret på den traditionelle beskrivelse meget tvivlsom. Et yderligere komplicerende aspekt er, at der stadig gives enkelte eksempler på, at den traditionelle beskrivelse godt kan benyttes, og samtidig flere eksempler på det modsatte.
Der er flere af forudsætningerne i den traditionelle beskrivelse, som der har været fokuseret på. Disse observationer vil kort blive opridset i det følgende:
3.2 1 Isotermtype
Som alternativ til den lineære isoterm benyttes oftest enten Freundlich-isotermen eller Langmuir-isotermen i de tilfælde, hvor der opnås "krumme" sammenhænge mellem koncentrationen på den faste fase og væskekoncentrationen (se Bilag A). På figur 3.2 ses der et eksempel, hvor der blev benyttet en Freundlich-isoterm for at beskrive sorptionssammenhængen. Der er dog også flere nyere eksempler, hvor en lineær beskrivelse er benyttet med succes.
Figur 3.2
Sammenhæng mellem koncentrationen i væske og koncentrationen i den faste fase
modelleret ved hjælp af Freundlich-isotern.
3.2.2 Fordeling til naturligt organisk stof
Sammenhængen:
![]()
hvor foc er fraktionen af naturligt organisk kulstof i jorden (på vægtbasis), og Koc er fordelingskoefficienten mellem det naturlige organiske kulstof i jorden og vand, har været benyttet i mange undersøgelser. Der er eksempler på, at beskrivelse af "sorptionskapaciteten" alene ved jordens indhold af organisk kulstof er utilstrækkelig. I stedet har det naturlige organiske stofs bestanddele af andre grundstoffer end kulstof (f.eks. ilt og kvælstof) været benyttet – se figur 3.3, som viser, at fordelingskoefficienten til det organiske stof vokser med stigende andel af kulstof. Det faktum, at fordelingskoefficienten til det naturlige organiske stof Koc ikke kan betragtes som en konstant for alle jorde (for et specifikt organisk stof), er formentlig hovedårsagen til, at estimationer af Koc ved hjælp af empiriske korrelationssammenhænge med oktanol-vand-fordelingskoefficienten, Kow (som er baseret på et antal sorptionsforsøg med forskellige jorde og organiske stoffer - se afsnit 7.4.2 i (Kjeldsen 1996)), giver vidt forskellige resultater afhængigt af, hvilken empirisk sammenhæng der benyttes. De seneste forsøg på at inddrage flere forsøgsresultater i udvikling af estimationssammenhænge har ikke ændret meget på den store usikkerhed, som disse estimationsformler afstedkommer (Sabljic et al. 1995, Gawlik et al. 1997, Müller 1997).
Figur 3.3
Fordelingskoefficienten til naturligt organisk stof, Kom, som funktion
af (O+N)/C-forholdet i jorden (Rutherford et al. 1992).
3.2.3 Indstilling af sorptionsligevægt
Tabel 3.1 er en sammenstilling af forskellige forsøg lavet for at belyse, hvor hurtigt sorptions- og desorptionsligevægte indstiller sig. Tabellen viser, at ligevægtsindstillingstiden kan variere meget. Det er også tydeligt, at en væsentlig andel af sorptionen oftest sker ganske hurtigt, men at en signifikant andel sorberer langsomt. Det kan faktisk være yderst vanskeligt ved sådanne forsøg at sikre sig, at ligevægt er nået, idet analyseusikkerheder gør det meget vanskeligt at fastslå, om der sker ændringer i den sorberede stofmængde i den sidste del af forsøget.
Den traditionelle beskrivelse benyttes til både at beskrive sorptionen af stof fra væsken til jorden og desorption af stof tilbage igen. Ved vurdering af udvaskning af organiske stoffer fra jord er det primært desorptionen, som skal betragtes. Som nævnt forudsættes det, at begge processer kan beskrives med den samme Kd-værdi, dvs. at der er fuld reversibilitet. Denne forudsætning har via laboratorieforsøg været undersøgt i en lang række tilfælde. Der er tilfælde, hvor der er fundet fuld reversibilitet, men også mange, hvor en
Tabel 3.1 Se her!
Nylige eksempler på observeret langsom sorption eller desorption i naturlige
sorbenter (baseret på sammenfatning af Pignatello og Xing 1996).
væsentlig del af stoffet ikke desorberede i samme grad eller med samme hastighed. I de senere år er også desorption af almindelige neutrale stoffer med beskeden hydrofobicitet (som TCE, benzen, toluen) blevet undersøgt. Figur 3.4 viser adsorptions- og desorptionsisotermer for naphthalen på et sandet overfladesediment med foc på 0,27% (Kan et al. 1994). Det ses af figuren, at adsorptionsisotermen er lineær, og at desorptionsisotermerne adskiller sig radikalt fra adsorptionsisotermerne. Desorptionsisotermerne viser, selv ved adskillige serielle udvaskninger, hvor udvaskningsvandet udskiftes med nyt vand, at der resterer væsentlige stofmængder i jorden. Der opnås således nogle relativt lave udvaskningskoncentrationer, selvom jorden stadig indeholder væsentlige stofmængder. Det er væsentligt at understrege, at det ikke var feltforurenede jordprøver, som benyttedes i forsøget, men jordprøver, hvor de organiske stoffer var tilsat jorden via indledende sorptionsforsøg, hvor stofferne indledningsvist tilsættes til væsken. Den store forskel mellem sorptionen og desorptionen skyldtes således ikke ældningsprocesser (se afsnit 3.6.6). Lignende observationer er gjort i andre forsøg (Miller og Pedit 1992, Harmon og Roberts 1994). Graden af reversibilitet vurderes ikke at have nogen væsentlig indflydelse på selve udførelsen af en udvaskningstest.
Figur 3.4
Sorptions- og desorptionsisotermer for naphthalen på et sediment efter enten 1
døgns (ð ) eller 7 døgns (o) kontakttid i hvert step (Kan et
al. 1994).
3.3 Konceptuel model
Det forrige afsnit viste meget tydeligt, at man stadig er langt fra den fulde forståelse af, hvad der styrer sorptionen og desorptionen af organiske stoffer i jord. En væsentlig del af de seneste års intense forskning har haft til formål at opnå en bedre forståelse af de processer, der styrer stofudvekslingen ved opstilling af konceptuelle modelbeskrivelser af processerne. Disse konceptuelle beskrivelser udvikles til stadighed, efterhånden som nye og flere resultater opnås. Her skal præsenteres en konceptuel model, som i overordnede træk er accepteret af de fleste forskergrupper, der arbejder med dette fagområde.
En beskrivelse af den konceptuelle model kræver, at vi betragter jorden på tre størrelsesskalaer, som vist på figur 3.5. På figur 3.5a ses jorden i forstørrelse med forskellige jordpartikler og porehulrummet imellem partiklerne. På denne skala kaldes porerne makroporer. På partiklerne anes der belægninger, som kan bestå af naturligt organisk stof, evt. i blanding med lermineraler, metaloxider/hydroxider og andet (Pignatello og Xing 1996). Eventuelt indeholder jorden små partikler bestående helt af organisk kulstof eller "ren" kulstof hidrørende fra sod, trækul eller stenkul (Luthy et al. 1997). Jorden kan også indeholde fri fase eller ikke vandblandbar væske (NAPL) i form af olie eller opløsningsmidler, siddende som små "afsnørede" klumper (residual NAPL-fase). Fra disse klumper kan der opløses stoffer, som NAPL’en oprindelig var bestående af (se kapitel 2), men udefra kommende organiske stoffer kan bindes til klumpen – og frigives igen. NAPL kan således virke som en menneskeskabt sorptionskapacitet som supplement til jordens indhold af naturligt organisk stof (Luthy et al. 1997).
I figur 3.5b er en af jordpartiklerne vist i yderligere forstørrelse. Forstørrelsen viser, at partiklen, som det ofte er tilfældet, er et aggregat sammensat af flere forskellige partikler, som er sammenkittet af diverse materialer (organisk stof, metaloxider, m.m.). Figuren viser, at stoffer, som udveksles mellem den faste fase og væskefasen, må passere en stillestående vandfilm. Figuren viser også, at sådanne partikelaggregater ofte er gennemskåret af indre hulrum af forskellig størrelse. Porerne inddeles ofte i to typer, nemlig mesoporer (typisk diameter større end 2 nm) og mikroporer (typisk diameter under 2 nm) (Pignatello og Xing 1996). Disse porer vil oftest være vandfyldte. Transporten i disse porer sker ved diffusion, og diffusionshastigheden kan være kraftigt reduceret, idet porerne kan indeholde naturligt organisk stof, som begrænser stofudvekslingen. Diffusionen begrænses også af porernes tortuositet, som er et mål for, hvor "kringlet" en vej molekylerne skal diffundere (se også afsnit 3.5). I mikroporerne kan tortuositeten være endnu mere udtalt, og der kan samtidig på grund af den begrænsede porediameter være signifikante adsorptionseffekter fra vægge til flere sider og "sterisk hindring" (det faktum, at molekylets størrelse ikke tillader en indtrængning i poren, idet dennes diameter er for lille) (som vist på figur 3.5d) (Pignatello og Xing 1996).
På figur 3.5c er der vist en forstørrelse af et område indeholdende naturligt organisk stof, hvor transporten af det organiske forureningsstof ind i det naturlige organiske stof sker ved diffusionsprocesser i den faste fase. Der har været anvendt analogier til diffusion i polymerer, som der i de seneste år også har været forsket intenst i. Flere forestiller sig således, at det naturlige organiske stof kan være af en "gummiagtig" struktur (fleksibel, amorf ) (A på figur 3.5c) eller af en mere "glasagtig" struktur (kondenseret, hård) (B på figur 3.5c). (Carroll et al. 1994, Young og Weber 1995, Weber et al. 1992, Xing et al. 1996). Herudover har man i polymerer set tegn på adsorptionsprocesser af Langmuir-typen (se Bilag A) til små mikroskopiske hulrum i den glasagtige struktur (C på figur 3.5c) . Man forestiller sig i øvrigt, at alle tre typer kan være til stede samtidig i et område af naturligt organisk stof.
Jorde kan således indeholde forskellige sorptionssites, som enten kan være placeret tæt på den mobile væskefase eller være "gemt" inden i aggregater, hvortil stofferne først skal transporteres via diffusionsprocesser, som kan være yderst langsommelige på grund af forskellige tilbageholdelsesprocesser.
Tabel 3.2 er en sammenstilling af de forskellige sorptionsmekanismer og deres forskellige karakteristika med hensyn til isotermtype, kinetik og konkurrence (Luthy et al. 1997). I det følgende vil de forskellige sorptionsmekanismer blive beskrevet yderligere.
Figur 3.5 Se her!
Konceptuel model for sorption/desorption i jord (efter (Luthy et al. 1997) og
(Pignatello og Xing 1996)) - for nærmere forklaring se teksten.
Tabel 3.2
Kvalitativ sammenligning mellem mekanismer og makroskopiske observationer (efter Luthy
et al. 1997).
Absorption ind i amorft (blødt fast organisk stof eller NAPL |
|
| Isoterm | Lineær |
| Kinetik | Hurtig (<minutter hvis partiklerne ikke er sammensat i aggregater) |
| Hysterese | Nej |
| Konkurrence | Nej |
Absorption ind i kondenseret (hårdt fast organisk stof |
|
| Isoterm | Ofte ikke lineær. Kan være lineær efter ligevægt |
| Kinetik | Langsom (> dage) |
| Hysterese | Ja |
| Konkurrence | Ja |
Adsorption på overflader af fast organisk stof |
|
| Isoterm | ikke lineær |
| Kinetik | Hurtig (<minutter hvis partiklerne ikke er sammensat i aggregater) |
| Hysterese | ? |
| Konkurrence | Ja |
Sorption ind i meso- og mikroporer til stede i jordpartiklerne |
|
| Isoterm | ikke lineær (hvis porerne er af variabel størrelse) |
| Kinetik | Langsom (> dage ) |
| Hysterese | Ja |
| Konkurrence | Ja (pga. begrænset antal sorption sites) |
Adsorption på hulrum i kondenseret fast organisk stof |
|
| Isoterm | ikke lineær |
| Kinetik | Hurtig (<minutter (efter langsom diffusion gennem det omgivende organiske stof)) |
| Hysterese | Ja |
| Konkurrence | Ja |
3.4 Sorptions-/desorptionsprocesser
Som illustreret i forrige afsnit er der flere fysisk-kemisk forskellige processer, som giver anledning til sorption/desorption af organiske stoffer i jord, og som bidrager til, at sorptionsbeskrivelsen adskiller sig fra den traditionelle beskrivelse (se afsnit 3.1). Et organisk molekyle kan således undergå flere diffusionsprocesser, før der sker en endelig sorption, eller før stoffet frigives (desorberer) fra partiklen til det mobile vand. Stoffet må (ved sorptionsprocessen) først transporteres fra den mobile væskefase gennem den stillestående vandfilm, som omgiver jordaggregatet (film-diffusion), eventuelt gennem porer i aggregatet (pore-diffusion) og endelig diffusion gennem det naturlige organiske stof (matrix-diffusion) (Pignatello og Xing 1996).
Diffusionsprocesserne er, som det fremgår af den efterfølgende gennemgang, af forskellig hastighed og vil afhænge af stoffets hydrofobicitet samt af jordtypen. Dette betyder at udvaskningen af de organiske stoffer kan ske meget hurtigt i nogle tilfælde og meget langsomt i andre. Det er p.t. ikke muligt at noget præcist om hvornår man har det ene eller det andet tilfælde. En udvaskningstest hvor væskekoncentrationen måles til forskellige tidspunkter kan afsløre den enkelte jords udvaskningshastighed (se i øvrigt kapitel 9).
3.4.1 Film-diffusion
I de fleste tilfælde vil film-diffusionen ikke være hastighedsbegrænsende for den langsigtede proces (Weber og Miller 1988), men kan formentlig godt være det for den initiale hurtige stofudveksling (Pignatello og Xing 1996). De afgørende parametre for diffusiv stoftransport er dels diffusionskoeffienten, dels den længdeskala, hvorover den diffusive transport foregår (se afsnit 3.5). Diffusionskoefficienten i væskefilmen er langt større end diffusionskoeffienter gældende for transport internt i partikler/aggregaters porer eller i selve det naturlige organiske stof. Ved en batchudvaskningstest, hvor prøven agiteres, kunne det godt tænkes, at tykkelsen eller sammensætningen af væskefilmen bliver påvirket, således at den diffusive transport gennem denne bliver anderledes (formentlig hurtigere), end den ville være i et naturligt system eller i et kolonneudvaskningssystem.
3.4.2 Pore-diffusion
Studier af de hastighedsbegrænsende processer, der er afgørende for den langsigtede udvaskning af organiske stoffer fra jorden, er krævende at udføre, idet de foregår på en mikroskopisk skala. Generelt er det nødvendigt at udvikle nye analyseteknikker, før man kan opnå mere direkte beviser på, hvilke processer der er hastighedsbegrænsende i et konkret tilfælde. Nogle af de indicier, der indtil videre er præsenteret, på at pore-diffusion er en væsentlig proces, er, at man i flere forsøg har opnået hurtigere rater, efter at jorden er blevet pulveriseret (Steinberg et al. 1987, Ball og Roberts 1991, Pignatello 1990). En syrebehandling af jordene har også givet hurtigere rater, hvilket er tolket som et resultat af opløsning af uorganiske oxider, der har virket som "cement" for mindre partikler, og deraf følgende mindre aggregatstørrelser (dvs. mindre diffusionsafstande) (Pignatello 1990).
Et enkelt jordaggregat kan – som vist på figur 3.5b – indeholde porer af helt forskellige størrelser, og porerne kan indeholde mere eller mindre naturligt organisk stof, hvilket resulterer i, at der inden for det enkelte aggregat kan være områder med en relativt høj diffusiv transport, hvor stofafgivelsen i en udvaskningssituation vil ske inden for minutter til timer, og andre områder, hvor den diffusive transport går ekstremt langsomt, og hvor stofafgivelsen vil ske over dage til uger (eller måske over endnu længere tid) (Luthy et al. 1997).
3.4.3 Matrix-diffusion i naturligt organisk stof
Flere forskere har i de seneste år set resultater, som tyder på, at diffusionsprocesser på meget lille skala i belægninger eller diskrete korn af naturligt organisk stof er hastighedsbegrænsende for stofudvekslingen (Brusseau og Rao 1989, Brusseau et al. 1991). Man har bl.a. set eksempler på lavere stofudvekslingsrater ved stigende indhold af naturligt organisk stof (Pignatello et al. 1993, Carroll et al. 1994, Pignatello 1990, Nkedi-Kizza et al. 1989) samt højere udvekslingsrater, når der tilsættes organiske cosolventer, som kan få det naturlige organiske stof til at svelle og blive mere åbent (Brusseau et al. 1991).
Som allerede beskrevet og vist på figur 3.5c kan det naturlige organiske stof bestå af en "blød" og en "hård" del. Der har været benyttet forskellige oxidationsmetoder til at skelne mellem de to typer. Den "bløde" del har været karakteriseret som den andel af det naturlige organiske stof, som kan fjernes ved vådoxidation med persulfat ved lav temperatur. Den "hårde" del er herefter blevet karakteriseret som den andel, der fjernes ved en forbrænding ved høj temperatur, men ikke ved den ovennævnte vådoxidation (Huang et al. 1998).
Som vist i tabel 3.2 betragtes sorptionen til den "bløde" del som en traditionel fasefordeling (lineær isoterm, hurtig diffusiv transport og dermed hurtig indstilling af ligevægt samt ingen konkurrenceeffekter eller forskel mellem sorption og desorption (hysterese). Derimod er sorptionen/desorptionen i den "hårde" del mere kompleks. Isotermerne menes typisk at være ikke-lineære, hvilket betyder, at den effektive diffusionskoefficient bliver mindre med faldende koncentrationer. Der er også set udtalt hystereseeffekt samt konkurrence mellem organiske stoffer til stede på samme tid. Den observerede ikke linearitet har været tolket som et resultat af en "two-site-sorptionsproces", idet den egentlige fasefordeling til det "hårde" organiske stof betragtes som traditionel lineær. Ud over denne kapacitet er det foreslået, at der findes diskrete sorptionssites i mikroskopiske huller i det organiske stof (type C på figur 3.5c), og hvor sorptionen er ikke-lineær af Langmuir typen (som forudsætter, at der er en maksimal sorptionskapacitet til stede). Den samlede sorptionsproces udviser derfor ikke linearitet (af Freundlich-typen) (Luthy et al. 1997).
3.4.4 Adsorption på overflader af naturligt organisk stof
De forudgående beskrevne sorptions-/desorptionsprocesser har generelt været absorptionsprocesser, hvor sorbenten (makroskopisk set) optages i de faste partikler. Herudover kan der også være en sorptionskapacitet i form af en adsorption til heterogene organiske overflader. Udvekslingen vil gå meget hurtigt (som de fleste adsorptionsprocesser), men isotermen vil være ikke-lineær (formentlig af Langmuir-typen på grund af den begrænsede mængde sorptionssites) (Luthy et al. 1997). Det betyder blandt andet, at man for materialer, hvor dette spiller en rolle, skal være forsigtig ved ekstrapolation eller omregning af resultater af udvaskningsforsøg udført ved ét koncentrationsniveau til andre koncentrationsniveauer.
3.4.5 Adsorption til mineraloverflader
Jorde indeholder primært uorganiske partikler som i størrelse kan spænde fra fine partikler i lerfraktionen til grovsand og grus. De fine partikler har meget store specifikke overfladearealer og kan samtidig være elektrisk ladede. Lermineraler er således overvejende negativt ladede og kan derfor have betydning for organiske baser, som i et vist pH-område er positivt ladede; derfor tiltrækkes de af negativt ladede lerpartikler, hvor de ionbytter med naturlige kationer. For neutrale organiske stoffer er sorptionen til mineraloverflader under mættede forhold oftest ikke af betydning (se Kjeldsen 1996, for flere detaljer). Under umættede forhold har de senere års forskning vist, at mineraloverfladerne kan spille en langt større rolle i sorptionen, end det er tilfældet under mættede forhold. Sorption til mineraloverflader er dog kun signifikant for jorde med et meget lavt indhold af naturligt organisk stof (foc < 0,001, Miljøstyrelsen 1996). Hvis jordpartiklerne er dækket af mindre end 5-10 lag vandmolekyler, vil de flygtige organiske stoffer indeholdt i poreluften begynde at konkurrere med vandmolekylerne om at adsorbere direkte til mineraloverfladerne (Ong og Lion 1991, Valsara og Thibodeaux 1992) – se også afsnit 3.6.2.
3.4.6 Sorptions-/desorptionshysterese
Der er flere eksempler på, at stoffer, som er sorberet til jorde, ikke frigives i samme grad, når stofferne forsøges desorberet igen, et begreb, der kaldes hysterese. Fænomenet resulterer i, at sorptions- og desorptionsisotermen ikke er sammenfaldende. Der kan være flere årsager til, at der observeres hysterese i udførte laboratorieforsøg (Huang et al. 1998):
- eksperimentelle artifacter, som giver en øjensynlig hysterese
- irreversibel binding til specifikke sorptionssites
- langsomme desorptionsrater
- "indespærring" af sorberede molekyler.
ad 1. En vigtig artifact er, at der ikke opnås ligevægt i det indledende sorptionsforsøg (hvis det naturlige organiske stof indeholder væsentlige hårde bestanddele, kan det tage op til måneder at opnå ligevægt) (Weber et al. 1998). I det efterfølgende desorptionsstep kan diffusionen foregå både længere inde i aggregatet og ude i bulkvæsken. Den observerede hysterese kan også skyldes tab af stof til forsøgsudstyret eller tilstedeværelse af partikler, som ikke separeres ud ved centrifugering.
ad 2. Der er flere eksempler på, at visse stofgrupper (bl.a. phenoler) bindes kemisk til det naturlige organiske stof og derefter kun meget vanskeligt frigives igen (Burgos et al. 1996, Sabbah og Rebhun 1997). En sådan reaktion, også kaldet kemisorption, adskiller sig fra den almindelige definition på sorption, idet den indbefatter covalent binding. En forskergruppe fra Rice University har i flere artikler beskrevet irreversibel sorption af neutrale organiske stoffer (Kan et al. 1994, Kan et al. 1997, Kan et al. 1998). For alle de jorde, som de studerede, fandtes én fraktion af sorptionen at ske som reversibel sorption. For hver kombination af jord/organisk stof fandt man en maksimal koncentration af irreversibelt sorberet stof, qirrmax, som enten kunne fyldes op på en gang eller ved flere sorptionsstep. En typisk værdi for qirrmax angives til at være (Kan et al. 1998):
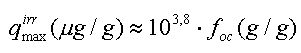
Man fandt også, at fordelingskoefficienten for den irreversible andel næsten er konstant for de ret forskellige organiske stoffer, der blev benyttet, idet man finder Kocirr=105,53± 0,48 l/kg. Efter at den maksimale irreversible sorptionskapacitet, qirrmax, er nået, er der fuld reversibilitet mellem sorption og desorption (hældningerne på sorptions- og desorptionsisotermerne er ens). Generelt findes det, at alle sorptions- og desorptionsdata kan modelleres ved hjælp af én enkel sorptionsisoterm. Isotermligningen består af to led, et lineært led, som repræsenterer den reversible sorption, og et led af Langmuir-typen, som repræsenterer den irreversible del (Kan et al. 1998).
ad. 3+4. En anden forskergruppe omkring Dr. Weber på University of Michigan har også studeret sorptionshysterese og sætter fænomenet i forbindelse med tilstedeværelse af "hårde" bestanddele i det naturlige organiske stof (se afsnit 3.4.3). De hårde bestanddele har en tre-dimensional struktur, som gør, at organiske stoffer kan blive "fanget" (Weber et al. 1998). Ud fra energibetragtninger (Gibbs frie energi) vil det være – energimæssigt – mindre fordelagtigt for sorberede molekyler at desorbere igen, hvilket giver hystereseeffekten med de meget langsomt desorberende stoffer.
Generelt må det konkluderes, at man på nuværende tidspunkt ikke har den fulde forståelse af, hvilke processer og faktorer der er styrende for sorptionshysterese. Det er dog åbenlyst, at der er mange eksempler på, at væsentlige fraktioner af sorberede stoffer er bundet på en form, som gør desorptionsprocessen meget langsommelig. Dette betyder også, at estimerede udvaskningskoncentrationer baseret på måling af totalindholdet i jorden og en fordelingskoefficient baseret på en hurtig udveksling følgende den lineære isoterm i mange tilfælde groft vil overestimere den reelle udvaskning (Kan et al. 1998), hvilket netop nødvendiggør udførelse af udvaskningstests med feltrealistiske opholdstider (se 3.6.8).
3.5 Modellering af langsom desorption
Ovenstående gennemgang af mekanismer for langsom desorption viser med stor tydelighed, at diffusionsprocesser på forskellig skala er afgørende for, hvorledes diffusionen forløber. Diffusionsprocesser modelleres oftest ved hjælp af Ficks lov:
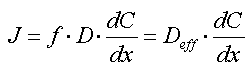
hvor
J er den diffusive flux (i f.eks. g m-2 s-1)
D er stoffets diffusionskoefficient i fasen (som oftest vand)
f er en reduktionsfaktor, som tager højde for geometriske og sorptionsmæssige forhold,
som medvirker til en reduktion af diffusionskoefficienten til den effektive
diffusionskoefficient, Deff
dC/dx er koncentrationsgradienten, således at store koncentrationsforskelle over små
afstande giver større diffusiv transport.
Denne diffusionsbeskrivelse har dannet grundlag for modellering af desorptionen af organiske stoffer fra jord, men vanskeliggøres af, at man på mikroskopiske skala skal kende typiske diffusionsafstande samt effektive diffusionskoefficienter. Den grundlæggende model af denne type er udviklet af Gschwend og medarbejdere (Wu og Gschwend 1986) og er i kortfattet udgave beskrevet i box 3.1 (for mere detaljeret beskrivelse se Kjeldsen (1996).
Som alternativ til diffusionsmodellen beskrevet i box 3.1 blev der af flere foreslået benyttet en mere empirisk tilgang til beskrivelse af desorptionsforløbet. Flere havde observeret, at en del af stoffet blev frigivet nærmest øjeblikkeligt, mens restfraktionen blev frigivet med en tilnærmet eksponentielt aftagende rate. Denne model, også kaldet bicontinuummodellen, har tidligere været benyttet i den kemiske industrier til modellering af lignende problematikker. Figur 3.6 viser koncentrationen i vandet som funktion af tiden for bicontinuummodellen. Til sammenligning er der vist en tilsvarende kurve for diffusionsmodellen. Den store forskel ses især for små tider. De grundlæggende ligninger for bicontinuummodellen er vist i box 3.2 (van Genuchten og Wieranga 1976, Brusseau og Rao 1989). Her er der også udregnet nøgletal, som fremkommer, når bicontinuummodellen indbygges i den én-dimensionelle strømningsligning. Den meget udbredte én-dimensionelle model, CXT-fit, indeholder som én af mulighederne bicontinuummodellen. CXT-fit vil kunne bruges til at vurdere effekten af desorptionskinetikken for udvaskningskoncentrationer i feltrealistiske scenarier, hvor de indgående parametre (F og k2) er bestemt via serier af batchforsøg.
En simpel sammenhæng mellem diffusions- og bicontinuummodellen kan opnås ved at udtrykke hastighedskonstanten som funktion af diffusionskoefficienten (Gong og Depinto 1998):
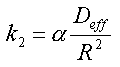
hvor
a er en korrektionsfaktor, som afhænger af tiden og
partikelstørrelsen
Deff er den effektive diffusionskoefficient i partiklen
R er retardationsfaktoren.
Box 3.1
Model for desorption baseret på diffusionsteori
| Forudsætninger De sorberende partikler forudsættes at være kugleformede, homogene partikler med samme radius. En opstilling af en massebalance for en partikel leder frem til følgende differentialligning:
hvor
hvor For et simpelt batchsystem bestående af en fast mængde jord (bestående af partikler med radius R) og en fast mængde vand, kan koncentrationen i vandet som funktion af tiden udregnes ved hjælp af ovenstående differentialligning. Løsningen opgives som et funktionsudtryk, der angiver den totale stofmængde i udvaskningsvandet til tiden t, w(t) i forhold til den endelige stofmængde ved ligevægt,w,¥ . Dette forhold vil afhænge af den dimensionsløse parameter, (D*t)/R2:
hvor F er en monotont voksende funktion med værdier mellem 0 og 1 Løsningen er vist grafisk i figur 3.6. Figuren viser funktionen F. Udover den dimensionsløse tid (D*t/R2) afhænger F af størrelsen Kdrsw,, hvor rsw, er forholdet mellem fast stof (i kg) og vand (i liter) i batchen. Benævnelsen "infinite bath" på figuren er det tilfælde, hvor mængden af partikler er så lille, at Cw kan betragtes som nul, dvs. det tilfælde, hvor den desorberede stofmængde ikke kan detekteres i udvaskningsvandet. Differentialligningen kan også danne grundlag for en model af udvaskningen fra et jordlag, idet diffusionsbeskrivelsen indbygges i en én-dimensional strømningsmodel. Ligningssystemet kan kun løses numerisk. |
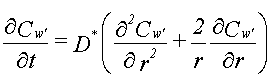
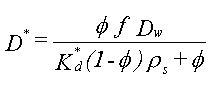
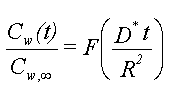
Box 3.2
Model for desorption baseret på bicontinuummodellen.
| Følgende ligninger opstilles:
hvor
Ved indbygning af ovenstående beskrivelse i den én-dimensionelle stoftransportligning fås følgende nøgletal: Damköhler tallet, w :
hvor
Damköhler tallet, w beskriver effekten af kinetik i sorptionsprocessen under givne strømningsbetingelser. De vides fra undersøgelser, at effekten af sorptionskinetik er negligibel, hvis Damköhler tallet, w , er større end 10. |
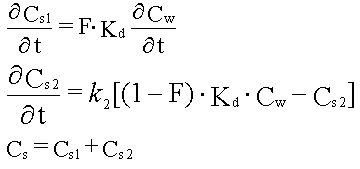
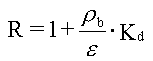
Figur 3.6
Desorptionsforløb beskrevet som væskekoncentrationen i batch som funktion af
tiden. Cw,¥ er ligevægtskoncentrationen. Forløb
både for diffusionsmodel (stiplet) og bicontinuummodel (ikke-stiplet) er vist.
Det er således tydeligt, at hastighedkoefficienten, k2, ikke er konstant over hele desorptionsforløbet. Dette forklarer, at man i flere kinetiske udvaskningsforsøg (hvor udvaskningskoncentrationens tidsafhængighed bestemmes) har haft vanskeligheder med at fitte hele kurveforløbet ved hjælp af bicontinuummodellen, (Wu og Gschwend 1986). Dette gælder også i de tilfælde, hvor diffusionsmodellen har været benyttet. Da desorptionen ofte er styret af forskellige diffusionsprocesser på forskellig skala (se figur 3.5 og tilhørende diskussion) hver med sine vidt forskellige beskrivende parametre, er det forståeligt nok, at diffusionsmodellen benyttende én partikelstørrelse og én diffusionskoefficient ikke kan modellere udvaskningen eksakt. Flere har prøvet at råde bod på dette ved at udbygge og forfine en af de to modeller. Tabel 3.3 viser et sammendrag af de forskellige modeller, som er udviklet med henblik på en mere sammenhængende beskrivelse af desorptionsforløbet. Mange af de beskrevne modeller indeholder flere parametre end de basale modeller, hvilket i sig selv ofte forbedrer en fitting til forsøgsresultater, idet der er flere parametre at "skrue" på. Den sidstnævnte modeltype i tabel 3.3 virker dog som en interessant nyudvikling. Grundtanken bag denne model er, at jorden indeholder en lang række sorptionssites, som har forskellige hastighedskonstanter. Man forestiller sig, at hastighedskonstanterne følger en fordelingsfunktion, idet man har arbejdet med modeller, hvor konstanterne følger en log-normalfordeling (Pedit og Miller 1994, Culver et al., 1997) eller en gammafordeling (Connaughton et al. 1993, Pedit og Miller 1994, Chen og Wagenet 1995, Culver et al. 1997). I begge tilfælde kan fordelingen af hastighedskonstanter beskrives ved to parametre i modsætning til andre multi-compartmentmodeller, som kræver to parametre pr. compartment. Sammenligninger af bicontinuummodellen og multi-compartment-modellen med fordelingsfunktionen faldt ud til den sidstnævnte models fordel, idet den gav en bedre overensstemmelse med forsøgsdata og samtidig var mere robust over for mindre ændringer i inddata (Culver et al. 1997).
Tabel 3.3 Se her!
Sammendrag af forskellige modeller til beskrivelse af desorptionsforløbet for
organiske stoffer fra jord (baseret delvis på Gong og Depinto 1998).
Figur 3.7 viser udløbskoncentrationen i et søjleforsøg som funktion af gennemstrømmede antal porevolumener. Forsøgsdata er med stor succes blevet simuleret med multi-compartmentmodel.
Figur 3.7
Udløbskoncentration fra søjleforsøg som funktion af gennemstrømmet antal
porevolumener simuleret med multi-compartmentmodel.
3.6 Faktorer af betydning for desorption
I dette afsnit vil nogle af de faktorer, som har eller kan have indflydelse på desorptionen blive diskuteret. En forbedret forståelse af disse faktorer vil forbedre muligheden for at forudsige, i hvilke tilfælde desorptionskinetikken vil have afgørende betydning for udvaskningen fra jord.
3.6.1 Temperatur
Flere undersøgelser har vist, at desorptionshastigheden generelt stiger med stigende temperatur. Piatt et al. (1996) fandt således en stigning i hastigheden på 1,2 - 2,6 gange ved en stigning på 22° C. Samtidig faldt fordelingskoefficienten 1,1 - 1,6 gange ved den samme temperaturstigning. Cornelissen et al. (1997) og Werth og Reinhard (1997a,b) fandt lignende sammenhænge. Generelt har temperaturen således ikke den store betydning i forhold til andre ukendte faktorer, og hensynet til desorptionsprocesserne giver derfor ikke anledning til at foreskrive en meget præcis temperaturkontrol ved udførelse af udvaskningstests. Måling af desorptionsforhold ved forskellige temperaturer kan dog benyttes til udregning af aktiveringsenergier for sorption-/desorptionsprocessen. Størrelsen af aktiveringsenergier kan belyse, på hvilken skala diffusionen foregår (pore-diffusion i meso- eller mikroporer eller matrix diffusion i naturligt organisk stof) (Cornelissen et al. 1997, Werth og Reinhard 1997b).
Forsøg til belysning af den meget langsomme desorption kan kræve meget lange forsøgstider. Hvis forsøgene gennemføres ved forhøjet temperatur (60° C), og hastighederne omregnes til feltrealistiske forhold, kan forsøgene klares på 5-10 gange kortere tid (Cornelissen et al. 1997), men andre uønskede ændringer er så til gengæld mulige.
3.6.2 Vandindhold
Ved udtørring af jorde stiger sorptionskapaciteten til den mineralske del af jorden ganske drastisk (se også afsnit 3.4.5). Det kræver dog et meget lavt vandindhold, som under danske forhold ikke er realistisk, undtagen i de allerøverste centimeter af jordprofilet i tørre somre. For normale vandindhold har vandindholdet en ringe effekt på sorptions- og desorptionsforhold (se i øvrigt Kjeldsen (1996) for en mere detaljeret diskussion af fænomenet). Vandindholdet kan således tilpasses testen uden at ændre på sorptions- og desorptionsforholdene.
3.6.3 Jordens surhedsgrad, pH
Jordens surhedsgrad, pH, kan påvirke strukturen af det naturlige organiske stof. Ved stigninger i pH ændres det naturlige organiske stof til en mere åben struktur, hvilket giver hurtigere desorptionsrater (Sahoo og Smith 1997). Der skal dog ændringer på flere pH-enheder til for at se en effekt. For mange organiske syrer og baser vil pH-ændringer direkte påvirke stoffernes ioniseringsgrad og dermed fordelingskoefficienten (se afsnit 3.7). I pH-statiske udvaskningstests på jord udført ved høje pH-værdier (f.eks. over pH = 11) ses ofte kraftig forøgelse af indholdet af totalt opløst kulstof. Det opløste organiske kulstof kan ved sorption og kompleksbinding medvirkedels til mobilisering/udvaskning af andre organiske komponenter, som ikke direkte mobiliseres af det høje pH, dels til mobilisering af uorganiske sporelementer, herunder blandt andet Cu.
Der er store forskelle i stoffers hydrofobicitet, et forhold, som har stor betydning for desorptionsrater. Desorptionen er som nævnt flere gange styret af diffusionsprocesser på flere skalaniveauer. Uafhængigt af skala kan processen beskrives som vist i box 3.1. En afgørende parameter for desorptionsraten er den effektive diffusionskoefficient beskrevet ved ligningen:
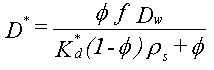
Hydrofobe stoffer har relativt store molekylevægte, hvilket giver lave diffusionskoefficienter i vand, Dw. Den resulterende effekt er meget lave effektive diffusionskoefficienter. Desorptionsrater for PAH’er og lignende er således meget mindre end for benzen eller trichlorethylen for den samme jord, hvilket der skal tages højde for ved fastsættelse af kontakttiden i forbindelse med udførelse af udvaskningstests for disse komponenter.
3.6.5 Konkurrence mellem stoffer til stede på samme tid
Som allerede diskuteret i afsnit 3.4 vil der forekomme konkurrence for nogle af de tilstedeværende sorptionssites. Især for den sorptionskapacitet, som karakteriseres med ikke-lineære isotermer. Dette gælder sorption til den hårde fraktion af det naturlige organiske stof (herunder Langmuir-adsorption til hulrum i det hårde organiske stof) samt adsorption til overflader af naturligt organisk stof (se tabel 3.2). Konkurrerende stoffer kan både være naturligt forekommende organiske stoffer i jorden (Xing og Pignatello 1998) og andre kemikalier (McGinley et al. 1996, Xing et al. 1996). For den del af sorptionen, der sker til den amorfe fraktion af det naturlige organiske stof, og hvor karakteristiske isotermer er lineære, ses der ingen konkurrenceeffekter (McGinley et al. 1996).
3.6.6 Kontakttid ("ældning")
Flere forsøg har i de seneste år vist, at den tid, hvor stofferne har været i kontakt med jorden, er afgørende for desorptionsraterne. Disse observationer er især gjort for hydrofobe stoffer (især PAH’er), hvor desorptionsrater generelt er lave. Carmichael et al. (1997) viste dette ved at tilsætte kulstof-14-mærkede PAH’er til en jord, som i forvejen var forurenet med de samme PAH’er (som naturlige/kulstof-12-stoffer). Jorden var prøvetaget fra en træimprægneringsanstalt, hvor træ var blevet behandlet med kreosot siden slutningen af 1800-tallet. En sammenligning viste, at desorptionsrater for de nyligt tilsatte PAH’er var væsentlig større end desorptionsrater for de oprindelige PAH’er (der sås op til en faktor 1000 til forskel). Lignende observationer er gjort i andre forsøg (Pignatello et al. 1993). Gong og Depinto (1998) så også faldende pore-diffusionskoefficienter med stigende kontakttider for PCB’er og så samtidig, at den øjeblikkelige ligevægtsfraktion (størrelsen F - se box 3.2) ligeledes faldt.
Årsagerne til de observerede ældningsfænomener kan være flere. Som nævnt i afsnit 3.5.4 er PAH’er meget hydrofobe stoffer, hvis diffusionskoefficienter – især i mikroporer eller i den hårde fraktion af det naturlige organiske stof – kan være så små, at det tager årtier at opnå fuldstændig ligevægt. Stoffer, som tilsættes, vil inden for det relativt korte tidsrum ikke have nået at diffundere helt ind i partiklernes indre, hvor diffusionshastighederne er meget små, og vil derfor desorbere hurtigere end det oprindelige stof. En anden årsag kan være, at der sker ændringer i partiklernes indre geometri, som fører til, at stofferne spærres inde.
Observationerne viser klart, at man ikke kan studere udvaskning af hydrofobe stoffer på kunstigt forurenede jorde – undersøgelserne skal ske på prøver udtaget i felten på den konkrete lokalitet.
3.6.7 Jordens alder og sammensætning
Flere har vist, at jordens geologiske alder samt dens oprindelse har stor betydning for sorptions-/desorptionsforhold. Unge jorde udviser generelt lavere fordelingskoefficienter til det naturlige organiske stof (Koc), og isotermerne er generelt lineære. Kile et al. (1995) så således for et stort antal overjorde med et indhold af organisk kulstof på 0,2-6% lineære isotermer og Koc-værdier for tetraklorkulstof fra 45-75 l/kg. De fandt i øvrigt generelt højere Koc-værdier for bundsedimenter. De fandt også meget højere Koc-værdier for jorde forurenet med diverse organiske kemikalier, herunder olie. Huang og Weber (1997) undersøgte 10 forskellige naturlige sorbenter (fra unge tørvejorde til kerogen isoleret fra geologisk gamle skiffer-prøver). Sorbenternes organiske stof blev karakteriseret ved forholdet mellem indholdet af ilt og kulstof, idet kulstoffraktionen i det naturlige organiske stof stiger med den geologiske alder af materialet. Figur 3.8 viser, at der opnås højere Koc-værdier ved faldende O/C-forhold (og højere alder) samt en højere grad af ikke-linearitet (lave Freundlich-eksponenter) ved faldende O/C-forhold.
Figur 3.8
a) Værdier for logKoc som funktion af indholdet af ilt (O) og kulstof (C) i
det naturlige organiske stof repræsenteret ved O/C-forholdet. Forsøgene er gennemført
ved høj koncentration (1000 m g/l) og lav koncentration (1 m g/l) b) Værdier for isotermens ikke-linearitet som funktion af
O/C-forholdet i det naturlige organiske stof (Huang og Weber 1997).
Ligeledes observeredes der udtalt hysterese for de gamle skifferprøver (se figur 3.9). Kleinedam et al. (1999) observerede også udtalt ikke-linearitet (lave Freundlich-eksponenter (1/n)) for geologisk gamle sorbenter. De nævnte observationer passer godt overens med forventningen om, at jorde med stigende geologisk alder får en større andel af hårdt, kondenseret organisk stof, et faktum, der kendes fra organisk stofs diagenetiske ændring til stenkul over en tidshorisont på millioner af år.
Figur 3.9
Phenanthrensorptions- og desorptionsisoterm for Norwood-skiffer. De to isotermer er
ikke sammenfaldende hvilket illustrerer en klar hystereseeffekt (Huang og Weber 1997).
Ud over O/C-forholdet er jorde blevet karakteriseret på forskellig vis med henblik på at bedre kunne forudsige sorptionsprocessen. Xing (1997) benyttede metoder til at karakterisere det naturlige organiske stofs polaritet og aromatiske indhold. Metoderne blev benyttet på fem jorde af forskellig alder og diagenetisk baggrund (omdannelsesstadier) med henblik på karakterisering af jordenes naphthalensorption. Forsøget viste, at fordelingskoefficienten, Koc, steg med stigende aromatisk indhold og faldende polaritet. Kleinedam et al. (1999) målte Koc-værdier for fraktioner af akvifermaterialer, idet materialet opdeltes efter de forskellige partiklers lithologiske oprindelse (sandsten, kalksten, granit, skiffer, m.m.), også kaldet lithokomponenter. Der blev fundet en faktor 590 mellem højeste og laveste Koc-værdi for sorption af phenanthren. De højeste værdier blev fundet i lithokomponenter, hvor det naturlige organiske stof udgjordes af kulpartikler, formentlig stammende fra skovbrande, som er blevet indlejret i de sedimentære bjergarter.
Forskellige fraktioneringsmetoder af jorde har været forsøgt for yderligere at karakterisere sorptionen. Barber (1994) fraktionerede akvifermaterialer efter partikelstørrelse. Undersøgelsen viste, at akvifermaterialernes indhold af naturligt organisk stof især var tilknyttet til fraktionerne med de mindste partikelstørrelser, som derved udviste de største fordelingskoefficienter, Kd.
Holmén, Gschwend (1997) fandt for tre akvifermaterialer, at sorptionskapaciteten var tilknyttet belægninger på akvifermaterialets partikler, som især bestod af kvarts. Belægningerne bestod især af metaloxider, der virkede som "bindemiddel" for underliggende naturligt organisk stof, der normalt antages at sidde yderst. Bestemmelse af fordelingskoefficienter, Kd, ved batch- og søjleforsøg viste divergerende resultater, idet man fandt meget lavere Kd-værdier i søjleforsøget. Dette blev tolket som et resultat af erosion af belægningerne ved batchforsøgene, hvorved naturligt organisk stof, som oprindeligt var utilgængeligt for sorption af stoffer, var blevet frilagt og derved deltog i sorptionsprocessen. Sorptionskinetikken blev med succes modelleret med en pore-diffusionsmodel, hvor det blev forudsat, at diffusionen kun skete i belægningerne, som alene indeholdt sorptionskapacitet.
3.6.8 Strømningshastighed
De meget store ligevægtstider, som er blevet observeret i et stort antal laboratorieforsøg, viser, at der vil være mange tilfælde, hvor den desorptionsbetingede udvaskning i felten må forventes at være betydeligt mindre, end hvis man forudsætter ligevægt mellem jorden og det porevand, som gennemstrømmer det forurenede jordlag. Derfor vil en analyse af typiske opholdstider for porevandet i det forurenede jordlag (herunder en vurdering af fordelingen af typiske opholdstider i et konkret forurenet jordlag) være en nødvendig aktivitet for at kunne vurdere, under hvilke kontakttider eventuelle laboratorieudvaskningstests skal gennemføres, såfremt de præcist skal simulere de pågældende feltforhold. Under alle omstændigheder er det væsentligt at vide, om kontakttiden i et givet naturligt system er større eller mindre end den påkrævede tid for opnåelse af ligevægt for de komponenter, hvis udvaskning ønskes undersøgt. Ved valg eller indretning af testmetode kan der så eventuelt tages hensyn hertil eller oplysningen kan som et minimum indgå i fortolkningen af testresultatet.
I Miljøstyrelsen (1996) angives typiske strømningshastigheder på 10-1000 m/år i den mættede zone og 0,5-2 m/år i den umættede zone (den aktuelle hastighed på en lokaliet kan estimeres udfra kendskab til den aktuelle nedbør samt jordbundsforhold). Opholdstiden i et jordelement på 1 m bliver med disse strømningshastigheder 0,5-2 år i den umættede zone og 0,001-0,1 år i den mættede zone, hvilket indikerer, at opholdstiden i nogle naturlige systemer kan være så lille, at der ikke vil være opnået sorptionsligevægt.
3.7 Specielle forhold for polære/ladede stoffer
De seneste fem års sorptionsforskning har næsten udelukkende beskæftiget sig med neutralt ladede stoffer. Der er således meget få nye erkendelser for ikke-neutrale stoffer (organiske syrer og baser) i det nævnte tidsrum, og der kan derfor henvises til Kjeldsen (1996) – afsnit 7.2.5 og 7.2.6 – for en gennemgang af sorptionen for disse stoffer. Det skal dog nævnes, at man er blevet mere opmærksom på, at phenoler kan indgå en kemisk binding (oxidativ kobling) med det naturlige organiske stof. I dette tilfælde vil stofferne praktisk talt ikke udvaskes på grund af den meget hårde binding (se afsnit 3.4.6).
Desorptionen af organiske syrer og baser styres, udover af de i tidligere afsnit beskrevne faktorer, af pH og stoffets pKa-værdi. Syrekonstanten, pKa (= -logKa), angiver den pH-værdi, hvor halvdelen af stoffet er ioniseret, og den anden halvdel er på neutral form. For en organisk syre vil størstedelen af stoffet være negativt ladet ved højt pH og neutral ved lavt pH. Dette betyder, at desorptionen vil stige ved stigende pH (over pKa-værdien). Mange organiske syrer, som er miljørelevante, har dog relativt høje pKa-værdier, hvorfor disse stoffer praktisk talt vil være fuldt neutrale ved miljørelevante pH-værdier. Dette gælder dog ikke for de fleste chlorphenoler, især de flersubstituerede, som har pKa-værdier i det miljørelevante pH-område - se også (Nielsen et al. 1996).
4. Betydningen af kolloidtransport
| 4.1 | Grundlæggende teori |
| 4.2 | Eksempler på observeret kolloidfaciliteret transport |
| 4.3 | Væsentlige faktorer for kolloidfaciliteret transport |
| 4.4 | Kolloider og testning af stofudvaskning |
4.1 Grundlæggende teori
Kolloider er betegnelsen for mikroskopiske eller submikroskopiske organiske eller uorganiske partikler suspenderet i en væskefase. Kolloider i porøse medier af interesse for udvaskningsundersøgelser spænder fra partikler af biologisk oprindelse (bakterier, virus, organisk materiale) og mineralske partikler (lermineraler, mineralske udfældninger, metaloxider og -hydro-xider) til kombinationer af de to (f.eks. lermineraler med humuskomponenter adsorberet). Kolloider er normalt defineret på basis af deres størrelse. Partikler større end 10 mm sedimenterer hurtigt og bruges som øvre grænse for kolloider. Nedadtil er kolloider afgrænset ved overgangen til rent opløste molekyler ved ca. 1-10 nm (Stumm 1992).
Kolloider udgør således en separat ekstra fase i et vandmættet porøst medie, der traditionelt betragtes som bestående af det faste medie og porevæsken (her ses der bort fra en eventuel fri fase af organisk forurening). På grund af det relativt store overfladeareal per masseenhed er sorptionskapaciteten af den kolloide fraktion over for miljøfremmede stoffer ofte meget større end for det porøse medie som helhed. Dette skyldes til dels også, at kolloiderne generelt har et højere indhold af naturligt organisk materiale (se afsnit 3.6.7). Naturligt forekommende kolloider kan dermed have følgende effekter på transport og udvaskning af organiske forureningsstoffer:
| En større fraktion af stoffet findes i væskefasen (via en binding til suspenderede kolloider) end ellers antaget ud fra en to-fase betragtning | |
| Stofferne er potentielt mere mobile, idet de med de suspenderede kolloider kan blive ført med porevandet. Denne proces kaldes kolloidfaciliteret transport | |
| Transport af stofferne er ikke udelukkende betinget af stoffernes frigivelse fra den faste fase til væskefasen, men er i høj grad bestemt af de processer, der styrer kolloidernes stabilitets- og transportegenskaber. |
4.2 Eksempler på observeret kolloidfaciliteret transport
Inden for de sidste snes år er der blevet forsket en del i kolloiders betydning for transport af forureningsstoffer. Der er flere studier, der viser, at kolloider kan forøge mobiliteten af organiske forureningsstoffer (PAH, pesticider, PCB) med høj bindingsevne under kontrollerede laboratorieforhold (Villholth et al. 1997). Det er straks vanskeligere at påvise, at kolloiderne er en væsentlig faktor i transporten under feltforhold. Dette hænger sammen med metodiske og fortolkningsmæssige problemer. F.eks. kræver det stor omhyggelighed at udtage prøver af jord- og grundvand med et naturligt forekommende uforstyrret indhold af kolloider ("kunstige" kolloider kan let medrives). Desuden er det nødvendigt at separere kolloiderne fra vandfasen for at bestemme indholdet af forurenende stof i den kolloide fase, hvilket kan volde metodiske problemer. Endelig er det svært at dokumentere, at eventuelt kolloidbundet stof i en udtaget væskeprøve også er blevet transporteret via kolloiderne og ikke bare er blevet bundet efter transport i opløst form. De påvisninger, der er gjort i felten, er derfor baseret på mere indirekte observationer (se review af Villholth et al. 1997):
| Kolloider forekommer naturligt i alle jord- og grundvandsmiljøer (McCarthy og Degueldre 1993) | |
| Det sorberende forureningsstof er transporteret hurtigere eller over længere afstand end muligt ud fra en antagelse om udelukkende opløst transport | |
| Transport af injicerede, detekterbare kolloider er påvist | |
| Forureningsstoffer, der er bundet til naturlige kolloider i jord og grundvand, er fundet nedstrøms forureningskilden. |
Som eksempel på det sidste fandt Villholth (1999) under feltforhold, at op til 35 % af individuelle 4- til 5-ringede PAHer var associeret med naturligt forekommende organisk-holdige kolloider i et forurenet grundvandsmagasin. Fraktionen af PAH bundet til kolloider (her defineret som > 0,1 mm) var positivt korreleret med hydrofobiciteten af det enkelte PAH stof. Disse resultater viser, at en ikke uvæsentlig del af de stærkt bindende stoffer i vandfasen potentielt kan findes som kolloidbundne.
4.3 Væsentlige faktorer for kolloidfaciliteret transport
For at kolloidfaciliteret transport i porøse medier skal have miljømæssig betydning kræves det:
| at der forekommer mobile/stabile kolloider i tilstrækkelig koncentration | |
| at der findes transportveje, der tillader transport af kolloider | |
| at forureningsstoffer sorberes stærkt til de mobile kolloider |
Som naturlige bestanddele af porøse medier genereres kolloider ofte ved en løsrivelse fra det faste medie. Dette kan forekomme ved dispergeringsprocesser, hvor frigivelsen er betinget af elektrostatisk frastødning mellem kolloider og kornskelettets overflade forårsaget af ændringer i porevæskens kemi. Kolloider kan også løsrives ved mekanisk påvirkning, hvor strømningshastighederne er tilstrækkeligt høje. Andre mekanismer indbefatter en frigivelse ved pH- eller redoxinduceret opløsning af en cementerende fase på kornskelettet, f.eks. bestående af jernoxider eller kalciumkarbonat. Endelig kan kolloider dannes ved kemisk udfældning af mineralske faser og ved tilførsel udefra, f.eks. via infiltrerende vand, der indeholder overfladeeroderede jordpartikler eller organisk stof og mikroorganismer. At kolloidtransport er et naturligt fænomen, fremgår af resultatet af pedogene og jordbundsforvitrende processer, der kommer til udtryk ved illuvieringshorisonter i jordprofiler, hvor kolloider bestående af lerpartikler og organisk materiale er akkumuleret som følge af nedvaskning og efterfølgende immobilisering.
Transportforholdene for kolloider adskiller sig fra transportforholdene for opløste stoffer, idet kolloidernes større størrelse medfører en signifikant mindre diffusionshastighed og samtidig en udelukkelse fra de fineste poresystemer. Porestrukturen er således meget væsentlig for kolloidtransporten, og uforstyrrede jorde vil typisk være mere permeable for kolloider end forstyrrede, ompakkede jorde på grund af et naturligt indhold af makroporer og sprækker.
Sorptionsforholdene mellem kolloid og kontaminant er af afgørende betydning for kolloidfaciliteet transport. Der kan i princippet være tale om de samme sorptionsprocesser og -mekanismer, der kendes fra sorption til det stationære faste medie, og som er beskrevet i afsnit 3. Dog kan sorptionsparametre bestemt for det totale medie ikke nødvendigvis antages gældende for den kolloide fraktion. Derimod vil sorptionsforholdene bedre kunne fastlægges ud fra en undersøgelse af den fineste størrelsesfraktion (lerfraktionen) eller en separat isoleret kolloidfraktion. For organiske forureningsstoffer vil sorptionen, og dermed muligheden for kolloidfaciliteret transport, være størst for de hydrofobe stoffer og for stoffer, der danner stærke komplekser med mineralske komponenter eller naturligt organisk materiale. Men da bindinger af disse stoffer generelt også er stor til det faste medie, vil den overordnede effekt af kolloiderne i høj grad ofte være betinget af andre faktorer, nemlig kolloidernes eventuelle relativt højere sorptionskapacitet, eventuel irreversibilitet i sorptionen til kolloiderne og kolloidkoncentrationen.
4.4 Kolloider og testning af stofudvaskning
I forbindelse med udvikling af udvaskningstests for forureningsstoffer må det vurderes, i hvilke tilfælde det vil være relevant at tage højde for effekten af kolloidtransport, og hvordan en testmetodik kan tilgodese dette.
Med hensyn til de situationer, der kan være aktuelle, vil faktorer som det(de) pågældende stof(fer), det anvendte medie og de påtænkte udvaskningsforhold tages i betragtning. Retningslinier for, hvilke stoffer der kan komme på tale, må ideelt baseres på stoffernes bindingsevne og -reversibilitet til kolloidfraktionen i den pågældende jord. Mere generelle retningslinier kan baseres på forholdsvis simple og lettilgængelige data for stoffernes sorptionskapacitet, f.eks. ligevægtsfordelingskoefficienten for stofferne udtrykt ved Kd eller Koc. For hydrofobe organiske stoffer, f.eks. PAH'er i jord og grundvandsmedier, vil det erfaringsmæssigt være relevant at inddrage de fire-ringede komponenter, svarende til logKoc større end 4,5. For kompleksdannende organiske stoffer vil mere komplekse forhold spille ind, så som de konkrete bindingsmekanismer, kolloidernes sammensætning og pH i udvaskningsmediet (se afsnit 3.7).
Med hensyn til prøvemediet og de gældende udvaskningsforhold vil de faktorer, der kan have indflydelse på kolloiddannelsen og -transporten, være de væsentligste. F.eks. vil der forventes genereret højere kolloidkoncentrationer i ikke-stabiliserede granulære medier og ved anvendelse af de-ioniseret udvaskningsvand, som på grund af meget lav ionstyrke fremmer kolloidstabiliteten og dermed -frigivelsen. Batch- og kolonnetests må forventes at give forskellige kolloidkoncentrationer, idet der i batchtests på grund af den store mekaniske påvirkning kan frigives et større og måske ikke repræsentativt indhold af kolloider. Tilsvarende kan forbehandling af prøver, der inkluderer tørring, nedknusning og sortering, bevirke, at kolloidindholdet under testning forøges væsentligt.
De mere kvantitative aspekter af testmetodikkens indflydelse på kolloidkoncentrationer i eluatet og dermed indirekte på evt. kolloidfaciliteret transport er ikke blevet undersøgt systematisk, og derfor er det på nuværende tidspunkt ikke muligt at give mere håndfaste retningslinier for, hvilke fremgangsmåder der er mest hensigtsmæssige med hensyn til både enkelthed og repræsentativitet. I traditionelle udvaskningstests er en del af problematikken undveget og simplificeret ved operationelt at definere 0,45 mm som afskæringsstørrelsen mellem potentielt mobile og ikke-mobile partikler. Typisk filtreres således eluatet gennem 0,45 mm filtre, og der analyseres udelukkende på filtratet, altså fraktioner mindre end 0,45 mm. Der foreligger imidlertid ikke noget videnskabeligt bevisgrundlag for, at denne metode giver det bedste mål for faktisk udvaskeligt stof. Indledende undersøgelser fra kolonnetests tyder på, at udvaskningen afhænger af den filterporestørrelse, der anvendes, svarende til, at partikler større end 0,45 mm (op til 10 mm) også bidrager til udvaskningen (Wahlström et al. 1994, Bauw et al. 1991). Desuden er denne filtereffekt større for mere hydrofobe stoffer (Bauw et al. 1991). Separation via centrifugering er også blevet anvendt. Ved sammenligning med filtrering fandtes der højere koncentrationer af PCB i opløsning efter centrifugering end efter filtrering, og den gule farve af eluat var bibeholdt efter centrifugering i modsætning til efter filtrering (Rood et al. 1994). Dette blev tolket som en manglende faseseparering af opløst organisk stof ved centrifugering, som dermed gav anledning til binding af mere PCB i den "opløste" fase.
Afslutningsvis kan det siges, at der resterer en del forskning for at udvide kendskabet til testmetodikkens indflydelse på kolloideffekten ved udvaskning af stærkt sorberende miljøfremmede stoffer. Der er kun udført én forsøgsserie, som beskriver denne effekt direkte (Broholm et al. 2000). Batchudvaskningsforsøg med PAH-forurenet jord viste, at der blev dannet en stor mængde partikler i størrelsen 0.45 –10 mm. Fraktionen var større ved en kontaktid på 10 dage end ved en kontakttid på 28 timer, hvorimod der i en kolonneudvaskningstest med en tilsvarende jord ikke blev observeret nogen væsentlig partikeldannelse. Store mængder af især tunge PAH’er var associeret til partikelfraktionen fra batchforsøgene. Disse forsøg viser, at den agitation, som i accelerede batchforsøg anvendes for at sikre opblanding og ligevægt, skaber partikler, som efterfølgende i betydeligt omfang må fjernes, hvis resultaterne af et batchforsøg skal kunne anvendes til at beskrive (aspekter af) udvaskiningen under feltforhold.
På det eksisterende grundlag må det således anbefales, at resultater fra allerede udførte tests bør vurderes kritisk med hensyn til forsøgsbetingelser for kvalitativt at vurdere, om der er tale om repræsentative udvaskningsforhold med hensyn til kollidfrigivelse og mobilitet. Hvis testresultater foreligger for tests udført under repræsentative forhold med hensyn til kolloidfrigivelse og –mobilitet (se senere), vil en eventuel korrektion af den beregnede udvaskning, der tager hensyn til bidrag fra kolloider, tilnærmelsesvist kunne laves ved at tage hensyn til det i standardmetoden fjernede (ved filtrering eller centrifugering) partikelmateriale og dets sorptionskapacitet overfor forureningsstoffet. Et groft skøn kan opnås ved at antage, at der er ligevægt mellem forureningsstoffet i ’vandfasen’ (typisk <0,45 mm) og partikelfasen, eller det organiske stof i partikelfasen i tilfældet af hydrofobe organiske forureningskomponenter. Bidraget til udvaskningen fra partikelfasen skal herefter adderes til bidraget fra ’vandfasen’. Denne korrektionsmåde vil ikke være anvendelig, hvis testen har givet anledning til et ikke-repræsentativt frafiltreret partikelindhold, som f.eks. efter en batchtest.
Med hensyn til udvikling og udførelse af nye tests for stoffer af relevans for kolloidproblemstillingen bør der sigtes på:
| at anvende medieprøver, der er så lidt forbehandlede som muligt | |
| at tests udføres som kolonnetests, helst med uforstyrrede medieprøver, dvs. prøver i deres naturlige pakning | |
| at udvaskningsvandet i kemisk sammensætning tilnærmes det i felten forekommende | |
| at strømningshastigheden er realistisk | |
| at minimal fraktionering (eller kun ved høj afskæring, f.eks. 10 m m) af eluat foretages før analyse af udvasket stofkoncentration. |
Det forventes på basis af den foregående gennemgåede teori, at sådanne testbetingelser vil give det mest realistiske estimat af udvaskningen, hvor kolloideffekten er repræsentativt inkluderet. Hvis der ydermere ønskes et konkret mål for, hvor stor denne effekt er, kan det opnåede eluat efterfølgende fraktioneres til en langt mindre afskæring, f.eks. 10 nm. En analyse af fraktionen mindre end 10 nm kan således antages som et udtryk for den opløste fraktion, og fraktionen 10 nm - 10 mm som den kolloide.
Endelig er det i forbindelse med evaluering af kolloiders betydning essentielt, at analysemetoden for udvaskningskoncentrationen i eluatet er i stand til kvantitativt at bestemme den andel af forureningsstoffet, der er bundet til den kolloide fraktion.
5. Udvaskning af organisk stof fra restprodukter
5.1 Generelt
Den øvrige del af rapporten har taget udgangspunkt i udvaskning af organiske stoffer fra (forurenet) jord. Det skyldes ikke mindst, at de fleste erfaringer med udvaskning af organiske stoffer er gjort med jord. Der er imidlertid også et behov for at kunne teste og beskrive udvaskningen af organiske stoffer fra restprodukter såsom slagger og flyveaske fra affaldsforbrænding og kulfyrede kraftværker, knust beton, m.v. i forbindelse med vurdering af de potentielle miljømæssige effekter af at nyttiggøre eller deponere disse. I denne sammenhæng vil kun udvaskning af organiske stoffer fra sådanne overvejende mineralske restprodukter på granulær form med et begrænset indhold af organisk materiale blive diskuteret.
Som allerede nævnt i indledningen vil de fleste af de forhold, som påvirker og har betydning for udvaskningen af organiske stoffer fra jord/forurenet jord, kunne forventes at have en tilsvarende betydning for udvaskningen af organiske stoffer fra restprodukter. Nogle restprodukter, f.eks. forbrændingsslagger, adskiller sig fra jord ved at være mere heterogene, både hvad angår sammensætning og tekstur, mens andre, som f.eks. flyveasker, snarere er mere ensartede end mange jordtyper. For mange restprodukter vil det gælde, at indholdet af organiske forureningskomponenter er en mere eller mindre integreret del af materialet, mens der for jords vedkommende normalt er tale om forureningskomponenter, som er tilført jorden udefra, og som derfor fortrinsvis er associeret med overfladen af jordpartiklerne eller porerne i disse. I modsætning til de fleste jordtyper reagerer en del restprodukter kemisk med vand, når de befugtes eller gennemstrømmes, og dette kan eksempelvis give anledning til mere ekstreme pH-værdier i vandfasen, end man normalt ser for jord. Dette gælder bl.a. ofte for restprodukter fra højtemperaturprocesser, som kan indeholde oxider, der hydrolyseres og skaber stærkt alkaliske forhold. Under alkaliske forhold forøges frigivelsen og opløseligheden af organisk kulstof fra det residuale organiske stof i restprodukterne, og eluatets indhold af opløst organisk kulstof (DOC, dissolved organic carbon) øges. Det er påvist, at et forøget indhold af DOC i eluatet gennem kompleksdannelse eller chelatering kan øge opløseligheden af flere spor-elelementer, herunder bl.a. Cu (van der Sloot et al. 1997), og der er sand-synligt, at større mængder frigjort DOC kan optræde som transportmiddel for adsorberede/absorberede hydrofobe stoffer som f.eks. PAH’er. En tysk undersøgelse har vist, at den dominerende gruppe af organiske stoffer i affaldsforbrændingsslagger er fedtsyrer, d.v.s. de primære og letopløselige nedbrydningsprodukter af det oprindelige organiske materiale (Prieste et al. 1996). Risikoen for yderligere biologisk nedbrydning af disse fedtsyrer ved længerevarende udvaskningstests vil sandsynligvis være betydelig, hvilket må tages i betragtning, når sådanne forsøg planlægges.
5.2 Anvendelse af eksisterende testmetoder til testning af udvaskning af DOC/NVOC
Selv om der er tale om organiske stoffer, er der ofte blevet foretaget bestemmelse af samleparameteren DOC (målt som ikke-flygtigt organisk kulstof, NVOC (non-volatile organic carbon)) på det filtrerede eluat fra udvaskningstests beregnet til bestemmelse af udvaskningen af uorganiske komponenter uden speciel hensyntagen til valg af materialer til test- og filtreringsudstyr. Ved kolonneudvaskningsforsøg og serielle batchudvaskningsforsøg med danske affaldsforbrændingsslagger er der typisk fundet udvaskede mængder af NVOC på 100 – 200 mg/kg (Hjelmar 1999) med koncentrationsniveauer i de enkelte eluater variende fra 150 – 200 mg/l til omkring 0,5 – 1,0 mg/l. Ved femdobbelte serielle batchudvaskningsforsøg med en flyveaske fra et forbrændingsanlæg udført i polypropylenflasker i henhold til prEN 12457-3, er der under antagelse af normalfordelte resultater fundet en gennemsnitsværdi for den udvaskede mængde NVOC ved L/S = 2 l/kg på 2,96 mg/kg med en standardafvigelse på 0,95 mg/kg, svarende til en relativ standardafvigelse på 32% (Hjelmar 1999). Eluatkoncentrationerne varierede mellem 1,0 og 2,0 mg/l. For flyveaskens vedkommende er der tale om forholdsvis lave værdier af NVOC, og det er ikke muligt at sige, om de fundne resultater alene er udtryk for tilfældige variationer, eller de også indeholder en systematisk adsorption/afgivelse af NVOC på/fra test- og/eller filtreringsudstyret. For forbrændingsslaggens vedkommende er koncentrationsniveauet af NVOC derimod så højt, at det forekommer rimeligt at antage, at de fundne værdier udgør en reel angivelse af størrelsesordenen af de udvaskede mængder NVOC. Det vil være hensigtsmæssigt og forholdsvis enkelt at undersøge disse forhold nærmere, således at der kan opnås en kvalificeret vurdering af, i hvor høj grad man kan tillade sig at lade NVOC indgå i analyseprogrammet for eluater fra udvaskningsforsøg udført med henblik på bestemmelse af udvaskningen af uorganiske stoffer.
5.3 Udvaskning af specifikke organiske stoffer
For udvaskningsforsøg med henblik på bestemmelse af udvaskningen af specifikke organiske stoffer fra restprodukter gælder i øvrigt stort set de samme betragtninger og overvejelser, som i rapportens øvrige kapitler er gjort vedrørende udvaskning af organiske stoffer fra jord, dog med hensyntagen til de ovenfor nævnte forhold, hvor restprodukter ofte har egenskaber, der afviger fra normal jords egenskaber.
Som nævnt tidligere er de fleste erfaringer med udvaskning af organiske stoffer knyttet til testning af jord, og der findes rent faktisk kun ganske få egentlige undersøgelser af restprodukter beskrevet. Dette bekræftes blandt andet af Larsson (1996). Sidstnævnte refererer blandt andet undersøgelser foretaget af andet van Houdt et al. (1991), Kjeldsen og Grundtvig (1995) og Wahlström et al. (1994):
Van Houdt et al. (1991) undersøgte udvaskningsegenskaberne for forskellige typer vejbygningsmaterialer som bl.a. indeholdt PAH’er (der analyseredes for de 16 PAH’er, som foreskrives af US EPA). Materialerne blev ved hjælp af fryseteknik nedknust til < 0,2 mm, og de blev derefter underkastet rysteforsøg med vand ved L/S = 5 l/kg i 4 timer, hvorefter vandfasen ved centrifugering blev fraskilt og analyseret. Processen blev gentaget, hvorved der fandtes yderligere udvaskeligt PAH, d.v.s. resultatet af den først udvaskning var altså ikke udtryk for den maksimalt udvaskelige mængde PAH. Materialerne indeholdt 0,1-32 mg PAH/kg tørstof og gav 7-170 mg PAH/l (0,035-0,85 mg/kg tørstof) i første udvaskning og 2-56 m g PAH/l (0,01-0,28 mg/kg tørstof) i anden udvaskning. Resultaterne viser, at det kan være svært at opnå fuldstændig udvaskning ved batchforsøg, med mindre der udføres flere i sekvens.
Noget tilsvarende blev fundet af Kjeldsen og Grundtvig (1995), som anvendte en lidt anden metodik til at undersøge udvaskningen af BTEX fra industrielt affald fra gamle deponier. Der udførtes et antal serielle batchudvaskningsforsøg i glasflasker med teflonbelagte låg, der først påfyldtes iltfrit vand, som for at forhindre mikrobiel nedbrydning var tilsat 2g natriumazid/l. Herefter tilsattes de faste prøver i våd tilstand, efter at tørstofindholdet var bestemt på separate prøver. Mængderne af væske og faststof justeredes, så head space blev minimeret for at undgå fordampningstab af materiale til luftfasen. Udvaskningsforsøgene udførtes som 5 henholdsvis 24 serielle ekstraktioner ved L/S = 2 l/kg og L/S = 20 l/kg og 20 timers kontakttid (rotation om tværaksen ved 1 rpm), hvor væske- og faststoffase mellem hver ekstraktion blev adskilt ved centrifugering (14 cm rotationsradius, 30-90 min ved 1500 rpm, udtagning med pipette). Væskefaserne ekstraheredes med pentan og analyseredes ved GC-FID for BTEX og TOC (se også Lyngkilde og Christensen 1992a og b). Metodens reproducerbarhed blev vurderet som god. Koncentrationsniveauet af toluen i eluaterne fra batchudvasknings forsøgene faldt dog meget hurtigere end forudsagt på grundlag af teoretiske beregninger baseret på en målt Kd-værdi. Dette blev tilskrevet fordampningstab ved håndtering af prøverne under udskiftning af vandfasen mellem hver ekstraktion, og det anbefaledes, at proceduren ved tilsvarende fremtidige forsøg blev ændret, så fordampningstabet minimeres (bl.a. gennem anvendelse af "zero head space" teknik gennem hele proceduren). Parallelt med batchforsøgene gennemførtes kolonneudvaskningsforsøg på det samme materiale. I nogle tilfælde blev det observeret, at der selv efter gennemstrømning af en vandmængde svarende til L/S = 550 l/kg ikke var sket en fuldstændig udvaskning af de organiske komponenter. Dette skyldtes bl.a., at det udvaskede materiale, som havde en hydraulisk ledningsevne på ca. 4 x 10-7 m/s, indeholdt gammelt deponeret aktivt kul, hvilket modvirkede desorption af BTEX.
Wahlström et al. (1994) har sammenfattet erfaringer med udvaskning af organiske stoffer ved fem europæiske forskningsinstitutter (RIVM og ECN i Holland, VTT i Finland, IVL i Sverige og DHI i Danmark) i starten af 1990’erne. Undersøgelserne har først og fremmest omfattet udvaskning fra jord, men enkelte restprodukter er også indgået (aske, sandblæsningssand, asfalt, spildevandsslam). Det konkluderes bl.a., at man næppe vil kunne bruge den samme udvaskningsmetode til alle typer organiske stoffer. Endvidere forventer man at kunne anvende grundprincipperne fra udvaskningstests for uorganiske komponenter til etablering af metoder til undersøgelse af udvaskningen af organiske stoffer, men metoderne skal tilpasses bl.a. med henblik på at minimere fordampning, adsorption og nedbrydning. Endvidere drages der følgende konklusioner (gældende både for restprodukter og jord):
| Centrifugering er at foretrække frem for filtrering af eluat indeholdende PAH’er. | |
| Højt pH (=12) give forøget udvaskning af PAH, ikke mindst når materialet indeholder humusstoffer. | |
| Eluat fra udvaskning af olieforurenet sand bør centrifugeres og ikke filtreres i glasmikrofiltre, da man fandt en stor del af kulbrinterne adsorberet på filtre af typen 12 mm glasmikrofibre. | |
| Der fandtes ingen adsorption af klorfenoler (1400 mg/kg i jord) på 0,7 m m glasmikrofiberfilter; 13 % af klorfenolerne udvaskedes fra jorden efter 64 timer (pH = 11-12). | |
| Kontakttiden og agitationsmetoden (rystebord, rotation om tværakse) havde ikke nævneværdig indflydelse på udvaskningen af kulbrinter fra olieforurenet sand ved batchforsøg. | |
| BTEX (10-100 mg/kg tørstof) syntes vanskeligt at udvaske (< 1% ved L/S = 0,4 l/kg) fra jord ved kolonneforsøg udført i mørke ved 5 ° C på forurenet jord, der var behandlet termisk. |
5.4 Etablering af metoder til testning af udvaskningen af organiske stoffer fra restprodukter
På grundlag af ovenstående vil det være rimeligt at antage, at metoder til testning af udvaskningen af organiske stoffer fra granulære, mineralske restprodukter i vidt omfang kan baseres på de samme principper og hensyn, som metoder til udvaskning af organiske stoffer fra jord. Dog vil der for restprodukternes vedkommende i mange tilfælde skulle tages specielle forholdsregler med hensyn til forbehandling af prøverne, ligesom restprodukternes potentielle reaktivitet med vand og de ofte ret ekstreme pH- og redoxforhold, som reaktionerne med vand kan medføre, må tages i betragtning. Det bør undersøges nøjere, om man i nogle tilfælde kan udvide analyseprogrammet for udvaskningstests til bestemmelse af udvaskningen af uorganiske komponenter fra restprodukter til også at omfatte DOC/NVOC.
Blandt de konklusioner, som fremkom i en af de ovenfor refererede undersøgelser, var, at man ikke kan regne med at kunne anvende den samme metode til undersøgelse af udvaskningen af alle forskellige organiske stoffer og stofgrupper. Til dette kunne man føje, at man heller ikke vil kunne forvente at kunne beskrive alle de udvaskningssituationer, som man vil være interesseret i at belyse, ved hjælp af en enkelt udvaskningstest. Det skal nævnes, at der i det meste af denne rapport af hensyn til den kommende bekendtgørelse om anvendelse af jord og restprodukter har været fokuseret meget på tilpasning af en enkelt testmetode, nemlig batchudvaskningstesten prEN 12457-1, udført ved L/S = 2 l/kg.
6. Testsystemer - betingelser og metodik
6.1 Spørgsmål og svar
Når man som med etablering af testmetoder til undersøgelse af udvaskningen af organiske stoffer næsten starter forfra, vil det være hensigtsmæssigt at gøre tingene i en logisk rækkefølge. Det vil i denne sammenhæng sige, at man starter med at stille de spørgsmål, man gerne vil have besvaret, og derefter søger at finde eller udvikle metoder, der er egnede til at tilvejebringe svarene.
Blandt de spørgsmål, man kunne stille, kunne for eksempel være følgende:
| Hvor meget vil der i alt kunne udvaskes af et givet organisk stof (uanset hvor lang tid det tager)? | |
| Hvilken hastighed udvaskes et givet organisk stof med under forhold, der svarer til feltforhold (scenariebetinget)? | |
| Hvad er koncentrationen i porevandet til et givent tidspunkt? | |
| Er der tale om ligevægt mellem stoffet i væskefasen og faststoffasen? | |
| Hvis der ikke er tale om ligevægt, hvorledes er da kinetikken? | |
| Hvorledes påvirkes udvaskningen af et givet organisk stof af en række forhold som f.eks. pH, ilt, temperatur, tryk, biologisk aktivitet, m.v.? |
Den testtype, som man vil vælge, vil afhænge af, hvilke(t) af spørgsmålene, man ønsker at besvare.
Til besvarelse af det første spørgsmål vil man formentlig skulle vælge eller udvikle en eller anden form for tilgængelighedstest, d.v.s. en test, som under forhold, der svarer til de "værste", man realistisk forestiller sig, at materialet kan blive udsat for og under betingelser, som i øvrigt er optimale med hensyn til stoftransport, beskriver den samlede udvaskede mængde. En tilgængelighedstest for organiske stoffer (testen kan være forskellig for forskellige organiske stofgrupper) vil næppe være den samme som en tilgængelighedstest for udvaskning af uorganiske komponenter.
Til besvarelse af det andet spørgsmål ville man nok i højere grad søge at vælge eller udvikle en test, som i videst mulige omfang simulerer et givet scenarie, f.eks. en kolonneudvaskningstest udført under nærmere fastsatte betingelser til simulering af udvaskningen fra jord eller restprodukter, der er nyttiggjort eller deponeret. Blandt andet som grundlag for fastsættelse af sådanne betingelser vil det være nødvendigt at designe specielle forsøg, som kan besvare de øvrige spørgsmål, som er stillet ovenfor (listen af spørgsmål er kun ment som kun eksempler, og skal ikke betragtes som udtømmende).
Det kan ikke udelukkes, at det viser sig at være umuligt eller alt for omstændeligt at besvare de spørgsmål, som man f.eks. i forhold til lovgivning eller en given risikovurdering ønsker besvaret. I så fald kan man være nødt til at lade sig nøje med det grovere og langt mindre nuanceret billede af virkeligheden, som et forholdsvis simpel test kan give. Opgaven vil så være at optimere udbyttet af den tilvejebragte information på det grundlag, som nu måtte foreligge. I den sammenhæng er det vigtigt, at resultaterne ikke overfortolkes, men måske snarere anvendes som en slags screeningsundersøgelser, der afgør, om yderligere testning er nødvendig.
6.2 Testsystemer og materialevalg til organiske stoffer
Da udvaskningstests gennemføres med henblik på at undersøge stofoverførslen fra en faststofprøve (evt. med porevand og frie faser af forureningsstoffer) til en væskefase, er det vigtigt at tage hensyn til risikoen for stofudveksling mellem det undersøgte udvaskningssystem og omgivelserne, primært det anvendte apparatur/testbeholder og atmosfæren.
Stofudveksling mellem på den ene side testmedierne i form af faststoffasen og udvasknings-/ekstraktionsmediet og på den anden side det anvendte apparatur og testbeholder er generelt uacceptabel, og der må derfor ved valg af materialer tages hensyn til hele udvaskningssystemets karakter, herunder specielt de stofkomponenter, som ønskes testet. For testsystemer til uorganiske stoffer kan der normalt anvendes forskellige former for plast, se f.eks. Hjelmar og Traberg (1995), mens undersøgelse af organiske stoffer normalt forudsætter anvendelse af glas eller rustfrit stål. Anvendelse af polytetrafluoroethylen (teflon) vil i mange tilfælde være acceptabel for både organiske og uorganiske stoffer.
Stofudvekslingen mellem udvaskningssystemet og atmosfæren er vanskelig at vurdere - og for meget flygtige stoffer at tage hensyn til. En vurdering af, hvorvidt en sådan stofudveksling er acceptabel, afhænger af udvaskningssystemets karakter og formålet med testen. I afsnit 7.3 er der angivet beregnede værdier for stofmængden i luften ved ligevægt i standardudvaskningssystemet.
Stofudvekslingen kan have karakter af materialeafgivelse eller stoftab til testbeholderen/udstyret. Med hensyn til materialeafgivelse skal det vurderes, om de benyttede materialer indeholder stoffer, der kan gå i opløsning ved kontakt med prøven eller udvaskningsmediet, eller om stof fra atmosfæren/omgivelserne kan diffundere igennem materialet og ind til testmedierne.
Nedenfor er der ud fra erfaringer beskrevet i litteraturen gennemgået forskellige materialetypers egnethed i relation til stofafgivelse og forskellige stofgruppers tab til disse materialer.
6.2.1 Materialeafgivelse af organiske stoffer
Afgivelse af organiske stoffer fra plastmaterialer antages primært at foregå ved diffusion imellem udvaskningsmediet og plastmaterialet. Mekanisk påvirkning af materialet kan også bevirke en stofafgivelse, således at forøgelse af tryk og strømningshastighed kan antages at forøge risikoen for stofafgivelse. Endvidere øges risikoen for afgivelse af stoffer, når kontakttiden og temperaturen øges.
Endelig kan indholdet af f.eks. opløsningsmidler i prøven og udvaskningsmediet virke som et ekstraktionsmiddel på materialer og derigennem øge frigivelse af visse organiske stoffer fra plastmaterialer, der er i kontakt med prøve og udvaskningsmedie.
Kjeldsen et al. (1989) gennemførte en sammenstilling af litteraturen vedrørende frigivelse af organiske stoffer fra plastmaterialer, plastbelægninger og forskellige typer af plastlim til vandprøver. De hyppigst forekommende og mængdemæssigt mest betydende stoffer er: blødgøringsmidler (f.eks. phthalater), opløsningsmidler (f.eks. tetrahydrofuran og tetrachlorethylen) og stabilisatorer (f.eks. N-butylbenzensulfonamid). I Kjeldsen et al. (1989) er der anført eksempler på stoffrigivelser svarende til fra ca. 1 µg specifikt organisk stof/l op til mere end 10 mg C /l målt som totalt organisk kulstof i vandprøver, der har været i kontakt med de forskellige materialer. Eksempelvis er der fra teflon og stive PVC-materialer rapporteret om afgivelser svarende til nogle få µg stof/l. Fra blød PVC, polyetylen og polypropylen typisk op til 1000 µg stof/l, og fra silikonegummi og PVC lim ligger de fleste eksempler fra litteraturen på et niveau svarende til mere end 1000 µg stof/l vandprøve.
Afgivelsen af organiske stoffer fra materialer til prøve- og testmedie er primært afhængig af materialets restindhold af frigivelige forbindelser. Der kan endvidere forekomme variationer i restindhold af organiske stoffer imellem de enkelte produktioner ("batch") af en bestemt type materiale af et givet fabrikat. Frigivelige stoffer kan dannes via nedbrydning af plasten og dens tilsætningsstoffer som følge af for høje temperaturer under plastbearbejdelsen eller fra hjælpestoffer benyttet under processer i tilknytning til den egentlige fremstilling som f.eks. limning, overfladerengøring med opløsningsmidler m.v.
Som nævnt indeholder bløde plastmaterialer og lim generelt større mængder af afgivelige organiske forbindelser end stive plastmaterialer. Stoffer med lille fordelingsforhold imellem materiale og vand vil frigives med de højeste koncentrationer til følge, men vil sandsynligvis kunne fjernes mest effektivt inden anvendelsen af materialet.
6.2.2 Tab af organiske stoffer til materialet
Tab af organiske stoffer til plastmaterialer foregår primært ved sorption til overfladerne eventuelt efterfulgt af egentlig absorption af stofferne via diffusion ind i selve materialet. Ved absorption af organiske stoffer i materialerne kan disse i modsætning til binding ved adsorptionsprocesser ikke forventes at kunne mættes ved konditionering med en prøve indeholdende stofferne. Stofferne vil desuden efterfølgende kunne frigives vedvarende til medier med et lavere indhold, f.eks. ved udskiftning af udvaskningsmediet i en sekventiel udvaskningstest.
Tabet af stoffer afhænger således af materialernes karakter og stoffernes fysisk-kemiske egenskaber. F.eks. vil mere apolære hydrofobe stoffer generelt have større affinitet for binding til plastmaterialer. Denne polaritets-egenskab kan til kvalitative formål illustreres ved stoffernes oktanol/vand-fordelingskoefficient Kow.
I Kjeldsen et al. (1989) findes der detaljerede oversigter over bindingen af forskellige organiske stoffer til udvalgte materialer. For de flygtige organiske stoffer, primært klorerede opløsningsmidler, rapporteres der om væsentlige stoftab (20-100 % af de tilsatte mængder) til en række af de hyppigst anvendte plastmaterialer, herunder teflon, stiv og blød PVC, polyetylen og polypropylen i accelererede laboratorieforsøg. I den undersøgelse af rustfrit stål, der indgår i oversigten, fandtes der ikke et målbart tab fra prøven (Kjeldsen et al. 1989). Med hensyn til ikke-flygtige organiske stoffer, primært detergenter og BTX-stoffer viser oversigten et betydeligt stoftab (10-80 % af de tilsatte mængder) til de bløde plastmaterialer og et relativt mindre stoftab (1-20 % af de tilsatte mængder) til stiv PVC, teflon og glas. Til rustfrit stål udgjorde stoftabet mindre end 5 % af de tilsatte mængder.
I det omfang, der anvendes filtermaterialer til separation af eluat (væskefase) fra faststoffasen i testsystemet, er afgivelse af stoffer til filtermaterialer et kritisk punkt i testningen. For de fleste af de hydrofobe/apolære stoffer, som f.eks. PAH-forbindelser, vil faststoffasen udgøre en større pulje for frigivelse af stoffer til eluatet, der vil være reguleret af ligevægten mellem de to faser. Når eluatet separeres fra den faste fase, fjernes muligheden for at kompensere et eventuelt tab fra eluatet til filtermaterialer, selv et mindre stoftab kan have væsentlig indflydelse på stofkoncentrationen i eluatet.
I relation til valg af materialer i det hollandske forslag til standard udvaskningstest (f.eks. Bauw et al. 1991 og afsnit 6.4) testede Rood et al. (1994) udstyr, herunder forskellige typer af filter- og forfiltermaterialer, for sorption af PCB (polyklorerede bifenyler) og klorerede organiske pesticider. Sorptionstests viste, at det var bedre at anvende glasmaterialer end teflon og teflonlignende materialer. Indledende sammenligninger af resultater opnået ved filtrering og centrifugering af eluater fra udvaskningstesten indikerede, at centrifugering af eluaterne resulterede i højere koncentrationer af PCBer og bedre reproducerbarhed.
Bauw et al. (1991) undersøgte endvidere stoftabet af PAH'er til forskellige glasfiberfiltre og -forfiltre samt genfindingen af PAH'er efter tilsætning til eluater fra jord. Adsorptionstabet varierede fra 0 til 25% mellem de forskellige filtre og PAH-stoffer. Til brug for standardtesten (kolonneforsøg) blev det på baggrund af de gennemførte filtertests anbefalet at fortsætte med et 0,8-1,5 µm glasfiber filter. For ingen af de 16 testede PAH-stoffer oversteg stoftabet 10% i dette filter.
6.3 Testbetingelser og metodik
6.3.1 Forbehandling af prøve
Før iværksættelsen af udvaskningstesten kan det være nødvendigt at forbehandle prøven, f.eks. i form af tørring, delprøveudtagning, sortering, nedknusning, kompaktering eller ældning af prøvematerialet. Typisk kan jord være så våd, at det kan være nødvendigt at foretage en tørring, hvilket skal foregå under hensyntagen til de organiske stoffer, der ønskes testet. For langt de fleste organiske stoffer vil risikoen for fordampningstab bevirke, at tørring er uønsket. Ved testning af stofudvaskningen fra jord benyttes tørstofindholdet oftest til opsætning af testen med en velkendt tørstofmængde af jorden i forhold til et bestemt volumen af udvaskningsmediet. Til dette formål kan man normalt blot udtage en eller flere mindre delprøver af jorden, som kun anvendes til tørstofbestemmelsen, og således antage, at denne er repræsentativ for hele prøven. I tilfælde af, at prøven må tørres, bør dette foregå ved den lavest mulige temperatur, f.eks. ved stuetemperatur eller i varmeskab ved 30-40°C. Mange gange vil det også være nødvendigt at sortere eller reducere partikelstørrelsen af jorden/restproduktet. I denne forbindelse skal risikoen for kontaminering eller tab af forureningskomponenter fra prøven minimeres under behandlingen. Ældning af prøven kan foregå enten som en uundgåelig proces af praktiske årsager eller med vilje for at fremskynde ønskede kemiske eller biologiske reaktioner. Ældningen kan forårsage ændringer i stoffernes tilgængelighed for udvaskning, i indhold af nedbrydelige eller flygtige stoffer samt i f.eks. prøvens redoxstatus og pH. Som diskuteret i afsnit 3 vil ældning af organiske typisk resultere i en mindre udvaskning af stofferne som følge af afdampning, nedbrydning og diffusion ind i partikelstrukturer. Hvis formålet med den aktuelle test er at beskrive stofudvaskningen fra materialet på langt sigt, kan det i nogle tilfælde tænkes at være hensigtsmæssigt at gennemføre en accelereret kunstig ældning af prøven. Der findes ikke standardiserede metoder til kunstig ældning af jord, og en passende fremgangsmåde må derfor udvikles i det konkrete tilfælde.
6.3.2 Udvaskningsmedie
Som udvaskningsmedie kan benyttes ikke-aggressive udvaskningsmedier som demineraliseret vand, vand med en elektrolyt i form af salt eller demineraliseret vand, som initielt er indstillet til et lavere pH (f.eks. pH = 4), ofte med henblik på at simulere udvaskning med regnvand. I mange tilfælde vil sammensætningen af udvaskningsmediet ved lave og moderate væske-/faststofforhold være styret af faststoffasen og være forholdsvis uafhængig af udvaskningsmediets initielle sammensætning. Ved høje værdier af L/S, eller hvis der anvendes et mere aggressivt udvaskningsmedie, kan stofudvaskningen påvirkes i betydelig grad.
I forbindelse med udvaskning af tungmetaller og andre uorganiske forureningsstoffer er der udviklet en lang række opskrifter på udvaskningsmedier til udvaskningstests, som skal simulere stofudvaskningen under nærmere hensyn til specificerede forhold. Opløsninger med kalciumklorid benyttes ofte til at simulere jordvæske med hensyn til den dominerende kation. For organiske stoffer eksisterer der ingen fast praksis, men sammensætningen af udvaskningsmediet kan være væsentlig i tilfælde, hvor kemiske processer, f.eks. styret af pH og opløsningens ionstyrke, har indflydelse på desorptionen af forureningsstoffer og frigivelse af kolloider.
6.3.3 Kontaktmetode og kontaktforhold mellem væske og faststof
Da udvaskningstests fortrinsvis gennemføres med henblik på undersøgelse af stofudvaskningen fra en faststoffase (prøven) til en væskefase, er det dels nødvendigt at tage hensyn til forsøgsbetingelser, som kan påvirke den hastighed, hvormed stofoverførslen sker, dels at tage hensyn til risikoen for stofudveksling mellem det undersøgte udvaskningssystem og omgivelserne, primært det anvendte apparatur og atmosfæren.
Ved mange batchudvaskningstests på jord og affaldsmaterialer, hvor man undersøger udvaskningen af uorganiske komponenter, agiteres udvaskningssystemet med henblik på at opretholde en maksimal gradient ved grænsefladen mellem væsken og de enkelte faststofpartikler og dermed sikre, at der hurtigst muligt opnås ligevægt i systemet. Agitationen kan ske ved hjælp af rysteapparater og ryste-/vippeapparater, ved rotation af en lukket udvaskningsbeholder om sin tværakse, ved hjælp af magnetomrøring (kun ved høje værdier af væske-/faststof-forholdet (L/S)), propelomrøring eller ved hjælp af gennembobling af gas eller luft. Uanset hvilken metode der anvendes, er det vigtigt, at der sikres en konstant god opblanding mellem væske- og faststoffaserne. Dog kan omrøring i batchtests forventes at frigive et større indhold af kolloider til eluatet end kollonnetests, hvorfor denne faktor også bør inddrages ved valg af metode- og kontaktforhold. For kolonneudvaskningstests skal det i videst mulige omfag sikres, at der ved den anvendte strømningshastighed opnås lokal ligevægt. Generelt er det vanskeligere at opnå ligevægt i kolonneforsøg, fordi der under accelererede laboratorieforhold ikke i fuldt omfang kan opnås optimale kontaktforhold mellem væske og faststof.
De fleste udvaskningssystemer beskriver forholdene i vandmættede systemer. Udvaskning ved gennemstrømning under umættede forhold kan principelt simuleres ved kolonneforsøg med nedadrettet flow, men sådanne forsøg kan være vanskelige at kontrollere og reproducere. I praksis anvendes ofte udvaskningsresultater fra forsøg udført under mættede forhold på grundlag af L/S-forholdet. Dette kan dog på grund af definitionen af L/S give anledning til unøjagtigheder, ligesom der ikke tages hensyn til de kemiske effekter af tilstedeværelsen af en gasfase i systemet eller til de effekter, som skift mellem våd og tør tilstand i naturlige umættede systemer kan tænkes at medføre. I mange tilfælde er det jo netop stofudvaskningen fra jord eller restprodukter, der gennemsives af infiltrerende nedbør under umættede forhold, som man ønsker at belyse ved hjælp af en udvaskningstest. Hvis man i den situation tester udvaskningen under vandmættede forhold, bør man med udgangspunkt i den konkrete situation og de konkrete materialer og forureningskomponenter foretage en vurdering af, hvilken betydning dette kan have for resultatet, og eventuelt beregningsmæssigt forsøge at kompensere for det (se afsnit 9).
Stofudveksling mellem på ene side prøven og udvaskningsmediet og på den anden side det anvendte testsystem (beholder, filter m.v.) og apparatur er generelt uacceptabel, og der må derfor ved valg af materialer tages hensyn til dette, hvilket er gennemgået i det foregående afsnit. For organiske stoffer er det endvidere generelt uacceptabelt med stofudveksling mellem udvaskningssystemet og atmosfæren, hvilket er gennemgået i afsnittet om stofudveksling med omgivelserne.
6.3.4 Væske-/faststofforhold (L/S)
Væske-/faststofforholdet eller L/S-forholdet er defineret som forholdet mellem den mængde udvaskningsmedium (liquid, L), som bringes i kontakt med den mængde faststof (solid, S), som testes. I denne rapport angives L som et volumen (i liter), og S angives som tørvægten (i kg) af faststoffasen, der tages i arbejde i udvaskningstesten. I nogle tilfælde ses L/S angivet som kg væske pr. kg faststof, og for en væske med en vægtfylde på 1 kg/l vil de to talangivelser være ens.
Som nævnt under afsnittet om forbehandling af prøven beregnes L/S for en våd prøve på grundlag af prøvens tørstofindhold, og ved tilsætning af udvaskningsmedium til en batchtest bør der kompenseres for den vandmængde, som prøven allerede indeholder.
Ved en kolonnetest beregnes L/S normalt på grundlag af den producerede perkolatmængde og tørvægten af faststoffasen i kolonnen. Dette giver for samme tilsatte væskemængde en forskel mellem L/S beregnet for forsøg udført under mættede forhold (med opstrøms flow) og for forsøg udført med samme kolonne under umættede forhold (med nedstrøms flow) en forskel mellem L/S beregnet for henholdsvis et kolonneforsøg og et batchforsøg, hvor hele den tilsatte væskemængde indgår i beregningen for batchforsøgets vedkommende. Forskellen mellem det beregnede L/S for henholdsvis batch- og kolonneforsøg er størst ved små værdier af L/S og betydningsløs ved større L/S-værdier.
6.3.5 Kontakttid
Længden af det tidsrum, hvor væskefasen er i kontakt med faststoffasen (prøven), vil påvirke størrelsen af stofudvaskningen, såfremt der ikke i løbet af dette tidsrum opnås en ligevægtslignende tilstand. I batchforsøg svarer kontakttiden til varigheden af testen, mens kontakttiden ved kolonneforsøg desuden er en funktion af gennemstrømningshastigheden.
Ved udvaskningstests, som er baseret på opnåelse af termodynamisk ligevægt eller ligevægtslignende tilstande, bør det tilstræbes, at kontakttiden i hvert trin af testen er tilstrækkelig lang til at sikre, at den ønskede ligevægts-tilstand opnås. På den anden side må kontakttiden ikke være så lang, at der opnås uønskede ændringer i forsøgsbetingelserne.
For batchforsøg varierer de foreskrevne kontakttider typisk mellem nogle timer og ca. et døgn pr. ekstraktion. I mange tilfælde, specielt for apolære stoffer og inhomogene prøver samt testmetoder, hvor der ikke stilles krav til partikelstørrelsen, må det antages, at det ikke vil være muligt at opnå ligevægt i løbet af den angivne kontakttid. Dette kan bl.a. afspejle sig i vanskeligheder med at reproducere og tolke testresultaterne. For kolonneforsøg kan strømningshastigheden foruden for kontakttiden også have betydning for løsrivelse af kolloider.
6.3.6 Temperatur
Temperaturen har indflydelse på forløbet af en udvaskningstest. Både termodynamiske ligevægtskonstanter i relation til opløselighed og flygtighed samt kinetiske processer som sorption og diffusion, der er relevante for stofudvaskningen, afhænger eksponentielt af temperaturen. For organiske stoffer er endvidere biokemiske processer, der er involveret i omdannelsen og nedbrydningen af disse stoffer, af betydning. Udvaskningsforsøg, hvor der ikke skal tages hensyn til nedbrydningen af stof, udføres som regel ved 20-25oC.
6.3.7 Separation af væskefase og faststoffase
De fleste udvaskningstests afsluttes med en separation af væskefasen fra faststoffasen ved tryk- eller vakuumfiltrering gennem et 0,45 µm membranfilter og/eller ved centrifugering svarende til den ønskede partikelafskæring. Komponenter, som kan passere et 0,45 µm filter, betragtes normalt pr. konvention som værende på opløst form. Små kolloide partikler kan imidlertid godt passere et 0,45 µm filter, og hvis de er til stede og ønskes tilbageholdt, må der anvendes et filter med mindre partikelafskæring. På den anden side kan der argumenteres for at medtage de partikler (måske op til flere µm i størrelse), som også vil transporteres igennem jordmediet (se afsnit 4.4).
Ved filtrering af udvaskningsmedier, som kan indeholde hydrofobe, tungt opløselige organiske molekyler, kan det være nødvendigt at anvende filtre af glasfiber, da de organiske forbindelser kan sorbere på f.eks. membranfiltre af celluloseacetat, som er bedre egnet til uorganiske stoffer. I relation til udvikling af udvaskningtests bekræftes dette af Rood et al. (1994) i forhold til sorptionstab af PCBer og klorerede organiske pesticider. Undersøgelserne af Bauw et al. (1991) viser, at filtrering er meget kritisk for PAH'er, og der kan være væsentlig forskel i sorptionstabet mellem forskellige typer af filtre på trods af, at de alle er af glasfiber, og diameteren for partikelafskæringen ligger inden for den samme størrelsesorden.
6.3.8 Stofudveksling med omgivelserne
Ud over tabet af stof til udstyr og materialer, som indgår i testningen, kan der også forekomme stoftab til atmosfæren. Her tænkes der især på tab af flygtige stoffer, idet fordampning kan give anledning til tab fra væske-/faststofsystemet til headspace (luftvolumenet over væske-/faststoffasen) i systemet og/eller til omgivelser uden for testsystemet.
Afdampningen vil afhænge af mange faktorer ud over stoffernes flygtighed, f.eks. om der optræder ikke-vandige faser i de organiske stoffer i prøvematerialet, samt af volumenet af headspace, kontaktmetode samt materialernes tæthed. Herudover er der naturligvis risikoen for stoftab under prøvetagningen, forbehandlingen af prøven, efterbehandlingen og analysen af eluatet som nævnt i afsnittet om testbetingelser og testmetodik.
Ved design af udvaskningstests er det ud over valget af materialer og håndtering af prøve og eluat før, under og efter testningen specielt volumenet af headspace, der kan fastlægges. Ofte kan man af hensyn til agitationen og muligheden for ændringer af det samlede volumen af prøve og udvaskningsmedie ikke vælge at teste helt uden headspace. Dette bør derfor minimeres, da man ud fra simple beregninger kan vise, at væsentlige stofmængder af meget flygtige forbindelser som klorerede opløsningsmidler kan tabes i systemer med store headspace volumener.Det skal dog bemærkes, at konkrete beregninger for den standardudvaskningsprocedure, som er blevet anvendt i dette projekt, faktisk har vist, at andelen af selv de mest flygtige stoffer i headspace udgør mindre end 3% af den totale mængde (se afsnit 7.3). Det kan samtidig anføres, at stoffer, der er så flygtige, ofte ikke udgør et udvaskningsproblem, hvis de fortsat findes i prøven, samt at de øvrige nødvendige håndteringer af prøven gør det nærmest umuligt at teste stofudvaskningen af disse i praksis.
6.3.9 Analyse af væskefase, prøvevolumener og detektionsgrænser
Et udvaskningsforsøg er først afsluttet, når der er foretaget en karakterisering af det frembragte eluat eller perkolat. Karakteriseringen består ofte i gennemførelse af et kemisk analyseprogram, men kan eksempelvis også omfatte økotoksikologisk testning. Behovet for relativt store prøvevolumener til kemiske analyser er et centralt punkt i forhold til testning af stofudvaskningen af organiske stoffer. Mange organiske stofgrupper kræver et prøvevolumen på ca. 1 liter, hvilket kan være problematisk dels i den tidligere nævnte separation af eluat fra prøvematerialer, dels i forhold til håndtering af et passende testsystem. Testning af prøver med blandingsforureninger af forskellige stofgrupper kan endvidere forøge volumenbehovet til analyse yderligere. Dette kan indfries enten ved at opskalere testsystemet eller opsætte flere parallelle testsystemer af den oprindelige størrelse. En tredje mulighed, som er beskrevet af Bauw et al. (1991), er at foretage en opkoncentrering af de organiske stoffer på fast fase efter udvaskning med relativt høje L/S-værdier. Ved en sådan procedure opnåede Bauw et al. (1991) gode genfindinger af 16 PAH'er tilsat til eluater fra jord.
Af hensyn til kvaliteten af analyserne anbefales der oftest nedkøling og opbevaring i mørke af prøver, som ikke kan analyseres umiddelbart efter gennemførelse af testen.
6.4 Oversigt over beskrevne/standardiserede udvaskningstests til organiske stoffer
Gennemgang af litteraturen viser, at der kun findes få standardiserede metoder til karakterisering af udvaskningen af organiske stoffer fra forurenet jord og restprodukter/affald. I det følgende er der foretaget en opsamling af de standardiserede udvaskningstests og andre velbeskrevne udvaskningstests fra Vesteuropa og Nordamerika, som det har været muligt at finde referencer på. Til udarbejdelse af oversigten er der anvendt oplysninger fra Larsson (1996) og Wahlström et al. (1994).
I Tyskland er der udsendt en præstandard til bestemmelse af koncentrationen af organiske stoffer i jordvæske (DIN 1998). Standarden er baseret på kolonneudvaskningsforsøg, der udføres i mindre kolonner (6 cm i diameter og en længde på 15 cm) med et volumen på 450 ml til prøvemateriale. Som udvaskningsmedie anvendes der vand, der sendes gennem kolonnen med opstrøms gennemstrømning med et flow på ca. 1 ml/min. Eluatet ledes direkte, uden filtrering, til en flaske med ekstraktionsmiddel, hvor det ekstraheres med henblik på analyse. Der er ikke defineret et bestemt akkumuleret væske-/faststofforhold eller en kontakttid, som bestemmer varigheden af testen, men der udtages flere prøver over 7-30 dage. Dog udelades de første 5-15 porevolumener, som antages at have et forhøjet indhold af kolloider/partikler. Baseret på målinger af koncentrationen i eluatet over tiden vurderes det, hvor vidt udvaskningen af de organiske stoffer primært er styret af opløsning fra fri fase eller ved desorption. Denne information anvendes i den videre risikovurdering.
I Holland har man afprøvet en standard udvaskningstest (SLT test), som er udarbejdet til testning af emission af metaller fra metalforurenet affald, på hydrofobe organiske stoffer (Bauw et al. 1991). Testresultaterne anvendes til at klassificere affaldsmaterialer i relation til hollandske retningslinier for nyttiggørelse og deponering af affald. Bauw et al. (1991) beskriver resultaterne af en afprøvning af testen på 2 PAH-forurenede materialer, henholdsvis en jordprøve og en flyveaske fra affaldsforbrænding.
SLT-testen omfatter dels en "kaskadetest" bestående af 5 serielle batchudvaskninger med et samlet L/S på 100 l/kg, dels en kolonnetest ved L/S 0-10 l/kg. Vand justeret til pH 4 anvendes som udvaskningsmedie. Hver af de 5 batchtests udføres ved et L/S-forhold på 20 l/kg og en kontakttid på 3 timer. Efterfølgende foretages der en filtrering af eluatet. Til brug for testningen af de organiske stoffer foretog Bauw et al. (1991) en modifikation af SLT-testen. Modifikationen bestod i, at eulatets indhold af organisk stof blev opkoncentreret ved hjælp af fast fase-ekstraktion (SPE), dvs. opkoncentrering på fast fase med efterfølgende ekstraktion før analyse.
Den amerikanske TCLP (Toxicity Characteristic Leaching Procedure) test anvendes til både uorganiske og organiske stoffer i forbindelse med klassificering af affald (US EPA 1991). Testen foreskriver udvaskning med eddikesyre/acetat, hvilket skal simulere, at det affaldsmateriale, som skal testes, bringes i kontakt med lossepladsperkolat. Testen udføres som en ét-trins batch rystetest ved et L/S på 20 l/kg og en kontakttid på 16-20 timer. Eluatet filtreres gennem et filter med en partikelafskæring på 0,6-0,8 m m. Med hensyn til testning af udvaskningen af organiske stoffer anbefaler metodeforskriften, at der anvendes glas, polytetrafluoretylen (PTFE, teflon) eller rustfrit stål, men derimod ingen former for PVC.
7. Klassificering og gennemgang af organiske stofgrupper
| 7.1 | Indledning |
| 7.2 | Stoffernes egenskaber |
| 7.3 | Fasefordelingsberegninger og vurderinger |
| 7.4 | Klassifikation af stofgrupper i relation til stofudvaskningsegenskaber |
7.1 Indledning
I det følgende er der med udgangspunkt i de organiske stoffer og stofgrupper, som der er fastsat kvalitetskriterier for i jord og grundvand (Miljøstyrelsen 1998a), foretaget fasefordelingsberegninger i et kombineret luft-/vand-/jordsystem med henblik på at opdele stofferne efter deres egenskaber og fordeling i systemet. Opdelingen er benyttet til at foretage en indledende klassifikation af de enkelte stoffer/stofgrupper i forhold til mulighederne og relevansen af at teste deres udvaskning fra jord og restprodukter.
7.2 Stoffernes egenskaber
Udvalgte fysisk/kemiske egenskaber er for de organiske stoffer vist i tabel 7.1.
Tabel 7.1
Stofegenskaber. S er opløseligheden i vand, H er Henrys konstant (fordeling mellem
luft og vand) og Kow er octanol-/vandfordelingskoefficienten. Fra
Miljøstyrelsen (1995) og (1996) samt Shiu et al. (1994). Temperatur = 20 – 25 ° C.
| Stof | S (mg/l) | H (-) | Log(Kow) (-) |
| Benzen | 1760 | 0,22 | 2,1 |
| Toluen | 555 | 0,27 | 2,7 |
| Xylener | 160-200 | 0,22-0,29 | 3,1-3,2 |
| Styren | 3200 | - | 2,95 |
| Naphthalen | 31 | 0,017 | 3,36 |
| Benz(a)pyren | 0,0038 | 2 x 10-5 | 6,5 |
| Dibenz(a,h)anthracen | 0,0005 | 8,3 x 10-8 | 6,5 |
| Phenol | 84000 | - | 1,5 |
| Pentachlorphenol | 14 | - | 5,0 |
| Vinylchlorid | 5700 | 1,1 | 1,38 |
| 1,2-Dichlorethylen | 3500-6260 | 0,17-0,38 | 1,86-1,93 |
| 1,1-Dichlorethylen | 3344 | 1,1 | 2,13 |
| Trichlorethylen | 1400 | 0,39 | 2,53 |
| Tetrachlorethylen | 240 | 0,72 | 2,88 |
| 1,1,1-Trichloroethan | 1250 | 0,7 | 2,49 |
| Chloroform | 8700 | 0,15 | 1,97 |
| Dichlormethan | 13200 | 0,09 | 1,25 |
| Tetrachlormethan | 780 | 1,2 | 2,64 |
| Acetone | blandbart | - | -0,24 |
| Butylacetater | 6300-8600 | - | 1,6-1,8 |
| Diethylether | 60000 | - | 0,83 |
| Isopropanol | blandbart | - | 0,05 |
| Methylisobutylketon | 17000 | - | 1,38 |
| Methyltertiærbutylether MTBE | 50000 | 0,02 | 1,2 |
| DDT | 100 | - | 5,94 |
| DEHP | 0,3-0,4 | - | 3-5 |
Her er der kun medtaget stoffernes opløselighed i vand, Henrys konstant (fordeling mellem
luft og vand) og octanol-/vandfordelingskoefficient, da det er de fysisk/kemiske
egenskaber, der kan antages styrende for, hvorledes stofferne fordeler sig i et forenklet
luft-/vand-/jordsystem. De organiske stoffer, der er vist i tabel 7.1, inkluderer kun
enkeltstoffer og ikke stofblandinger, som benzin, diesel og tjære. Benzin og tjære er
dog repræsenteret med BTEX, methyltertiærbutylether MTBE, naphthalen, benz(a)pyren og
dibenz(a,h)anthracen.
I relation til gennemførelse af udvaskningstests med stofferne kan stoffernes nedbrydelighed primært under tilstedeværelsen af ilt være af væsentlig betydning. Betydningen vil naturligvis afhænge af forsøgstiden og temperaturen, men generelt vil en hel eller delvis nedbrydning af et teststof inden for et døgn eller nogle få dage være kritisk og kræve et tiltag enten i form af at begrænse denne nedbrydning eller at tage højde for den ved tolkningen af resultaterne. I tabel 7.2 er stoffernes nedbrydelighed under aerobe forhold angivet. De fleste af stofferne med undtagelse af de klorerede opløsningsmidler med 3 eller 4 kloratomer i molekylet og pentaklorphenol er nedbrydelige under aerobe forhold. Det ses, at det primært er BTX-stofferne, der er nedbrydelige inden for de tidsperioder, der er relevante for hurtige udvaskningstests.
Tabel 7.2
Stoffernes nedbrydelighed under aerobe forhold, hvor de optræder som primær
substrat, hvilket vil sige, at et stof er nedbrydeligt, uden at det er nødvendigt, at
andre stoffer er til stede (Miljøstyrelsen 1995 og 1996).
| Stof | Nedbrydelighed |
| Benzen | +++ |
| Toluen | +++ |
| Xylener | ++ |
| Styren | (+) |
| Naphthalen | ++ |
| Benz(a)pyren | + |
| Dibenz(a,h)anthracen | + |
| Phenol | +++ |
| Pentachlorphenol | - |
| Vinylchlorid | + |
| 1,2-Dichlorethylen | + |
| 1,1-Dichlorethylen | + |
| Trichlorethylen | - |
| Tetrachlorethylen | - |
| 1,1,1-Trichloroethan | - |
| Chloroform | - |
| Dichlormethan | + |
| Tetrachlormethan | - |
| Acetone | (+) |
| Butylacetater | |
| Diethylether | (+) |
| Isopropanol | |
| Methylisobutylketon | (+) |
| Methyltertiærbutylether | + |
| DDT | + |
| DEHP | ++ |
-: ikke påvist nedbrydelig
+: langsomt nedbrydeligt
(+): nedbrydeligt, men uvist hvor hurtigt
++: hurtigt nedbrydeligt
+++: meget hurtigt nedbrydeligt (definition: at der muligvis kan ske en væsentlig
nedbrydning i løbet af et par dage).
7.3 Fasefordelingsberegninger og vurderinger
I det efterfølgende er fordelingen af de organiske stoffer eksemplificeret ved en serie af fasefordelingsberegninger. Beregningerne er baseret på simple massebalancer og under antagelse af ligevægt. Eksempler på de udførte beregninger er vist i bilag B. Som det fremgår af sammenstillingen af oplysningerne fra litteraturen i kapitel 2 og 3, vil denne form for fasefordelingsberegninger i mange tilfælde ikke kunne simulere de faktiske forhold under mere feltlignende forhold, men i denne sammenhæng anvendes de indledningsvis til at vurdere stoffernes fysisk/kemiske egenskaber og den generelle betydning af disse for fordelingen af stofferne i et luft-/vand-/jordsystem.
For at beregne fordelingen af stofferne mellem jord og vand er der foretaget et valg med hensyn til beskrivelse af sorptionsligevægten i beregningen. Ud fra en sammenhæng mellem henholdsvis Koc og Kow og henholdsvis Kd, foc og Koc er følgende sammenhæng anvendt (Miljøstyrelsen 1996):
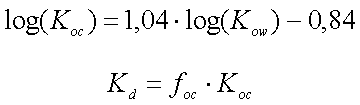
hvor
Koc er fordelingskoefficienten mellem det organiske stof i jorden og vand
Kow er fordelingskoefficienten mellem octanol og vand
Kd er fordelingskoefficienten mellem jord og vand
foc udtrykker indeholdet af naturligt organisk stof i jorden..
Der er foretaget 4 beregninger. Beregning nr. 1 er udført for få et overblik over, hvordan stofferne relativt fordeler sig imellem faserne (jord, vand og luft). Den er udført på samtlige stoffer, der er relevante (Tabel 7.1). Beregningen er udført med et system med 0,94 kg jord, 1,88 l vand og 0,05 l luft. Jordkoncentrationerne er valgt arbitrært, så der ikke er fri fase i jorden. Beregning nr. 2 er udført for at få overblik over hvilken eluatkoncentrationer, et ligevægtssystem med jordkoncentrationer, der svarer til jordkvalitetskriterierne, resulterer i. Systemet er det samme, som er benyttet i beregning nr. 1, men antallet af stoffer er begrænset til dem, for hvilke der er fastsat jordkvalitetskriterier. Formålet med beregning nr. 2 er at undersøge, hvilken følsomhed der opnås for de stoffer, der skal kunne testes. Beregning nr. 3 har samme formål, som beregning nr. 2, nemlig at teste systemets følsomhed. Der er imidlertid taget udgangspunkt i stoffernes detektionsgrænse i eluatet, og beregningerne viser de dertil svarende jordkoncentrationer. Beregning nr. 4 er magen tilberegning nr. 2, bortset fra at de L/S-forhold, som i beregning nr. 2 var 2 l/kg, i beregning nr. 4 er ændret til 10 l/kg.
Resultatet af beregning nr. 1 er vist i figur 7.1. Beregningen viser den relative fordeling af de organiske stoffer mellem luft, vand og jord i et system, hvorom det er antages, at koncentrationerne er så lave, at der ikke optræder frie faser. De benyttede stofkonstanter er angivet i tabel 7.1. Fasefordelingsberegningen er udført på et system med 0,94 kg jord TS, 1,88 l vand og 0,050 l luft, hvilket svarer til de mængder, der er benyttet som standard i udvaskningsforsøgene. Systemet har således et væske-/faststofforhold L/S på 2 l/kg TS. Jorden indeholder 0,1% naturligt organisk stof.
Beregning nr. 1 viser, at der for eksempelvis BTEX findes mere end 80% af stofmængden i vandfasen, mens mere end 99% af mængden af benz(a)-pyren derimod findes sorberet til jorden. Forskellene i disse stoffers fordeling kan forklares ud fra forskellene i stoffernes Kow-værdier (tabel 6.1). Beregning nr. 1 viser ligeledes, at i det anvendte system udgør mængden af stofferne i headspace 1-3% for vinylchlorid, 1,1-DCE, PCE, 1,1,1-TCA og tetrachlormethan, og mindre end 1% for de øvrige stoffer.
Beregning nr. 2 er gennemført for det samme system som beregning nr. 1, men udelukkende for de enkeltstoffer, som der findes jordkvalitetskriterier for (Miljøstyrelsen 1998a). I beregningen er der anvendt en stofkoncentration i jorden, der netop svarer til kriteriet, og resultatet i form af den tilhørende koncentration i væsken (eluatet) er vist i figur 7.2. Der ses en betydelig forskel i koncentrationen i væsken mellem de forskellige stoffer. Den laveste koncentration ses for benz(a)pyren, der skal sættes i relation til detektionsgrænsen for stoffet på vandig form. Det betyder, at der fra udvaskningsforsøget skal kunne udtages prøver af væske til analyse med mindst denne følsomhed.
Da det danske grundvandskvalitetskriterie er på 0,2 µg/l for summen af fluoranthen, benz(b+k)fluoranthen, benz(a)pyren, indeno(1,2,3-cd)pyren og dibenzo(g,h,i)perylen (Miljøstyrelsen 1998a), er det nødvendigt, at testen er følsom for disse stoffer. Af figur 7.2 fremgår det, at de beregnede væskekoncentrationer af benz(a)pyren og dibenz(a,h)anthracen ligger i størrelsesordenen (0,01 µg/l) for kriterieværdierne, men for de øvrige parametre skulle der ikke umiddelbart være problemer i forhold til stoffernes detektionsgrænser. Dog vil de nødvendige prøvevolumener til kemiske analyser være store i forhold til udvikling af testsystemer, der kan håndteres via normale laboratorieprocedurer og med til rådighed værende udstyr. Som en indledende vurdering af mulighederne for at optimere og tilpasse nuværende kemiske analyser til at kunne gennemføres med mindre prøvevolumener er der i tabel 7.3 med udgangspunkt i oplysninger fra DHIs kemiske afdeling vist detektionsgrænser for analyse af de forskellige stoffer i vand og det tilhørende nødvendige prøvevolumen. Højre halvdel af tabellen er et resume af vurderingen af mulighederne for at gennemføre analyser med mindre volumener, samt hvilken betydning dette alt andet lige vil få for detektionsgrænserne. Af tabellen fremgår det, at der er muligheder for flere af de meget prøvekrævende (1000 ml) analyser, men at detektionsgrænserne vil blive hævet proportionalt. Et alternativ til de mindre volumener til analyse ville være at benytte større flasker til udvaskningsforsøgene. Dette ville være mere tidskrævende, fordi separationen af eluat og faststof ville tage længere tid. Derud kan det resultere i problemer med at sikre en god kontakt mellem faststof og væskefase under udvaskningen.
Beregning nr. 3 er foretaget på samme system som beregning nr. 1 og 2, men her er jordkoncentrationen beregnet udfra stoffernes detektionsgrænser i vandfasen. Resultatet af beregningen er vist i figur 7.3. Eksempelvis for benz(a)pyren skal jordprøven indeholde mere end 0,04 mg/kg TS for, at eluatet i udvaskningstesten indeholder en koncentration, der er højere end detektionsgrænsen. For de fleste stoffer er den mindste jordkoncentration, hvor systemet resulterer i målelige koncentrationer i eluatet, 3-4 størrelsesordener mindre end jordkvalitetskriterierne. For benz(a)pyren og dibenz(a,h)anthracen er den mindste jordkoncentration, hvor systemet resulterer i målelige koncentrationer i eluatet, imidlertid kun en faktor 2 mindre end jordkvalitetskriterierne.
I beregning nr. 4 er der taget udgangspunkt i opsætningen for beregning nr. 2, men ændret på væske-/faststofforholdet i systemet. Det samlede volumen er bibeholdt, svarende til volumenet af glasflasken fra "standard" udvaskningsforsøget, men med en mindre jordmængde (0,2 kg) og større vandmængde (2,0 l), hvilket giver et væske-/faststofforhold på 10 l/kg. Resultatet af beregningen er vist i figur 7.4. Det ses, at koncentrationen af de stoffer, som i forvejen optræder i relativt lave koncentrationer, ikke reduceres væsentligt yderligere. Dette skyldes stoffernes store affinitet for jorden og herigennem fastholdelse af koncentrationsniveauet i væsken, som de er i lige-
Figur 7.1 Se her!
Beregninger af den relative fordeling (i %) mellem organiske stoffer i et vand
(1,88 l) jord (0,94 kg TS) luft (0,050 l) system, der svarer til det benyttede standard
udvaskningssystem. Der optræder ikke frie organiske faser i systemet. De sorte søjler
angiver andelen i vand, den hvide i luft og den grå på jorden
Figur 7.2 Se her!
Beregninger af koncentrationerne i væskefasen af organiske stoffer i et vand (1,88
l) jord (0,94 kg TS) luft (0,050 l) system, der svarer til det benyttede standard
udvaskningssystem. Stofmængderne i systemet svarer til jordkvalitetskriteriet for de
enkelte stoffer.
Figur 7.3 Se her!
Beregninger af koncentrationerne på jorden af organiske stoffer i et vand (1,88 l)
jord (0,94 kg TS) luft (0,050 l) system, der svarer til det benyttede standard
udvaskningssystem. Væskekoncentrationerne i systemet svarer til detektionsgrænsen for de
enkelte stoffer.
Figur 7.4 Se her!
Beregninger af koncentrationerne i væskefasen af organiske stoffer i et vand (2,0
l) jord (0,2 kg TS) luft (0,050 l) system, der svarer til det benyttede standard
udvaskningssystem i total volumen, men med et væske-/faststofforhold på 10 l/kg som
alternativ til 2 l/kg. Stofmængderne i systemet svarer til jordkvalitetskriteriet for de
enkelte stoffer.
Tabel 7.3 Se her!
Detektionsgrænser og prøvevolumen for de forskellige analyser. Desuden er
grundvandskvalitetskriteriet angivet tillige med muligheden for at reducere
prøvevolumenet til analysen og konsekvensen for detektionsgrænsen.
vægt med. Anvendelse af et højere væske-/faststofforhold kan således være en mulighed, der kan give større prøvevolumener til analyse (evt. ved opkoncentrering) og samtidig mindske ("fortynde") problemerne med partikler i testsystemet.
7.4 Klassifikation af stofgrupper i relation til stofudvaskningsegenskaber
Stofferne, der er beskrevet i det ovenstående, kan på basis af deres stofudvaskningsegenskaber foreslås opdelt i 3 grupper eller klasser:
Første gruppe indeholder stoffer, der sorberer kraftigt til jorden, har en lille opløselighed i vand og er ikke flygtige. Til denne gruppe hører de fleste PAH'er og dieselstoffer. Det er oplagt, at en metodik for testning af udvaskningen af organiske stoffer fra jord og restprodukter skal omfatte disse stoffer, der vil optræde og flyttes sammen med jorden, men i et vist omfang samtidig blive udvasket. Mange af stofferne har lave grænseværdier i vand. Som det er videre diskuteret i kapitel 8, vil det særligt være separationen af fast fase fra eluatet, herunder hensyntagen til kolloidbundet stof og forsøgstiden, der vil være kritisk for testningen.
Den anden gruppe omfatter flygtige stoffer med en forholdsvis høj opløselighed i vand. Stofferne i denne gruppe er bl.a. de klorerede opløsningsmidler og BTEX. Mange af disse stoffer vil det være hensigtsmæssigt at kunne teste for, da de har lave kriterieværdier i vand. Mange af disse stoffer har langt højere diffusionskoefficienter i luft end i vand og vil derfor spredes hurtigere i luft. I forhold til det konkrete arbejde, hvor den potentielle anvendelse af jord og restprodukter primært vil bestå i bygge- og anlægsarbejer vil stofferne dog sjældent udgøre et luft(indeklima) problem, da der er krav om befæstning, så kontakten afskæres. Det kritiske forhold ved testning af stofudvaskningen vil for denne gruppe af stoffer derfor være deres spredning i luft (flygtighed). Ved at minimere luftvolumenet i testsystemet kan dette håndteres i en batch, da kun en mindre del af den totale stofmængde vil findes på luftform, men i forhold til håndtering af prøven før og efter testningen vil flygtigheden være vanskelig at kontrollere. Specielt separationen af faststoffasen og eluatet vil være kritisk for de flygtige stoffer og dermed testens anvendelighed. Det kan være relevant at undersøge om det er mere hensigtsmæssigt at vurdere udvaskningen af disse flygtige stoffer ud fra måling af stofkoncentrationerne i poreluften.
Den tredje gruppe af stoffer med kriterieværdier indeholder stoffer med meget høj opløselighed eller blandbarhed i vand, og de sorberer meget lidt til jord. Til denne gruppe hører bl.a. methyltertiærbutylether (MTBE). Phenolerne tilhører også denne gruppe, idet de har en høj opløselighed i vand, men de sorberer kraftigere end f.eks. MTBE. Stoffernes spredning kan næppe reguleres gennem testning af udvaskningen fra jord og restprodukter, da stofferne i meget begrænset omfang vil findes associeret med de faste faser. Direkte prøvetagning af pore- eller grundvand virker som en umiddelbar mere velegnet metode til at identificere og vurdere disse stoffers udvaskning fra en potentiel kilde.
8. Eksperimentelle undersøgelser
8.1 Forsøgsplan og testbetingelser
Som et led i undersøgelserne indgår afprøvning af en udvalgt testmetodik på fem forskellige jordprøver, som er forurenet med organiske stoffer. De fem forskellige forurenede jordprøver er udvalgt, således at de dels repræsenterer typiske danske forureningstyper, dels omfatter en bredde i forureningstyper med hensyn til stoffer med forskellige fysisk-kemiske egenskaber, herunder flygtige stoffer. Med henvisning til afsnit 7.3 er alle 3 klasser af organiske stoffer repræsenteret.
Tabel 8.1 viser en oversigt over de fem jordprøver, der indgår i de eksperimentelle undersøgelser.
Tabel 8.1 Se her!
Oversigt over jordprøver og forsøgsplan til eksperimentel afprøvning af
testmetodik.
Jordprøven, der er forurenet med MTBE og benzin, indeholder ved tilstedeværelsen af MTBE'er et stof, der stort set ikke sorberer til jorden. Jordprøven fra Vapokongrunden indeholder opløsningsmidler (bl.a. tri- og tetrachlorethylen), som repræsenterer de meget flygtige stoffer. Dieselolieforurenet jord repræsenterer en meget typisk forurening, der har en kompleks sammensætning. Tjæreforurenet jord findes der også en del af i Danmark. I denne rapport er det kun PAH’erne, der er fokuseret på, fordi de kan belyse nogle problemer omkring stoffer, der bindes kraftigt til jorden. Ydermere kan de bindes til kolloider i væskefasen, hvilket gør dem interessante i forhold til design af udvaskningstests. Nogle af PAH'erne er kræftfremkaldende. Dette gælder bl.a. benz(a)pyren, som typisk findes i lave koncentrationer i jorden, og hvor grundvandskriteriet er meget lavt (summen af fluoranthen, benz(b+k)fluoranthen, benz(a)pyren, indeno(1,2,3-cd)pyren og dibenzo(g,h,i)perylen er 0,2 µg/l (Miljøstyrelsen 1998a)). Det er således ønskeligt, at udvaskningstesten er tilstrækkelig følsom til at kunne analysere koncentrationer i eluatet med en detektionsgrænse, som ligger under den anførte kriterieværdi.
Med hensyn til forureningsniveauet i de forskellige jordtyper er der bevidst valgt prøver, som er let til moderat forurenede samt lerholdige, dvs. vanskelige at teste og analysere. Ved således at teste vanskelige prøver er det hensigten at få et nærmere kendskab til eventuelle tekniske problemer ved gennemførelsen at udvaskningstests med organiske stoffer.
Med udgangspunkt i Miljøstyrelsens nye bekendtgørelse om genanvendelse af jord og restprodukter (Miljøstyrelsen 2000) og i forlængelse af arbejdet med de uorganiske stoffer er det valgt at afprøve et batchsystem, hvor der anvendes en 0,001 molær kalciumklorid-opløsning som udvaskningsmedie ved et væske-/faststofforhold på 2 l/kg. Dette er et lavt væske-/faststofforhold at udføre et batchforsøg ved, men der opnås et eluat, som i nogle tilfælde vil kunne simulere jordvæske. Ved at udføre testen med et relativt højt indhold af faststofprøve er det endvidere hensigten at opnå relativt høje og analyserbare koncentrationer i eluaterne. Sammenlignet med testproceduren beskrevet i forslaget til bekendtgørelse er det af hensyn til kravene til volumener til analyse ydermere nødvendigt at opskalere testsystemet, så der opnås et volumen af eluat på mere end 1 liter. Endelig viser erfaringerne med undersøgelser af organiske stoffer (se også kap. 6), at det er nødvendigt at undgå plastmaterialer i alt udstyr, som prøvematerialet og eluatet bringes i kontakt med under testproceduren. Som det fremgår af det videre, er der udelukkende anvendt glas og rusfrit stål samt teflon i mindre omfang. Ud fra litteraturen er det på forhånd klart, at visse stofgruppers flygtighed samt kontakttiden mellem prøve og eluat og den benyttede teknik til separation af eluat fra faststoffase er meget central og kritisk for udviklingen af en testprocedure. Derfor er der som supplement til testningen af de flygtige klorerede opløsningsmidler og de hydrofobe PAH-stoffer gennemført forsøg henholdsvis med helt lukkede testsystemer og ved forskellige kontakttider, hvilket er beskrevet nærmere i det følgende.
Ud over udvaskningsforsøgene er der gennemført en tørstofkarakterisering i form af kemisk analyse og partikelstørrelsesfordelingsanalyse af alle 5 jordprøver. Omfanget af den samlede forsøgsplan er vist i tabel 8.1.
8.2 Indsamling og karakterisering af jordprøver
8.2.1 Beskrivelse af prøvetagningslokaliteter
De fem undersøgte jordprøver blev indsamlet fra følgende lokaliteter:
| Servicestation i Dianalund | |
| Vapokon industrigrund | |
| Sandholm, jordrensning | |
| Frederiksberg Gasværk (Da den første prøve (Frederiksberg Gasværk 1) ikke var tilstrækkelig forurenet i forhold til de planlagte, blev der udtaget en ny jordprøve (Frederiksberg Gasværk 2) til yderligere forsøg). |
Servicestation i Dianalund
Lokaliteten er en DK-servicestation, hvor der stadig er et værksted, og hvor der foregår handel med benzinprodukter. Lokaliteten er i forbindelse med forureningsundersøgelser beskrevet nærmere i Dansk Geo-servEx a/s (1998) og Jord Miljø A/S (1999). Kilden til forureningen er MTBE-holdig blyfri benzin. Udbredelsen er begrænset til det øvre fyldlag, som overlejrer et lag af moræneler, der findes i ca. 2 meters dybde. Der foregår endnu ingen afværgeforanstaltninger på grunden. Vandprøver udtaget lige nedstrøms den formodede lækage indeholdt 150 mg/l totalkulbrinter, 122 mg/l BTEX og 729 mg/l MTBE. Jordprøven, som er anvendt i dette projekt, er udtaget 0,3 - 0,4 m fra boringen i en dybde på 1,2 - 1,6 meter under terræn, hvorfra også ovenstående vandprøve er udtaget.
Vapokon industrigrund
Vapokon Petrokemisk Industri er en ældre virksomhed til oparbejdning af opløsningsmidler. Forureningens udbredelse og de påtænkte afværgeforanstaltninger for både jord og grundvand er beskrevet af HOH Water Technology (1998). Forureningen består af klorerede opløsningsmidler og BTEX med koncentrationer op til 1100 mg klorerede opløsningsmidler/kg og 2100 mg BTEX/kg. Desuden er der forurenet med vandblandbare opløsningsmidler (acetone, isopropanol, ethylacetat). Den forurenede jord, der er blevet fjernet fra grunden, ligger til midlertidig opbevaring i et depot og er opdelt i tre forureningstyper: en meget forurenet jord, der er kilden; en mindre forurenet jord; og en mindre forurenet jord, som er blevet biologisk behandlet. Der er udtaget jordprøver af alle tre typer, men udvaskningsforsøget er udført med den ubehandlede, mindre forurenede jord.
Sandholm jordrensning
Jorden er en dieselforurening med totalkulbrintekoncentrationer på 3000-8000 mg/kg fra den tidligere telegrafkaserne i Høvelte. Jorden er blevet gravet op og transporteret til Sandholmlejren, hvor den forsøgsvis bliver biologisk renset on-site. Jordprøven til dette projekt er udtaget fra opbevaret jord, der ikke er renset.
Frederiksberg Gasværk
Frederiksberg gasværk blev etableret i 1895 til gasproduktion af stenkul indtil 1964. Efter lukning af gasproduktionen blev 2 ud af 3 tanke stadig anvendt til opbevaring af gas. Resten af bygningerne blev revet ned, men fundamenterne forblev der. Geologisk set er lokaliteten karakteriseret ved at bestå af moræneler med sandlinser, der overlejrer kalken, som udgør det primære grundvandsmagasin. Jordprøverne, der er udtaget til disse undersøgelser, forventes at være tjæreforurenet, men uden ammoniak og cyanider. De indeholdte ikke fri tjære og kun den ene prøve (Frederiksberg Gasværk 2) afgav en lugt af tjære. Den første prøve (Frederiksberg Gasværk 1) er udtaget fra moræneleret i en dybde på 0,3-0,5 m ,og den anden (Frederiksberg Gasværk 2) er udtaget fra en sandlinse i en dybde på 1,8-2,2 m.
8.2.2 Prøvetagning og forbehandling
Jordprøverne blev udtaget med rengjorte graveredskaber eller håndboreudstyr. I begge tilfælde blev prøverne overført direkte til særlige 10 liters metalbeholdere med tætsluttende låg. Beholderne blev opbevaret i køletaske med køleelementer under hjemtransporten og i kølerum ved 4°C efter hjemkomst til DHI. Inden forsøgene blev igangsat, blev prøverne delvist homogeniseret ved, at de større jordklumper blev presset/skåret i mindre dele. Efter genpåsætning af lågene blev spandene rystet kraftigt for at homogenisere prøverne.
8.2.3 Karakterisering af jordprøverne
Af de homogeniserede prøver blev der udtaget delprøver til karakterisering af jordprøverne samt til udvaskningsforsøgene. Jordprøverne blev karakteriseret med hensyn til indhold af de relevante organiske forureningsstoffer, ikke flygtigt organisk kulstof, pH og ledningsevne. De anvendte analysemetoder er beskrevet i Bilag C.
8.3 Udvaskningsforsøg
8.3.1 Forsøgsbeskrivelse
Hovedparten af forsøgene blev udført i 2 l Pyrex glasflasker (med et totalvolumen på 2,3 l) med skruelåg med teflonindlæg. I figur 8.1 er der vist et fotografi af de anvendte flasker.

Figur 8.1
Fotografi af de anvendte testbeholdere til udvaskningsforsøgene.
Ud fra en bestemmelse af tørstofindholdet i jordprøverne blev de beregnede jordmængder tilsat ved afvejning, hvorefter den fastlagte væskefase, der bestod af destilleret vand indeholdende 0,001 mol CaCl2/l, blev tilsat (begge svarende til L/S = 2 l/kg). De afvejede mængder fremgår af tabel 8.2. Under forsøget blev flaskerne agiteret i en lukket trækasse ved rotation omkring deres tværakse (8-9 rpm). Forsøget blev gennemført over en periode på 24 timer, hvorefter flaskerne henstod i ca. 1 time for at sedimentere partikler ud af eluatet. Herefter blev jordmaterialet skilt fra det dekanterede eluat i et trykfiltreringsudstyr (se afsnit 8.3.2).
Ovennævnte beskrivelse dækker "standardforsøget", der blev udført med alle jordprøverne undtagen Frederiksberg Gasværk 2. Herudover blev der som tidligere nævnt gennemført supplerende forsøg til nærmere undersøgelse af særligt kritiske tekniske faktorer.
Med henblik på at undersøge kontakttidens betydning blev der udført tre parallelle forsøg med agitationstiderne 6, 24 og 120 timer. Forsøgene blev gennemført med den PAH-forurenede jord (Frederiksberg Gasværk 1) på grund af kontakttidens væsentlige betydning for desorptionen/udvaskningen af disse stoffer fra jord. For at undgå/begrænse den aerobe bionedbrydning af de organiske stoffer (PAH’erne) i langtidsforsøgene over 120 timer blev ilten fjernet fra vandet ved at gennemboble det med kvælstof i mindst 16 timer. Desuden blev der tilsat 200 mg Na2SO3/l, der reducerer den resterende iltmængde, der findes i flaskerne. Koncentrationen af Na2SO3 kan reducere omkring 25 mg O2/l. For at opnå sammenlignelige resultater blev de to korttidsforsøg (6 og 24 timer) med samme type jordprøve udført med tilsvarende sammensætning af vandet som forsøget over 120 timer.
Med hensyn til den PAH-forurenede jord, blev betydningen af at anvende filtrering og centrifugering undersøgt ved at opdele eluatet udtaget efter 24 timers agitation i to delprøver, hvoraf den ene blev trykfiltreret som beskrevet under "standardforsøget", og den anden blev centrifugeret til en estimeret tilsvarende partikelafskæring.
Da det ikke var muligt at konkludere noget endeligt om kontaktidens indflydelse, dels fordi jordprøven indeholdt for lille en koncentration af PAH`er, dels fordi der blev tabt noget til filteret ved trykfiltringen og dels fordi trykfiltringen resultererede i for små prøvevolumener, hvormed detektionsgrænsen blev for høj, blev forsøget gentaget med en anden jordprøve (Frederiksberg Gasvæk 2). Forsøget blev udført efter den samme metode, som det tidligere forsøg til belysning af kontaktidens indflydelse, blot blev seperationen udført ved centrifugering i stedet for ved trykfiltrering, fordi centrifugering resulterede i en større prøvemængde.
Til nærmere undersøgelse af betydningen af stoffernes flygtighed blev der udført et supplerende forsøg med jordprøven fra Vapokonlokaliteten i en specialudviklet "zero headspace beholder" (Skúladóttir 1996). Beholderen havde et volumen på ca. 200 ml og er vist sammen med standard glasflasken på figur 8.1. Den var udført i rustfrit stål og bestod af en cylindrisk beholder, hvori jord og vand tilsattes. Derefter blev der trykket et tætsluttende låg med to O-ringe ned i beholderen, indtil luften var fortrængt, og der begyndte at komme vand op af en åbning i låget. Åbningen lukkedes med en swagelock fitting. Beholderen blev som i de øvrige forsøg agiteret ved rotation omkring dens tværakse, men centrifugeret direkte i centrifugen for at separere vand og jord. Efter centrifugeringen påsattes en teflonslange, som tillod udtagning af vandprøver ved at presse låget yderligere ned i beholderen. Hele forsøget udførtes således med minimal risiko for, at de flygtige stoffer i prøven fordampede.
I tabel 8.2 er forsøgsdetaljerne vedrørende de gennemførte udvaskningsforsøg sammenfattet.
Tabel 8.2 Se her!
Oversigt over de udførte forsøg med angivelse af de afvejede jord- og vandmængder,
det beregnede L/S-forhold, den anvendte kontakttid, metoden for efterbehandling af eluatet
og den tilsatte mængde natriumsulfit.
8.3.2 Indledende filtreringsforsøg
Inden selve forsøget blev igangsat, blev forskellige filtre og kombinationer af disse testet. Denne test blev udført med eluat fra forsøg med de samme jordtyper og udført magen til standardforsøgene. Målet var at få tilstrækkeligt vand (1000 ml til de fleste analyser bortset fra de klorerede opløsningsmidler, der kun kræver 100 ml) filtreret til analysen inden for 1-1,5 time, fordi selve testen skal være tidsmæssig praktisk gennemførlig.
Strategien var, at vandet skulle igennem et 0,7 µm Whatman glasfiberfilter. Dette kunne kræve et forfilter eller en forfiltrering. Følgende forfiltre blev testet: 100 µm stålfilter, 23 µm stålfilter og et Millepore glasfiber forfilter.
Samtidig filtrering igennem 0,7 µm glasfiberfilter og glasfiber forfilter gav for testen med Dianalund-prøven 400 ml eluat på ca. 2 timer, for Vapokon 140 ml eluat på ca. 1 time og 40 min og for Frederiksberg Gasværk 1 250 ml på ca. 1 time.
Filtrering kun igennem glasfiber forfilteret gav for Frederiksberg Gasværk 1 125 ml eluat på ca. 30 min og 190 ml på ca. 1 time og for Sandholm 300 ml på ca. 30 min.
En forfiltrering kun igennem 23 µm stålfilter gav for Sandholm 170 ml eluat på ca. 20 min, der efterfølgende blev filtreret igennem et 0,7 µm glasfiberfilter på ca. 30 sek.
En forfiltrering af vandprøverne igennem et 100 µm stålfilter varede for Sandholm kun nogle få minutter. Til gengæld resulterede det i et eluat, som stadig var meget grumset, hvilket tyder på, at filteret stort set ikke virkede.
Det fremgår af ovenstående, at det ved disse forforsøg ikke lykkedes at få filtreret 1000 ml prøve ved nogen af de afprøvede teknikker. De fire jordprøver er imidlertid meget lerede, hvilket betyder, at de repræsenterer den største udfordring ved en filtrering. For mere sandede jorde vil der kunne filtreres større mængder eluat indenfor den samme tidsperiode.
8.3.3 Efterbehandling af eluat
På baggrund af resultaterne af de indledende filtreringsforsøg blev det valgt, at trykfiltreringen skulle foregå gennem et 23 µm stålfilter efterfulgt af en trykfiltrering igennem et 0,7 µm glasfiberfilter.
Den specielt designede "zero headspace"-beholder blev centrifugeret ved 1500 rpm i 18 min., hvilket medførte, at partikler med en densitet på 1,5 kg/l og en diameter på 0,7 µm blev fjernet fra væskefasen. En delprøve af forsøget med tjæreforurenet jord (Frederiksberg Gasværk 1) blev centrifugeret ved 1500 rpm i 24 min, hvilket svarer til samme partikelafskæring som nævnt ovenfor (længere tid på grund af større volumen).
Separeringen af vandprøverne ved henholdsvis filtrering og centrifugering resulterede i de i tabel 8.3 anførte volumener af eluat. De tider, der er vist i tabel 8.3, svarer, hvis ikke andet er angivet, til tiden til næsten clogning af filteret. Som nærmere diskuteret i resultatafsnittet blev der kun for prøverne til analyse for klorerede opløsningsmidler opnået de ønskede minimumsvolumener, der kræves for at gennemføre analysen med den akkrediterede detektionsgrænse (se kap. 7). For de øvrige eluater betyder dette, at analyseresultaterne ikke kan angives med den ønskede detektionsgrænse. Eluaterne blev analyseret for de relevante stoffer (bilag C).
Da trykfiltrering både resulterede i tab af PAH`er til filteret og et lille prøvevolumen, blev separeringen i det andet kontakttidsforsøg med jorden Frederiksberg Gasværk 2 foretaget ved centrifugering.
Tabel 8.3 Se her!
De genererede eluatmængder efter fraseparering af partikler. Filtreringen foregik i
to omgange. Den angivne tid er den tid, det varede at filtrere prøven igennem det 23 µm
stålfilter. Den efterfølgende filtrering igennem det 0,7 µm glasfiberfilter tog mindre
end 10 min.
8.3.4 Sorptionsforsøg med filtermaterialer
Endelig blev der på baggrund af et øjensynligt tab af PAH-stoffer under gennemførelsen af udvaskningsforsøgene udført et efterfølgende sorptionsforsøg med de anvendte filtermaterialer i standardforsøget.
Der blev fremstillet 5 l vand med 0,001 mol CaCl2/l og 200 mg Na2SO3/l samt 24 PAH’er i en koncentration på hver ca. 3 µg/l. PAH’erne blev tilsat fra en stamopløsning i acetone. Af de 5 l vand blev der dels udtaget en vandprøve til analyse af begyndelseskoncentrationen, dels fyldt en 2,3 l Pyrex glasflaske, som standardforsøgene blev udført i. Flasken stod til omrøring i 24 timer, hvorefter halvdelen af vandmængden i flasken blev filtreret efter samme procedure (inkl. samme filtreringshastighed), som de rigtige jordprøver blev filtreret efter. Både det filtrerede vand og den resterende halvdel blev analyseret for PAH’er. De anvendte analysemetoder er beskrevet i bilag C.
8.4 Resultater og diskussion
8.4.1 Faststofkarakterisering
Resultatet af karakteriseringen af de fem jordprøver med hensyn til pH, ledningsevne, glødetab og indholdet af organisk stof NVOC er præsenteret i tabel 8.4a. I tabel 8.4b ses resultatet af bestemmelsen af kornstørrelsesfordelingen i de fem prøver.
Tabel 8.4a
pH, ledningsevne, glødetab og indholdet af organisk stof NVOC i de fem jordprøver.
| Jordprøve | pH |
Ledningsevne |
Glødetab |
NVOC |
| Vapokon | 8,1 1 |
285 1 |
17,1 |
5,1 |
| Dianalund | 7,4 1 |
92 1 |
13,1 |
2,6 |
| Sandholm | 7,9 1 |
175 1 |
9,9 |
4,4 |
| Frederiksberg 1 | 5,8 1 |
230 1 |
21,2 |
7,3 |
| Frederiksberg 2 | 8,8 2 |
909 2 |
7,4 |
2,3 |
1: Bestemt i en opslemning bestående af 20 g TS jord og 50 ml demineraliseret vand. 2: Bestemt i selve eluatet fra forsøget med 6 timers kontakttid.
Tabel 8.4b
Kornstørrelsesfordelingen for de fem jordprøver. Angivet som procentuel fordeling
baseret på vægt.
|
Kornstørrelsesfordeling (vægtprocent) |
||||||
Jordprøve |
>2 mm |
1-2 mm |
0,5-1 mm |
0,25-0,5 mm |
0,125-0,25 mm |
0,063-0,125 mm |
<0,063 mm |
Vapokon |
2,7 |
2,4 |
4,6 |
9,4 |
14,0 |
12,4 |
54,5 |
Dianalund |
25,4 |
9,8 |
12,6 |
14,4 |
12,8 |
5,5 |
19,6 |
Sandholm |
13,2 |
3,0 |
4,8 |
8,7 |
16,8 |
15,5 |
37,9 |
Frederiksberg 1 |
1,2 |
2,2 |
4,1 |
7,6 |
12,6 |
13,6 |
58,3 |
Frederiksberg 2 |
7,7 |
2,9 |
6,4 |
31,6 |
21,6 |
6,4 |
23,4 |
Som det fremgår af tabel 8.4a, varierer indholdet af organisk stof mellem 2,3 og 7,3 mg
C/g TS for de fem jordprøver. Analysen af organisk stof i form af NVOC inkluderer ud over
det naturlige organiske kulstof det ikke-flygtige kulstof, der er relateret til selve
forureningskomponenterne. På basis af de koncentrationer af organiske stoffer, der er
målt på jorden (tabel 8.5), er det kun jordprøven fra Sandholm, hvor bidraget fra de
organiske forureningskomponenter i form af indhold af total kulbrinter på 3 mg/kg TS, der
udgør en væsentlig fraktion af det målte NVOC-indhold. For de øvrige fire vurderes
det, at den målte NVOC-koncentrationen svarer omtrent til indholdet af det naturlige
organiske stof.
Kornstørrelsesfordelingen for de fem jordprøver viser, at samtlige jordprøver har et relativt højt indhold af partikler med en kornstørrelsesdiameter på mindre end 0,063 mm, hvilket svarer til fraktionen af ler og silt. Specielt prøverne fra Vapokon grunden og Frederiksberg Gasværk 1 har et meget højt indhold af fine partikler, hvilket svarer til forventningerne ud fra håndteringen af prøverne. Prøverne må således som helhed formodes at have et højere indhold af fine partikler, end det som gennemsnit vil ses i et repræsentativt udsnit af forurenede jordprøver fra Danmark.
Tabel 8.5a og b viser de fundne indhold af udvalgte organiske stoffer i de fem jordprøver. Resultaterne er diskuteret i sammenhæng med resultaterne af udvaskningsforsøgene i det efterfølgende afsnit.
8.4.2 Udvaskningsforsøg
Generelt indeholder de udtagne jordprøver de undersøgte stoffer i koncentrationer, der var høje nok til, at stofferne var målbare i de tilhørende eluater fra udvaskningsforsøgene. Dog gælder dette ikke prøven med PAH’er, Frederiksberg Gasværk 1, der må karakteriseres som let forurenet. I alt blev 22 af de 24 PAH'er, som analysen omfattede, fundet i jordprøven (tabel 8.5). Den højeste koncentration af enkeltkomponenterne var 0,8 mg/kg for fluoranthen. I de tre udvaskningsforsøg med PAH’erne, hvor eluaterne blev filtreret inden analyse, blev der kun målt naphthalen i en af de tre prøver. De resterende stoffer fandtes i koncentrationer lavere end detektionsgrænserne (0,05 - 0,25 µg/l), der som tidligere nævnt dog var forhøjede på grund af mangel på prøvevolumen. I eluatet, der blev centrifugeret før analyse, blev der detekteret 14 af de 24 PAH’er. Denne væsentlige forskel mellem de to typer separationstekniker kan skyldes, at PAH’erne tabes i udstyr, hvor eluatet filtreres. For at afklare dette nærmere blev der efterfølgende gennemført et sorptionsforsøg med filtrering af en vandig referenceprøve. Resultaterne af dette forsøg er præsenteret i tabel 8.6.
Forsøget viser, at størstedelen af den tilsatte stofmængde af PAH'er kan genfindes efter agitationen på 24 timer. Dog er genfindingen for nogle af de tungere 5- og 6-ringede stoffer nede på 64-80% i forhold til de tilsatte mængder ved start af forsøget. Dette tab kan kun tillægges kontakten med glas og teflon, da stofferne udelukkende har været i kontakt med disse materialer.
Under den efterfølgende filtrering af den PAH-holdige vandprøve ses et væsentligt tab af de tungere PAH'er, således at helt ned til mindre end 10 % af de oprindeligt tilsatte mængder genfindes i filtratet. For de lettere 2-ringede PAH'er er genfindingen typisk på ca. 80% af de tilsatte mængder. Denne graduering i genfinding for PAH’erne stemmer overens med hypotesen om, at PAH’erne i et væsentligt omfang kan sorbere på filtermaterialet, idet de tungere PAH’er har en større affinitet til at sorbere end de mindre.
Dette resultat forklarer sandsynligvis endvidere forskellene mellem de lavere koncentrationer i eluat, der er filtreret, sammenlignet med eluatet, der blev centrifugeret efter udvaskningsforsøget. Grundet formodet sorbtionstab har det ikke været muligt at teste, om filtrering og centrifugering giver væsentligt forskellige resultater på grund af forskellig partikelafskæring.
Tabel 8.5a Se her!
Koncentrationerne af de vigtigste organiske stoffer i fire jordprøver og i eluaterne
fra de gennemførte udvaskningsforsøg.
Tabel 8.5b Se her!
Koncentrationerne af de vigtigste organiske stoffer i jordprøven Frederiksberg 2 og i
eluaterne fra de gennemførte udvaskningsforsøg.
Jordprøven Frederiksberg Gasværk 1 med PAH’er indeholder totalt 3,36 mg PAH’er/kg TS som summen af de målte PAH´er. Summen af fluoranthen, benz(b+k+j)fluoranthen, benz(a)pyren, indeno(1,2,3-cd)pyren og dibenzo(a,h)anthracen i jordprøven er 1,37 mg/kg TS, hvor jordkvalitetskriteriet er 1,5 mg/kg TS (Miljøstyrelsen 1998a). Jordkvalitetskriterierne for naphthalen, benz(a)pyren og dibenz(a,h)anthracen er 1, 0,1 og 0,1 mg/kg TS (Miljøstyrelsen 1998a), hvor der i jordprøven er målt henholdsvis 0,02, 0,3 og 0,014 mg/kg, så i denne jordprøve er det kun benz(a)pyren, som overskrider jordkvalitetskriterierne. I det centrifugerede eluat måles der 0,19 µg naphthalen/l i sammenligning med et grundvandskvalitetskriterie på 1 µg/l (Miljøstyrelsen 1998a), og summen af fluoranthen, benz(b+k)fluoranthen, benz(a)pyren, indeno(1,2,3-cd)pyren og benzo(g,h,i)perylen i eluatet er 9,0 µg/l, hvor grundvandskvalitetskriteriet er 0,2 µg/l (Miljøstyrelsen 1998a). Den PAH-forurenede jordprøve overskrider således grænseværdien for benz(a)pyren med en faktor 3 og ikke summen af PAH’er i øvrigt. En direkte sammenligning af koncentrationerne målt i eluatet med grundvandskvalitetskriterierne giver anledning til overskridelser af de fastsatte grundvandskriterier på en faktor 45. En sådan direkte sammenligning skal naturligvis gøres med forbehold, idet eluatet fra et agiteret testsystem udført ved et væske-/faststofforhold på 2 l/kg ikke direkte kan sammenlignes med den koncentration, som vil opnås ved nedsivning af porevand fra forurenet jord til et grundvandsmagasin. Ved en direkte sammenligning kan koncentrationerne målt i testen på den ene side overestimere koncentrationerne i nedsivende porevand på grund af agitationen i laboratorietesten. På den anden side kan man tilsvarende underestimere koncentrationsniveauet i det nedsivende porevand for stoffer, hvis koncentration er langt fra deres maximale opløselighed. I den forbindelse kan det anføres, at en jord med en porøsitet på 0,3-0,4 og en partikelvægtfylde på 2,5-2,7 kg/l vil have en L/S-værdi på 0,16-0,27 l/kg, svarende til en mulig opkoncentreringsfaktor på 7,5-12,5. For jord udlagt i en lagtykkelse på 1 m svarer et års nettoinfiltration på 300 mm til L/S = 0,16 l/kg, og det tager 12,5 år at nå L/S = 2 l/kg. Resultatet af en udvaskningstest udført ved L/S = 2 l/kg repræsenterer altså i dette tilfælde et gennemsnit af situationen over en periode på ca. 13 år.
Tabel 8.6 Se her!
Resultat af sorptionsforsøg med PAH'er tilsat standard udvaskningsforsøget i en
vandig opløsning.
Resultaterne viser under alle omstændigheder, at testsystemet selv for svagt forurenede til uforurenede prøver er tilstrækkeligt følsomt til at påvise PAH-stoffernes tilstedeværelse i eluatet helt ned til grundvandskvalitetskriteriet.
I jordprøven Frederiksberg Gasværk 2 med PAH’er er det kun koncentrationen af naphthalen på 1,6 mg/kg TS, der overskrider jordkvalitetskriteriet på 1 mg/kg TS (Miljøstyrelsen, 1998a). Efter 24 timers agitation måles der i eluatet f.eks. 190 µg naphthalen/l, hvilket er en overskridelse af grundvandskvalitetskriterie på 1 µg/l med 190 gange (Miljøstyrelsen 1998a).
Udfra forsøget med jorden Frederiksberg Gasværk 1 kan effekten af kontakttiden på væskekoncentrationen ikke klarlægges på grund af det omtalte tab af stoffer under filtreringen af eluaterne.
Forsøget med jorden Frederiksberg Gasværk 2, hvor eluatet centrifugeres, resulterede imidlertid i data, hvorudfra det er muligt at vurdere kontakttidens betydning på koncentrationen i eluatet. Resultaterne er vist i figur 8.2 samt tabel 8.5b og 8.7. Naphthalenkoncentrationen i eluatet er efter 6 timers agitation 170 µg/l, efter 24 timer 190 µg/l og efter 120 timer 230 µg/l (Tabel 8.5b). For pyren måles der 2,1, 2,3 og 6,7 µg/l efter henholdsvis 6, 24 og 120 timers agitation. For naphthalen observeres en stigning med kontakttiden på 30-40 procent, hvorimod stigningen for pyren er på ca. 300 procent. Der er en stor effekt af kontakttiden på koncentrationen af PAH`er i eluatet, en effekt der er størst for de kraftigst sorberende PAH`er. Den store effekt af kontaktiden skyldes sandsynligvis, at det er en gammel forurening, så desorptionen af de organiske stoffer er langsom (se kapitel 3). Figur 8.2 viser også, at udvaskningen af PAH`er falder med stigende molekylvægt, hvilket skyldes, at de lette stoffer sorberer mindre til jorden end de tungere, hvorfor de er lettere at udvaske.Tallene for 1-methylphenanthren skal tages med forbehold, idet mængden, der udvaskes, svarer til f.eks. 48% efter 120 timer, hvilket er meget højt relativt til de øvrige PAH`er.
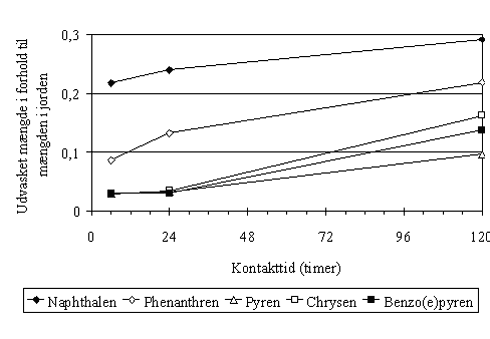
Figur 8.2
Resultater for 5 udvalgte PAH`er ved kontakttidsforsøg med jordprøven Frederiksberg
Gasværk 2.
I figur 8.3 er den udvaskelige mængde vist som funktion af log(Kow). Da jordprøverne har et relativt lavt indhold af organiske stoffer (der er ingen fri fase), vil sorptionen styre udvaskningen af PAH`er fra jorden. Sorptionen er alt andet lige propertional med stoffernes log(Kow)-værdier, hvorfor de udvaskelige mængder burde afhænge af stoffernes log(Kow)-værdier. Som det fremgår af figur 8.3, falder den udvaskelige mængde næsten lineært med log(Kow) (uafhængig af kontakttid) op til log(Kow) på 5, svarende til phenanthren. Derover er den udvaskelige mængde nærmest konstant. Hvad dette skyldes, er ikke klarlagt, men årsagen kunne være, at udvaskningen af PAH`er med log(Kow) højere end 5 influeres af kolloider. Men den tid og den hastighed, der er benyttet ved centrifugeringen, burde også fjerne kolliderne fra elutatet, inden det analyseres.
De målte koncentrationer af stofferne i eluatet fra udvaskningsforsøgene kan sammenholdes med en maksimal eller potentiel udvaskelig stofmængde beregnet ved en antagelse om, at hele den målte stofmængde i jorden udvaskes til eluatet. Resultaterne af disse beregninger er gengivet i tabel 8.7. For MTBE bør den beregnede maksimale koncentration og den målte være identiske, da MTBE ikke forventes at sorbere til jorden. Den beregnede maksimale koncentration er 10 µg MTBE/l, og der er målt 14 µg MTBE/l i eluatet (tabel 8.5), hvilket vurderes at være acceptabelt og således at vise, at udvaskningsforsøget med MTBE giver et anvendeligt resultat.
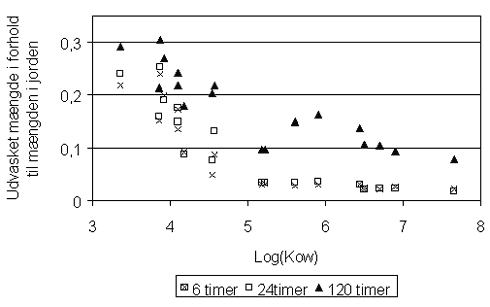
Figur 8.3
Den udvaskede mængde i forhold til mængden i jorden som funktion af stoffernes log(Kow)-værdier
for PAH`er i kontakttidsforsøg med jordprøven Frederiksberg Gasværk 2.
Fra den dieselforurenede jordprøve (Sandholm) udvaskes der kun 0,013% af den oprindelige mængde af forureningen. Den overvejende del af dieselolie består af tungere aliphater (flere end 10 kulstofatomer), der sorberer kraftigt til jorden, og derfor skyldes den relativt lave fundne koncentration i eluatet sandsynligvis sorption til jorden. De tungere aliphater har også en lille opløselighed, hvilket betyder, at koncentrationen af de organiske stoffer i eluatet kontrolleres af opløsning, hvis der er fri fase i jordprøven. Udvaskningen af dieselolie fra jordprøven med 3000 mg totalkulbrinter/kg TS repræsenterer en situation med en kraftig jordforurening (kvalitetskriteriet for dieselolie i jord er 100 mg/kg TS, Miljøstyrelsen 1998a)), der resulterer i en koncentration i eluatet på 190 µg/l (tabel 8.5). Grundvandskriteriet for dieselolie er 9 µg/l, og ved oprensninger af forurenet jord og grundvand benyttes ofte et oprensningskriterium for grundvand på 10 µg/l (Miljøstyrelsen 1998b). Det fundne tab af PAH'er ved filtrering af eluaterne kan tilsvarende muligvis også gøre sig gældende for de tungere aliphater, hvilket resultaterne af de foreliggende forsøg ikke kan afklare.
De to forsøg med Vapokonjorden i henholdsvis standard-forsøgsopstillingen og "zero headspace"-beholderen viste, at der blev fundet 1,5-6 gange højere koncentrationer af chloroform, 1,1,1-trichlorethan, tri- og tetrachlorethylen i forsøget med zero headspace beholderen end i standard-forsøgsopstillingen, se tabel 8.5. Dette viser tydeligt, at der ved standardprocedurerne tabes stof, som ikke fjernes ved anvendelse af den særlige testbeholder og -procedure til flygtige stoffer. Den mest oplagte forklaring er fordampning af stof under påfylding af eluat på trykfiltreringsudstyret samt under selve filtreringen, da der er et luftrum over vandet i selve beholderen til filtreringen. Da hverken chloroform, 1,1,1-trichlorethan, tri- eller tetrachlorethylen sorberer så kraftigt, som PAH’erne gør, er det ikke sandsynligt, at de sorberer til selve filteret, som PAH’erne muligvis gør. Med hensyn til den maksimale udvaskelige mængde svarer den målte koncentration i vandfasen til 1,6 - 3,1 af den maksimale udvaskelige mængde for no headspace forsøget og til 0,55 - 1,1 for standardforsøget. De høje forhold kan umiddelbart ikke forklares, men årsagen kan være enten, at der er tabt stof under proceduren for analyse af totalindhold i jordprøven (faststofanalyser er altid langt mindre præcise end analyser af stof opløst i vand), eller at den delprøve af jordprøven, som er udtaget til analyse, ikke har været homogen og repræsentativ for indholdet i hele jordprøven.
Tabel 8.7 Se her!
Beregnede maksimale stofkoncentrationer i eluaterne ud fra en antagelse om
fuldstændig stofoverførsel fra udvalgte jordprøver til eluatet. Endvidere er forholdet
mellem de målte og beregnede maksimale stofkoncentrationer beregnet, og den udvaskede
mængde er sat i forhold til mængden af stof i jorden.
Udover at sammenligne de målte koncentrationer i eluatet med en maksimal udvaskelig mængde kan den målte eluat koncentration sammenholdes med en beregnet porevandskoncentration. Den er beregnet udfra den målte jordkoncentration ved fasefordelingsberegning og ved brug af jordens naturlige vand- og luftindhold, samt det målte indhold af naturligt organisk stof. Princippet følger JAGG (Miljøstyrelsens risikovurderingsprogram), men her er beregningerne foretaget i det samme regneark, som er brugt til de øvrige fasefordelingsberegninger i denne rapport. Af jordparametrene er det kun vandindholdet i den oprindelige prøve og indholdet af organisk stof, der er bestemt analytisk. De øvrige er varieret i beregningerne. Porøsiteten er sat til 0,3 og 0,4, korndensiteten er sat til 2,5 og 2,7 kg/l. Udfra porøsiteten og korndensiteten er bulkdensiteten beregnet, og udfra det målte vandindhold, den beregnede bulkdensitet og porøsiteten er vandindholdet omregnet til henholdsvis vand- og luftfyldt porøsitet beregnet. Et eksempel på beregningerne er vist i tabel 8.8. Fasefordelingsberegningerne er udført med hver enkelt af de fire samhørende sæt jordparametre. Hovedresultaterne er gengivet i tabel 8.9, og detajlerne i beregningerne er vist i bilag D.
Resultatet af beregninger i tabel 8.9 viser, at forholdet mellem den målte eluatkoncentration og den beregnede porevandskoncentration varierer mellem 0,032 og 560. Forholdet burde være 0,065-0,08, fordi den eneste forskel er L/S-forholdet, der er 2 l/kg i selve udvaskningsforsøget og 0,13-0,16 l/kg (det naturlige L/S-forhold i jorden umiddelbart efter prøvetagningen, som kan beregnes som porevolumen x mætningsgraden/tørdensititen) i beregningen af porevandskoncentrationen. I beregningere af forholdet mellem den målte eluatkoncentration og den beregnede porevandskoncentration indgår analytisk bestemte parametre (eluatkoncentration, jordkoncentration og indholdet af naturligt organisk stof), som er behæftet med usikkerhed. Desuden er Kd-værdien estimeret udfra log(Kow) vha. en imperisk sammenhæng samt under den forudsætning, at det kun er indholdet af naturligt organisk stof, der betyder noget for sorptionen. Af den usikkerhed, der indgår i beregningerne, er det sammenhængen mellem Kd-værdien og log(Kow), der er den væsentligste usikkerhedsfaktor. De beregnede forhold skal derfor tages med et vist forbehold.
Tabel 8.8
Eksempel på målte og beregnede jordparametre. Tørstofindholdet er for jordprøven
Frederiksberg gasværk 1. Se tekst for yderligere forklaring.
| Porø- sitet |
Korn- densitet (kg/l) |
Bulk- densitet (kg/l) |
Tørstof- indhold (g/kg våd jord) |
Vand- indhold (g/kg våd jord) |
Vandfyldt porøsitet |
Luftfyldt porøsitet |
| 0,4 | 2,7 | 1,62 | 846 | 154 | 0,249 | 0,151 |
| 0,4 | 2,5 | 1,50 | 846 | 154 | 0,231 | 0,169 |
| 0,3 | 2,7 | 1,89 | 846 | 154 | 0,291 | 0,0089 |
| 0,3 | 2,5 | 1,75 | 846 | 154 | 0,270 | 0,0305 |
Tabel 8.9
De beregnede porevandskoncentrationer udfra en simpel fasefordelingsberegning ved
anvendelse af de fysiske forhold i tabel 8.8 og jordenes indhold af naturligt organisk
stof. Til sammenligning er angivet de målte jordkoncentrationer og eluatkoncentrationer
fra udvaskningsforsøget.
Prøve |
Stof |
Målt konc. |
Målt konc. |
Beregnet |
Forhold mel-lem målt eluat konc. og beregnet porevands- konc. |
Vapokon |
Trichlore- |
0,13 |
2001 |
257-280 |
0,71-0,78 |
Tetrachlore- |
0,20 |
3001 552 |
208-226 |
1,32-1,44 |
|
Dianalund |
MTBE |
0,02 |
14 |
142-145 |
0,097-0,098 |
Sandholm |
Totalkul- |
3000 |
190 |
-3 |
|
Frederiksberg gasværk 1 |
Naphthalen |
0,020 |
0,194 |
5,84 |
0,032 |
2-Methylnaphthalen |
0,006 |
<0,09 |
0,54 |
<0,17 |
|
1-Methylnaphthalen |
0,009 |
<0,09 |
0,79 |
<0,11 |
|
Acenaphthylen |
0,073 |
0,16 |
3,7 |
0,043 |
|
Acenaphthen |
0,007 |
<0,09 |
0,55 |
<0,16 |
|
Fluoren |
0,037 |
<0,09 |
1,6 |
<0,057 |
|
Phenanthren |
0,220 |
0,3 |
3,7 |
0,082 |
|
Anthracen |
0,110 |
0,44 |
2,0 |
0,22 |
|
1-Methylphenanthren |
0,009 |
<0,09 |
0,038 |
<2,3 |
|
Fluoranthen |
0,800 |
2,7 |
2,8 |
0,96 |
|
Pyren |
0,720 |
2,6 |
2,8 |
0,93 |
|
Benz(a)anthracen |
0,360 |
1,7 |
0,50 |
3,4 |
|
Chrysen |
0,290 |
1,8 |
0,20 |
9,2 |
|
Benz(b+k+j) |
0,041 |
2,6 |
0,042 |
62 |
|
Benz(e)pyren |
0,220 |
0,9 |
0,042 |
22 |
|
Benz(a)pyren |
0,300 |
1,5 |
0,049 |
30 |
|
Indeno(1,2,3-cd)pyren |
0,210 |
1,2 |
0,0022 |
560 |
|
Dibenzo(a,h)- anthracen |
0,014 |
0,17 |
0,0023 |
74 |
|
Dibenzo(g,h,i)perylen |
0,180 |
1,03 |
0,011 |
91 |
|
Frederiksberg gasværk 2 |
Naphthalen |
1,6 |
1905 |
1300 |
0,14 |
2-Methylnaphthalen |
0,19 |
15 |
53 |
0,28 |
|
1-Methylnaphthalen |
0,12 |
15 |
33 |
0,46 |
|
Biphenyl |
0,031 |
2,7 |
4,9 |
0,55 |
|
Acenaphthylen |
0,038 |
2,8 |
6,1 |
0,46 |
|
Acenaphthen |
0,033 |
3,1 |
8,0 |
0,39 |
|
Fluoren |
0,17 |
7,3 |
22 |
0,32 |
|
Phenanthren |
0,26 |
17 |
14 |
1,2 |
|
Anthracen |
0,12 |
4,5 |
6,8 |
0,66 |
|
Fluoranthen |
0,21 |
3,5 |
2,3 |
1,5 |
|
Pyren |
0,14 |
2,3 |
1,7 |
1,3 |
|
Benz(a)anthracen |
0,034 |
0,57 |
0,15 |
3,8 |
|
Chrysen |
0,03 |
0,52 |
0,064 |
8,1 |
|
Benz(b+k+j)fluor- anthen |
0,072 |
0,81 |
0,023 |
35 |
|
Benz(e)pyren |
0,031 |
0,46 |
0,019 |
25 |
|
Benz(a)pyren |
0,042 |
0,43 |
0,022 |
20 |
|
Indeno(1,2,3-cd)- pyren |
0,031 |
0,27 |
0,0010 |
270 |
|
Dibenzo(a,h)anthra- cen |
0,005 |
0,05 |
0,0026 |
19 |
|
Dibenzo(g,h,i)perylen |
0,024 |
0,27 |
0,0048 |
56 |
1: data fra forsøget med No-Headspace beholderen
2: data fra standard udvaskningsforsøget
3: ikke muligt at lave fasefordelingsberegninger på en stofblanding
4: data fra forsøget med 24 timers agitation og centrifugering
5: data fra forsøget med 24 timers agitation og centrifugering.
I forhold til de usikkerhedsmomenter, der er beskrevet ovenfor, er der en rimelig overensstemmelse mellem de beregnede porevandskoncentrationer og de målte eluatkoncentrationer, når der tages forbehold for forskellen i L/S-forhold mellem de to systemer, som nævnt i det foregående afsnit. En undtagelse er de meget store forhold, der er fundet for de 4-, 5- og 6-ringede PAH`er, som viser, at koncentationen i eluatet i udvaskningsforsøget er for høj, og at udvaskningen baseret på batchudvaskningsforsøgene måske overvurderes. Det er de samme stoffer, som "falder uden for kurven" i sammenhængen mellem den udvaskelige mængde og log(Kow). Som tidligere nævnt kan det være et problem ved separeringen af faststof fra eluatet.
Sammenfattende har de eksperimentelle afprøvninger vist, at det er teknisk muligt at gennemføre udvaskningsforsøg med organiske stoffer, men at der er en række problematiske forhold forbundet med testningen af PAH-stoffer. Disse forhold er rettet mod separationen af faststof fra eluat, der er vanskelig på grund af det høje indhold af fine partikler i prøverne og for de tungere PAH'er ikke giver de forventede resultater, fordi stofferne delvist tabes under filtreringen. Problemet kan ikke umiddelbart løses ved at centrifugere partiklerne ud af eluatet, da centrifugering kun vanskeligt kan foretages for de relativt store prøvevolumener, der er nødvendige af hensyn til den efterfølgende analyse. I så fald kræves der omhældning af prøven til flere mindre centrifugerør, hvorved prøven eksponeres yderligere med forøget risiko for stoftab. Et vist sorptionstab af tunge PAH'er til glas og glasfiltre er påvist. Alternativt bør rustfri stål testes.
Kontakttidsforsøget med jorden Frederiksberg Gasværk 2 viste, at agitationstiden var vigtig, idet de højeste koncentrationer i eluatet opnåedes efter 120 timer, og at for de tungere PAH`er opnåedes en stigning på 100-500% i eluatkoncentrationen fra 6 timers agitation til 120 timer. Om 120 timers kontaktid bedre udtrykker den udvaskelige mængde er ikke afklaret, men hvis man er interesseret i den maksimale udvaskelige mængde under de givne omstændigheder, skal forsøgene forløbe længere end 24 timer.
Afprøvningen på klorerede opløsningsmidler har vist, at det er vanskeligt ikke at tabe flygtige stoffer under testningen. Brug af særligt udstyr og procedure har dog vist, at tabet kan reduceres betragteligt. Igen kræver dette system særlig centrifugering og kan kun udføres med relativt små prøvevolumener (begrænset af størrelsen af laboratoriecentrifuger), hvilket ikke er hensigtsmæssigt i relation til den videre analyse.
9. Bestemmelse af udvaskningen af organiske stoffer
| 9.1 | Indledende forslag til testmetode |
| 9.2 | Eksempel på praktisk anvendelse af testmetoden |
9.1 Indledende forslag til testmetode
I forbindelse med vurderinger af risikoen for og konsekvenserne af udvaskning af organiske forureningskomponenter fra forurenet jord og udlagte restprodukter har Miljøstyrelsen behov for en testprocedure, som kan anvendes til at estimere porevandskoncentrationen i det resulterende perkolat. Herudover er der behov for at kunne beskrive massefluxen ud af jorden eller restproduktet.
Der er næpppe nogen tvivl om, at den mest velegnede laboratorietest til frembringelse af denne type information vil være en kolonneudvaskningstest, som i betydeligt omfang kan simulere udvaskningsforholdene ved det aktuelle scenarie (se f.eks. afsnit 6.1). Som det fremgår af afsnittene 5 og 6, er de praktiske erfaringer med kolonneudvaskning af organiske forureningskomponenter imidlertid stærkt begrænsede, mens erfaringerne med batchudvaskning af organiske stoffer er noget mere omfattende. Det har ligget udenfor rammerne af dette projekt at gennemføre den nødvendige udvikling af en kolonneudvaskningstest til organiske forureningskomponenter, og projektets eksperimentelle del har som beskrevet i afsnit 8 i stedet været fokuseret på afprøvning af en simplere batchudvaskningsprocedure. For at komme så tæt som muligt på en simulering af porevandssammensætningen er det valgt at udføre batchtesten ved L/S = 2 l/kg, hvilket normalt anses for at være det laveste praktisk anvendelige L/S-forhold i en batchudvaskningstest. Herved får testen for udvaskning af organiske stoffer en stor lighed med den udvaskningstest på overensstemmelsesniveau, prEN 12457-1, som i let modificeret form er foreskrevet til undersøgelse af udvaskningen af uorganiske forureningskomponenter fra jord og restprodukter i Miljøstyrelsens nye bekendtgørelse om genanvendelse af jord og restprodukter til bygge- og anlægsformål.
Selv om grundlaget er spinkelt og der fortsat er en række uafklarede forhold omkring de optimale forsøgsbetingelser, skal der i det følgende opstilles et indledende forslag til procedure til testning af udvaskningen af udvalgte organiske komponenter fra jord og restprodukter. Testmetoden er væsentligt simplere, hurtigere og billigere at udføre end en kolonneudvaskningstest er, men samtidig er den mindre velegnet end denne til at besvare de ovenfor stillede spørgsmål om porevandssammensætning og flux, og det opnåede svar vil være baseret på en grov og forenklet gennemsnitsbetragtning. Det må anbefales, at de gjorte forudsætninger i hvert enkelt tilfælde i videst muligt omfang søges verificeret, og at der sker en fortsat udvikling dels af denne metode, dels af en modsvarende kolonneudvaskningstest. Det indledende forslag er baseret på de opnåede projektresultater, herunder både de teoretiske overvejelser og de praktiske erfaringer og resultater fra projektets eksperimentelle del.
Metoden er foreløbig begrænset til testning af udvaskning af ikke-flygtige organiske stoffer som olieprodukter (benzin undtaget) og PAH’er. For at minimere adsorptionen til udstyret anvendes en glasflaske (Pyrex), som først renses for organisk materiale ved opvarmning til 550 ° C i 2 timer. Som et kompromis mellem ønsket om en (tilstrækkelig) stor mængde eluat til analysering og de problemer, som håndtering af en stor mængde eluat giver (f.eks. ved centrifugeringen), er det valgt at anvende en 2-liters flaske og at benytte 940 g jord (på tørstofbasis). For at simulere jordvæske udvaskes med en 0,001 M opløsning af CaCl2- i demineraliseret vand, og der anvendes 1880 ml heraf, således at der opnås et L/S på 2. Opløsningen af CaCl2 anbringes i flasken, og for at undgå biologisk nedbrydning af de organiske stoffer under udvaskningsforsøget gennemblases væsken i 24 timer med kvælstof, således at størstedelen af den opløste ilt fjernes. Herefter placeres jordprøven i flasken, den sidste ilt fjernes ved tilsætning af 200 mg Na2SO3/l (i alt ca. 375 mg), og flasken lukkes med en prop tildækket med Teflon. Der opnås herved et begrænset headspace, men der vil normalt stadig være luft nok i flasken til at sikre en god opblanding under agitationen.Den lukkede flaske agiteres herefter ved rotation om sin tværakse ved 5-10 rpm, fuldstændig svarende til den i prEN 12457-1 forskrevne agitation. Agitationen fortsættes i 24 timer eller 5 døgn, afhængigt af den aktuelle situation og formålet med testen (kontakttiden kan eventuelt tilpasses et givet scenarie). Flasken henstår herefter i 1 time, hvorved de størst partikler vil sedimentere. På grund af de observerede problemer med filtrering af eluatet (se f.eks. afsnit 8.3) er det valgt at adskille væske- og faststoffasen ved centrifugering. Vandfasen overføres herefter ved dekantering til centrifugebeholdere af glas eller stål, og der centrifugeres med en hastighed og tid svarende til en partikelafskæring på 0,7 m m. Denne værdi er valgt forholdsvis arbitrært, idet projektet ikke har omfattet en undersøgelse af betydningen af partikelafskæringen. Efter centrifugeringen overføres eluatet forsigtigt med en glaspipette til en analysebeholder, og de planlagte analyser for ikke-flygtige organiske stoffer gennemføres. En separat delprøve af eluatet analyseres for pH og ledningsevne.
Det indledende forslag til testproceduren er i Bilag E opsummeret i form af et indledende forslag til testprotokol.
9.2 Eksempel på praktisk anvendelse af testmetoden
Som nævnt ovenfor er det ofte ønskeligt at kunne benytte udvaskningstesten til over et givet tidsrum at estimere en gennemsnitlig eller maximal porevandskoncentration for udvaskede organiske forureningskomponenter i jorden lige under eller i underkanten af et lag af forurenet jord. Endvidere vil det ofte være ønskeligt at kunne bestemme den tilsvarende stofflux ud af den forurenede jord.
Blandt de forudsætninger, som bør være opfyldt, for at dette vil kunne lade sig gøre, er et krav om, at såvel udvaskningstesten som det betragtede scenarie repræsenterer systemer, hvor der der er ligevægt mellem væskefasen og faststoffasen. Det vil sige, at kontakttiden i udvaskningsforsøget skal være lang nok til, at der opnås ligevægt, og det forurenede jordlag skal være tilstrækkeligt homogent og tykt og strømningshastigheden tilstrækkelig langsom til, at der kan antages at være (lokal) ligevægt mellem væskefase og faststoffase for de aktuelle organiske forureningskomponenter. Som det fremgår af afsnit 3.6.8 vil det ofte kunne forventes, at den sidstnævnte forudsætning er opfyldt, mens det af resultaterne i afsnit 8.4 fremgår, at den første forudsætning ikke nødvendigvis er opfyldt ved anvendelse af kortere kontakttider i udvaskningstesten. I det følgende antages det dog, at begge betingelser er opfyldt.
Hvis der forekommer fri fase af en organisk forureningskomponent i jorden i hele den betragtede periode, vil porevandskoncentrationen lige under laget svare til den koncentration af komponenten, som er målt i udvaskningstesten, uanset hvilket L/S-forhold, denne er udført ved. Koncentrationen i vandfasen vil svare til stoffets vandopløselighed under de givne omstændigheder. Dette gælder, så længe der er tilstrækkelig meget fri fase i jorden til at sikre, at der er en optimal kontakt imellem vandet og den fri fase. Hvis den fri fase af en organisk forureningskomponent slipper op under udvaskningsforløbet, vil koncentration af denne falde gennem forløbet.
Hvis udvaskningen udelukkende kontrolleres af desorption, kan porevandskoncentrationen lige under laget i nogle tilfælde beregnes ved at forudsætte, at den eneste forskel på udvaskningsforsøget og strømningen i laget er L/S-forholdet. Hermed kan koncentrationen lige under laget ud fra en simpel fortyndings- eller opkoncentreringsbetragtning estimeres som koncentrationen i udvaskningsforsøget ganget med L/S-forholdet i udvaskningsforsøget divideret med L/S-forholdet i laget. De tilfælde, hvor dette princip fungerer, kan karakteriseres ved, at sorption/desorption på den aktuelle jord styres af præcis de samme processer uafhængigt af den resulterende stofkoncentration i jorden, hvilket medfører, at der kan omregenes imellem de forskellige L/S-forhold.
Tiden for udvaskningen kan ikke forudsiges. I begge tilfælde vil der være en ekstrem lang hale, idet diffusionen kontrollerer den sidste del af stofafgivelsen fra jorden eller den frie fase.
For at illustrere princippet i de ovenstående teoretiske betragtninger er der i det følgende vist et eksempel på estimering af porevandskoncentration i den nederste del af forurenet jourdlag ud fra målte eluatkoncentrationer af organiske forureningdkomponenter. Her er benyttet de resultater, som blev opnået i udvaskningsforsøget med jordprøven Frederiksberg gasværk 2 med et L/S-forhold på 2 l/kg og en agitationstid på 24 timer (se afsnit 8.4). De målte eluatkoncentrationer er gengivet i tabel 9.1.
Vandgennemstrømningen gennem det forurenede jordlag forudsættes at foregå i en umættet zone med samme karakteristika, som den testede jordprøve havde, da den blev udtaget (dvs. et L/S-forhold på 0,163). Under de forudsætninger, som er beskrevet ovenfor, beregnes koncentrationen i porevandet, CV, som eluatkoncentrationen, CE,, multipliceret med forholdet mellem de to systemers L/S-forhold, dvs:
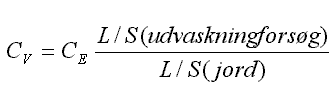
Eluatkoncentrationen skal derfor ganges med en faktor på 2,0/0,163 = 12,27. Resultatet af disse beregninger er ligeledes vist i tabel 9.1. For at kontrollere, at der ikke er fri fase i systemet, er opløseligheden af stofferne vist. Såfremt den beregnede koncentration for et stof overskrider stoffets vandopløselighed, erstattes den beregnede værdi med koncentrationen svarende til vandopløseligheden ("mætningsfunktion"). Dette sker faktisk for 6 af de beregnede porevandskoncentrationer i tabel 9.1. Her er der ikke taget hensyn til, at den reelle opløselighed af nogle stoffer i blandinger kan være mindre end deres opløselighed som enkeltstoffer.
Den estimerede porevandssammensætning kan betragtes som en gennemsnitsværdi for et tidsrum, hvis længde afhænger af tykkelsen af det betragtede jordlag og størrelsen af nettoinfiltrationen.
Den til de estimerede porevandskoncentrationer hørende flux, F, af stof igennem/ud af jordlaget kan beregnes udfra en nettoinfiltration eller en nedsivning, N, igennem laget ved brug af følgende formel: F = N·CV. I regneeksemplet er benyttet et N på 300 mm/år.
Det må ikke glemmes, at ovenstående estimat blot er et regneeksempel, som bygger på en række forudsætninger, som ikke nødvendigvis er opfyldt. Ved lignende anvendelser af testresultater bør der i hvert enkelt tilfælde foretages en vurdering af, i hvor høj grad forudsætningerne for anvendelsen er eller kan forventes at være opfyldt.
Tabel 9.1 Se her!
Estimering af porevandskoncentrationener under umættede forhold ud fra den målte
eluatkoncentration fra en udvaskningstest ved et L/S-forhold på 2 l/kg. Fluxen af stoffet
er ligeledes beregnet, og opløseligheden af stofferne i vand er vist.
10. Samlet metodeevaluering og anbefalinger
| 10.1 | Erfaringer |
| 10.2 | Anbefalinger |
10.1 Erfaringer
Erfaringerne med testning af udvaskningen af organiske stoffer fra forurenet jord og restprodukter er små, og der eksisterer kun få veletablerede udvaskningstests til undersøgelse af udvaskningen af organiske stoffer. Dog findes der i litteraturen et relativt veletableret og hastigt voksende fagligt grundlag vedrørende de processer og faktorer, som styrer fordelingen af flere organiske stofgrupper i jord og sedimenter. Indsamling og sammenstilling af erfaringer fra litteraturen kan bidrage til udvikling af en metodik for testning af udvaskningen af organiske stoffer, for så vidt angår forståelse af stoffernes opførsel i forurenet jord og fastlæggelse af testtekniske forhold som f.eks. valg af materialer og kontakttid, agitationsform og efterbehandling af eluatet fra udvaskningsforsøgene. Gennem opdeling af de relevante organiske stoffer i grupper efter deres fysisk/kemiske egenskaber er det hensigten at kunne identificere ligheder i relation til stoffernes fordeling i et kombineret luft-/vand-/jordsystem. En sådan opdeling kan anvendes til en indledningsvis klassifikation af stofferne i forhold til mulighederne og relevansen af at teste deres udvaskning fra jord og restprodukter.
Gennem sammenstilling af erfaringer fra litteraturen er der identificeret en række forhold, der er centrale for den videre udvikling af udvaskningstests til forurenet jord:
| I de situationer, hvor forurenet jord og restprodukter indeholder fri fase af organiske stoffer, vil opløsningen af de organiske stoffer være styrende for udvaskningen af stoffer. I tilfælde af, at forureningen ikke indeholder fri fase, eller den frie fase er opløst, vil udvaskningen af organiske stoffer være styret af desorption fra de faste partikler i jorden/rest-produktet. |
| Opløsning fra frie og residuale faser af organiske stoffer i jord er en relativt hurtig proces (minutter/timer). Udvaskningsforløbet ved tilstedeværelse af en fri fase kan forudsiges ud fra en kendt sammensætning af den frie fase. Dette kræver imidlertid kendskab til stofferne og jorden, og sådanne oplysninger er det vanskeligt at fremskaffe i praksis. |
| Desorptionen af organiske stoffer fra jord er i mange tilfælde langsom (dage/uger). Desuden er den vanskelig at forudsige, hvilket antagelig skyldes, at den er afhængig af såvel stof- som jordtype. Specielt ses meget lave desorptionsrater for hydrofobe stoffer. Med hensyn til jordtypernes betydning er der indikationer af, at unge jordtyper er karakteriseret ved relativt høje desorptionsrater og små hystereseeffekter, mens det omvendte er tilfældet for geologisk gamle jordtyper. Ud over bestemmelser af O/C-forholdet, som stiger med jordens alder, findes der ingen umiddelbare eller veletablerede metoder til karakterisering af jordens alder. Endvidere skal det bemærkes, at topjord godt kan bestå af eller indeholde geologisk set gamle materialer. |
| Bestemmelse af jordmaterialets indhold af naturligt organisk kulstof kan benyttes til at få et generelt indtryk af prøvens forventede sorptionskapacitet. Herudover menes forholdet mellem ilt og kulstof, det såkaldte O/C-forhold at være indikator for alderen af det faste organiske kulstof i prøven. Lave værdier af O/C-forholdet indikerer, at det organiske stof er gammelt, og at der derfor kan forventes lave desorptionsrater. |
| I forhold til testning af stofudvaskningen er det centralt, at udvaskningen af organiske stoffer fra jord i mange tilfælde vil være styret af desorptionskinetik. Dette stiller direkte krav til kontakttiden mellem materialet og udvaskningsmediet og således til den endelige testprotokol for testningen. I den forbindelse bør det også overvejes, hvad der kan betragtes som feltrealistiske kontakttider i de relevante fysiske scenarier. Umiddelbart er det oplagt at udvikle testsystemer, hvor der opnås ligevægtslignende forhold mellem de organiske stoffer i faststofprøven og udvaskningsmediet. I de tilfælde, hvor tidshorisonten for at opnå ligevægtslignende forhold er lang, vil denne ikke nødvendigvis være et hensigtsmæssigt kriterium for fastlæggelse af testens kontakttid. Dette skyldes dels praktiske forhold, dels hensyn til, at de samme ligevægtslignende forhold ikke altid opnås i felten. |
| De fleste af ovennævnte forhold, som påvirker og er af betydning for udvaskningen af organiske stoffer fra jord/forurenet jord, kan forventes at have en tilsvarende betydning for udvaskningen af organiske stoffer fra restprodukter. For mange restprodukter vil det dog gælde, at indholdet af organiske forureningskomponenter er en mere eller mindre integreret del af materialet, mens de for jords vedkommende normalt er associeret med overfladen af jordpartiklerne eller porerne i disse. I modsætning til jord vil en del restprodukter reagere kemisk med vand, når de befugtes eller gennemstrømmes, hvilket f.eks. kan give anledning til mere ekstreme pH-værdier i vandfasen, end man normalt ser for jord. |
På grundlag af de indsamlede erfaringer om testtekniske forhold samt de gennemførte eksperimentelle afprøvninger af testsystemer kan der fremhæves en række forhold, som er af betydning for udvikling af en udvasknings-test, der i princip og udformning ligner den, som Miljøstyrelsen har foreslået anvendt til sporelementer og salte:
| En vis forbehandling af prøven er nødvendig som minimum af hensyn til homogenisering af prøven. En sådan forbehandling må være skånsom og bør i mange tilfælde foregå i et lukket system for at mindske fordamp-ningstabet af organiske stoffer. Dette bevirker enten, at sigtning og evt. nedknusning af prøven i praksis er umulig, eller at fordampningstabet må accepteres. |
| Væske-/faststofforholdet er helt centralt for gennemførelsen og tolkningen af resultaterne af en udvaskningstest. De eksperimentelle afprøvninger er gennemført med et væske-/faststofforhold på 2 l/kg, hvilket i mange tilfælde er nær det laveste forhold, som en simpel batchtest i praksis kan udføres ved. De praktiske vanskeligheder med partikler i systemet kombineret med behovet for store volumener af eluat til videre analysering betyder, at det lave L/S-forhold, som er benyttet, ikke har været optimalt. I de gennemførte tests har volumenet af luft (headspace) i testbeholderen været minimeret for at mindske risikoen for afdampningstab af stoffer samt for at reducere tilførslen af ilt til testsystemet under testningen. |
| Separationen af faststoffasen fra eluatet har vist sig at være et særdeles kritisk punkt i forhold til udviklingen af en metode. Til sammenligning med arbejdet vedrørende testning af udvaskningen af salte og sporelementer fra jord og restprodukter vanskeliggøres disse forhold af, at organiske stoffer i langt højere grad end de uorganiske salte og sporelementer kan være flygtige, og det er tilsyneladende vanskeligere at undgå tab af stofferne til filtermaterialer m.v. Endvidere er der generelt behov for langt større prøvevolumener til analyse for organiske stoffer sammenlignet med de uorganiske. |
| Til de gennemførte forsøg er der for at mindske udvekslingen af organisk stof med de anvendte materialer primært anvendt glas og rustfrit stål samt i mindre omfang teflon. Filtermaterialer af glas har resulteret i stoftab, men herudover har testsystemet virket efter hensigten. Det ser endvidere ud til, at sammensætningen af udvaskningsmediet, temperaturen og agitationsformen ikke har givet anledning til problemer i forbindelse med afprøvningen på de udvalgte jordprøver. |
10.2 Anbefalinger
For mange af de relevante stofgrupper findes der et godt fagligt grundlag for at arbejde videre med en testmetodik for stofudvaskningen. Dette gælder for oliestoffer, klorerede opløsningsmidler og polycykliske aromatiske hydrokarboner (PAH), mens stoffer som MTBE og DEHP i mindre grad er undersøgt. Forholdene omkring partikler i prøverne og det resulterende eluat fra testen er undersøgt (og undersøgelser er i gang), for så vidt angår PAH-forbindelser. På grund af PAH'ernes affinitet for partikler vurderes denne stofgruppe at være den væsentlige for denne problemstilling, men når den endelige form for partikelafskæring er fastlagt, bør det afklares, hvorvidt tilstedeværelsen af partikler har betydning for testningen af andre prioriterede organiske stofgrupper. I denne undersøgelse er der af praktiske årsager bl.a. brugt en partikelafskæring på 0,7 mm i stedet for de 0.45 mm, som normalt anvendes i forbindelse med udvaskning af sporelementer og uorganiske salte. Ved at anvende den større partikelafskæring reduceres problemerne med håndtering af de forholdsvis store eluatmængder, som nogle af analyserne af organiske stoffer kræver, noget.
Baseret på forenklede beregninger af fordelingen mellem faserne i et kombineret luft-/vand-/jordsystem foreslås det at klassificere de organiske stoffer, som der er fastsat toksikologiske kvalitetskriterier for i jord og grundvand, i forhold til mulighederne for og relevansen af at teste deres udvaskning fra jord og restprodukter. De foreslås inddelt i tre grupper:
| Første gruppe indeholder stoffer, der sorberer kraftigt til jorden, har en lille opløselighed i vand og er ikke-flygtige. Til denne gruppe hører de fleste PAH'er og dieselstoffer. Det er oplagt, at en metodik for testning af udvaskningen af organiske stoffer fra jord og restprodukter skal omfatte disse stoffer, der vil optræde og flyttes sammen med jorden, men i et vist omfang samtidigt udvaskes. Det vil specielt være separationen af fast fase fra eluatet, herunder hensyntagen til kolloidbundet stof og forsøgstiden, der vil være kritisk for testningen. Der er udarbejdet et forslag til metode til testning af udvaskning af ikke-flygtige organiske stoffer fra forurenet jord, hvori der ikke er foreslået nogen endelig kontakttid og ikke taget stillling til problematikken omkring kolloider. |
| Den anden gruppe omfatter flygtige stoffer med en forholdsvis høj opløselighed i vand. Stofferne i denne gruppe er bl.a. de klorerede opløsningsmidler og BTEX. Mange af disse stoffer vil det være hensigtsmæssigt at kunne teste for, da de har lave kriterieværdier i vand. En del af dem har langt højere diffusionskoefficienter i luft end i vand og vil derfor spredes hurtigere i luft. I forhold til det konkrete arbejde, hvor den potentielle anvendelse af jord og restprodukter primært vil bestå i bygge- og anlægsarbejder, vil stofferne dog sjældent udgøre et luft(indeklima) problem, da der er krav om overdækning, så kontakten afskæres. Det kritiske forhold ved testning af stofudvaskningen vil for denne gruppe af stoffer derfor være at forhindre tab af stof til luften.Ved at minimere luftvolumenet i testsystemet kan dette håndteres i en batch, da kun en mindre del af den totale stofmængde vil findes på luftform, men i forhold til håndtering af prøven før og efter testningen vil flygtigheden være vanskelig at kontrollere. Specielt separationen af faststoffasen og eluatet vil være kritisk for de flygtige stoffer og dermed testens anvendelighed. |
| Den tredje gruppe af stoffer med kriterieværdier indeholder stoffer med meget høj opløselighed eller blandbarhed i vand, og de sorberer meget lidt til jord. Til denne gruppe hører bl.a. methyltertiærbutylether (MTBE). Phenolerne tilhører også denne gruppe, idet de har en høj opløselighed i vand, men de sorberer kraftigere end f.eks. MTBE. Andre stoffer i gruppen vil kun i meget begrænset omfang kunne findes associeret med de faste faser, og det vil derfor være vanskeligt eller irrelevant at søge at beskrive deres opførsel på grundlag af udvaskningstests. Stofferne i tredje gruppe kan, i det omfang det er relevant, også testes med den ovenfor foreslåede metode. For disse stoffer vil kolloider sandsynligvis ikke være af stor betydning for transporten, da de ikke sorberer så kraftigt som stofferne i første gruppe. |
For alle prioriterede stoffer vil det være påkrævet at foretage en videre udvikling af en testmetodik, også af den i dette projekt foreslåede testprotokol. Dette vil kræve tiltag på en række af de tidligere nævnte kritiske faktorer. Af høj prioritet er udvikling eller tilpasning af metoder til analyse af stofferne med opretholdelse af de detektionsgrænser, som er gældende for analyse af grundvand. Ved anvendelse af de eksisterende metoder bevirker kravene til prøvevolumener, at testsystemerne bliver uhensigtsmæssigt store (eller mange), og at separationen af eluatet fra faststofprøverne bliver vanskelig og tidskrævende.
Det er sandsynligvis mest oplagt at gå videre med udformningen af batchforsøg, men det er ikke uden problemer, og en eksperimentel sammenligning med små kolonneforsøg vil være hensigtsmæssig. En nærmere afklaring kan nås i forbindelse med fastlæggelse af, hvordan resultaterne fra en test forventes anvendt. Som nævnt ovenfor er problematikken vedrørende separation af partikler fra eluatet central. For nogle af de mindre hydrofobe stoffer er betydningen af partikelafskæringen antagelig minimal, mens den for mere hydrofobe stoffer kan have større betydning. Det anbefales at satse på sådanne teknikker til separation af eluat fra faststoffasen, hvor anvendelse af filtre ikke er nødvendig (f.eks. centrifugering). Disse teknikker bør udvikles/afprøves sammen med udviklingen af de omtalte analyseteknikker. Det er antagelig for optimistisk at designe testsystemerne, således at de kan tage hensyn til alle potentielle stofgrupper, da de så vil blive uhensigtsmæssigt komplicerede. Testsystemet bør kunne tage hensyn til de typisk forekommende blandingsforureninger, f.eks. BTEX og tungere oliestoffer, mens mere sjældent forekommende blandingsforureninger, f.eks. klorerede opløsningsmidler og benzinstoffer, om fornødent må testes i to parallelle tests. Muligheden for at gennemføre testningen ved et højere væske-/faststof-forhold end 2 l/kg, f.eks. 5 eller 10 l/kg bør også afprøves. Denne mulighed vil alt andet lige øge volumnet af eluat og muligvis reducere problemerne med separation af eluat fra faststof. Ulemperne kan være, at koncentrationerne af de organiske stoffer i eluatet reduceres, hvilket kræver mere følsom analyseteknik, samt at det hidtidigt valgte væske-/fast-stofforhold fraviges.. Dette vil i givet fald medføre, at de forudsætninger og antagelser, der ligger til grund for det hidtidige valg af væske-/faststofforholdet, bør revurderes. Et alternativ vil være at bruge samme L/S-forhold, men benytte større beholdere. Håndtering af større eluatvolumener kan også give betydelige problemer med hensyn til filtrering og/eller centrifugering.
Agitationen kan også vise sig at have væsentlig betydning, idet den påvirker jordpartiklerne fysisk. Aggregater og belægninger kan ødelægges, således at sorptionskapaciteten ændres. Hvis f.eks. den fysiske påvirkning resulterer i, at fast organisk stof bliver mere tilgængeligt, vil desorptionen underestimeres. Omvendt vil frigivelse af kolloider kunne resultere i overestimering af udvaskningen af sorberede stoffer. Det sidste er blevet vist eksperimentelt, hvor partikler i størrelsen 0,45 – 10 mm fandtes i eluatet efter agitation, og der væsentlige mængder PAH var associeret med disse partikler. I eluatet fra søjleforsøg (som i højere grad end batchforsøg må antages at simulere virkelige forhold) blev der ikke fundet samme mængde partikler, som i batchforsøgene.
De tidligere omtalte konstaterede forskelle i nødvendig kontakttid bør opfølges med videre forsøg kombineret med en vurdering i de konkrete tilfælde. Som en mulig strategi for opsætning af forsøg, der tager hensyn til kinetikken i udvaskningen, kan der foreslås opstillet parallelle batchtest, der prøvetages efter forskellige tidsperioder. De første to prøvetages f.eks. efter henholdsvis 1 og 3 timer, og hvis der ikke ses en væsentlig forskel mellem koncentrationerne af stofferne i disse to systemer, slutter testningen her. Hvis der ses en betydelig forskel, fortsætter testningen og analyseringen til f.eks. 24 timer og 5 døgn. I sidstnævnte tilfælde indlægges resultaterne i en model, som tager hensyn til feltrealistiske forhold.
Temperaturen er ikke af væsentlig betydning for sorptions- og desorptionsprocesser, mens den naturligvis har betydning for flygtigheden og nedbrydeligheden af visse stoffer. I det omfang, nedbrydeligheden og flygtigheden kan kontrolleres, anbefales det at udføre forsøgene ved stuetemperatur.
Det vil være rimeligt at antage, at metoder til testning af udvaskningen af organiske stoffer fra granulære, mineralske restprodukter i vidt omfang kan baseres på de samme principper og hensyn, som metoder til udvaskning af organiske stoffer fra jord. Dog vil der for restprodukternes vedkommende i mange tilfælde skulle tages specielle forholdsregler med hensyn til forbehandling af prøverne (f.eks. tørring og nedknusning), ligesom restprodukternes potentielle reaktivitet med vand og de i nogle tilfælde ret ekstreme pH- og redoxforhold, som reaktionerne med vand kan medføre, må tages i betragtning. Det bør eventuelt foretages en vurdering af værdien af de resultater man opnår, når man, som det i praksis ofte er tilfældet, udvider analyseprogrammet for udvaskningstests til bestemmelse af udvaskningen af uorganiske komponenter fra restprodukter til også at omfatte indholdet af opløst organisk stof.
Sammenfattende må det anføres, at den gennemførte undersøgelse har tilvejebragt et fagligt grundlag for at opstille forslag til en udvaskningstest til organiske stoffer. Det er lykkedes at identificere en række forhold, der er centrale for udviklingen af udvaskningstests til forurenet jord. Der er foretaget en klassifikation af de vigtigste organiske stoffer, som kan benyttes til at målrette eventuel videre udvikling. Det er imidlertid klart, at den præcise viden om de organiske stoffers egenskaber og forhold, der er kritiske for opstilling af en testprotokol til organiske stoffer, er mangelfuld. Det betyder, at på trods af, at eksperimentelle afprøvninger har bidraget med yderligere specifik information i relation til udvalgte jordtyper og stofgrupper, er det blevet klart, at flere forhold behøver videre afklaring og nærmere undersøgelse, før der kan opstilles en egentlig protokol for udvaskningstests til klassificering af udvaskningen af organiske stoffer fra jord og restprodukter i Danmark.
Der er udarbedet et indledende forslag til en testmetode til bestemmelse af udvaskningen af ikke-flygtige organiske stoffer som olieprodukter (benzin undtaget) og PAH’er fra jord. Testen foreslås udført som en enkelt batchtest ved L/S = 2 l/kg, og for at simulere jordvæske foreslås en 0,001 M CaCl2-opløsning i demineraliseret vand anvendt som udvaskningsmedium. Der anvendes en kontakttid på 1 eller 5 døgn, afhængigt af den aktuelle situation og formålet med testen. For at forebygge biologisk nedbrydning af de organiske stoffer fjernes ilten fra testsystemet med kvælstof og NaSO3.Det foreslås, at separationen af eluatet fra jordfasen sker ved centrifugering i stedet for filtrering. Der er efterfølgende givet et eksempel på, hvorledes resultatet af en udvaskningstest på en forurenet jord kan anvendes til at estimere indholdet af forureningskomponenter i porevandet i jorden lige under et lag af den samme forurenede jord, som gennemsives af infiltrerende nedbør.|
11. Litteratur
Ball, W.P., Roberts, P.V. (1991). Long-term sorption of halogenated organic-chemicals by aquifer material .2. Intraparticle diffusion. Environmental Science & Technology, 25, 1237-1249.
Barber, L.B. (1994). Sorption of chlorobenzenes to Cape-Cod aquifer sediments. Environmental Science & Technology, 28, 890-897.
Bauw, D.H., de Wilde, P.G.M., Rood, G.A., Aalbers, Th.G. (1991). A standard leaching test, including solid phase extraction, for the determination of PAH leachability from waste materials. Chemosphere, Vol. 22, No. 8, 713-722.
Broholm, K., Feenstra, S. (1995) Laboratory measurements of the aqueous solubility of mixtures of chlorinated solvents. Environmental Toxicology and Chemistry, 14 (1), 9-15.
Broholm, K., Wahlström, M., Larsson, L.B., Villholt, K.G., Hjelmar, O. (2000): Development of tests for characterising the leaching of organic compounds from contaminated soils and waste products. Rapport til Nordtest udarbejdet af DHI i samarbejde med VTT og SGI. DHI, Hørsholm.
Brusseau, M. L., Jessup, R. E., Rao, P.S.C. (1989). Modeling the transport of solutes influenced by multi process nonequilibrium. Water Resources Research, 25, 1971-1988.
Brusseau, M.L., Rao, P.S.C. (1989). Sorption nonideality during organic contaminant transport in porous media. Critical Reviews in Environmental Control, 19, 33-99.
Brusseau, M. L., Jessup, R. E., Rao, P.S.C. (1991). Nonequilibrium sorption of organic chemicals. Elucidation of rate-limiting processes. Environmental Science and Technology 25 (7), 1334.
Brusseau, M.L., Wood, A.L., Rao, P.S.C. (1991). Influence of organic cosolvents on the sorption kinetics of hydrophobic organic chemicals. Environmental Science and Technology 25 (5), 903.
Burgos, W.D., Novak, J.T., Berry, D.F. (1996). Reversible sorption and irreversible binding of naphthalene and alpha-naphthol to soil: Elucidation of processes. Environmental Science & Technology, 30, 1205-1211.
Burris, D.R., MacIntyre, W. G. (1985). Water solubility behavior of binary hydrocarbon mixtures. Environ. Toxicol. Chem., 4, 371-377.
Carmichael, L.M., Christman, R.F., Pfaender, F.K. (1997). Desorption and mineralization kinetics of phenanthrene and chrysene in contaminated soils. Environmental Science & Technology, 31, 126-132.
Carroll, K.M., Harkness, M.R., Bracco, A.A., Balcarcel, R.R (1994). Application of permeant polyme/ diffusional model to the desorption of polychlorinated-biphenyls from hudson river sediments. Environmental Science & Technology, 28, 253-258.
Chen, W.L., Wagenet, R.J. (1995). Solute transport in porous-media with sorption-site heterogeneity. Environmental Science & Technology, 29, 2725-2734.
Connaughton, D.F., Stedinger, J.R. , Lion, L.W., Shuler, M.L. (1993). Description of time-varying desorption-kinetics - release of naphthalene from contaminated soils. Environmental Science & Technology, 27, 2397-2403.
Cornelissen, G., vanNoort, P.M., Parsons, J.R., Govers, H.J. (1997). Temperature dependence of slow adsorption and desorption kinetics of organic compounds in sediments. Environmental Science & Technology, 31, 454-460.
Culver, T.B., Hallisey, S.P., Sahoo, D., Deitsch, J.J., Smith, J.A. (1997). Modeling the desorption of organic contaminants from long-term contaminated soil using distributed mass transfer rates. Environmental Science & Technology, 31, 1581-1588.
Dansk Geo-servEx a/s (1998). Vurdering af mulig forurening – registreringsundersøgelse. Olie-benzinforurening, Dr. Sellsvej 1, Dianalund, Dianalund Kommune, lokalitet nr. 303-104-109. Udført for Vestsjællands Amt.
De Europæiske Fællesskabers Tidende (1998). Rådet. Fælles holdning nr. 13/98 fastlagt af Rådet den 19. december 1997 med henblik på vedtagelse af Rådets direktiv 98 om kvaliteten af drikkevand, C91/1, 26/3-1998.
DIN (1998). Ableitung von Konzentrationen organischer Stoffe im Bodenwasser. Bodenbechaffenheit. Vornorm DIN V 19736. Oktober 1998.
Eganhouse, R.P., Calder, J.A. (1976). The solubility of medium weight aromatic hydrocarbons, and the effects of hydrocarbon co-solutes and salinity. Geochimica et Cosmochimica Acta, 40, 555-561.
Farrell, J., Reinhard, M. (1994). Desorption of halogenated organics from model solids, sediments, soil under unsaturated conditions .1. Isotherms. Environmental Science & Technology, 28, 53-62.
Fredenslund, Aa., Gmehling, J., Rasmussen, P. (1977). Vapor-liquid equilibria using UNIFAC, a group-contribution method. Elsevier Scientific Publishing Company, Amsterdam.
Gawlik, B.M., Sotiriou, N., Feicht, E.A., SchulteHostede, S., Kettrup, A. (1997). Alternatives for the determination of the soil adsorption coefficient, K-oc, of non-ionicorganic compounds - A review. Chemosphere, 34, 2525-2551.
Gmehling J., Rasmussen, P., Fredenslund, A. (1981). Vapor-liquid equilibria by UNIFAC group contribution. Revision and extension. 2. Industrial & Engineering Chemistry. Process, Design & Development, 21, 118-127.
Gong, Y.Y., Depinto, J.V. (1998). Desorption rates of two PCB congeners from suspended sediments - II. Model simulation. Water Research, 32, 2518-2532.
Harmon, T.C., Roberts, P.V. (1994). Comparison of intraparticle sorption and desorption rates for a halogenated alkene in a sandy aquifer material. Environmental Science & Technology, 28, 1650-1660.
Heiger-Bernays, W., Menzei, C., Montgomery, C., Edwards, D., Pauwels, S. (1995) In: Draft Report Environmentally Acceptable Endpoints in Soil; Gas Research Institute, Environment & Safety Research Group, Chapter 3.
Hjelmar, O., Holm, P.E., Lehmann, N.K.J., Asmussen, O., Rose, N. (1998). Grundlag for nyttiggørelse af forurenet jord og restprodukter. Miljøprojekt nr. 415, Miljø- og Energiministeriet, Miljøstyrelsen, København.
Hjelmar, O. (1999). Ikke-publicerede resultater. VKI, Hørsholm,
Hjelmar, O., Traberg, R. (1995). Vejledning i valg og fortolkning af udvaskningstests for affaldsmaterialer. Nordtest report. NT Techn Report 272.
HOH Water Technology (1998). Skitseprojekt for reaktiv væg, Vapokon, Søndersø. Fyns Amt.
Holm, P.E., Hjelmar, O., Lehmann, N.K.J., Asmussen, O.W. (1997). Undersøgelse af mulighederne for at lade testning af stofudvaskning indgå i grundlaget for vurdering og klassificering af forurenet jord. VKI sag 60.5420. Rapport til Miljøstyrelsen. Februar 1997.
Holmen, B.A., Gschwend, P.M. (1997). Estimating sorption rates of hydrophobic organic oxide- and aluminiumsilicate clay coated aquifer sand. Environmental Science & Technology, 31,105-113.
Huang, W.L., Weber, W.J. (1997). A distributed reactivity model for sorption by soils and sediments .10. Relationships between desorption, hysteresis, the chemical characteristics of organic domains. Environmental Science & Technology, 31, 2562-2569.
Huang, W.L., Yu, H., Weber, W.J. (1998). Hysteresis in the sorption and desorption of hydrophobic organic contaminants by soils and sediments - 1. A comparative analysis of experimental protocols. Journal Of Contaminant Hydrology, 31, 129-148.
Jord•Miljø A/S (1999). Forureningsundersøgelse. DK-servicestation, Dr. Sellsvej 1, Dianalund. Udført for Vestsjællands Amt.
Kan, A.T., Fu, G., Tomson, M.B. (1994). Adsorption/desorption hysteresis in organic pollutant and soil/sediment interaction. Environmental Science & Technology, 28, 859-867.
Kan, A.T., FU, G.M., Hunter, M.A., Tomson, M.B. (1997). Irreversible adsorption of naphthalene and tetrachlorobiphenyl to Lula and surrogate sediments. Environmental Science & Technology, 31, 2176-2185.
Kan, A.T., FU, G.M., Hunter, M.A., Chen, W., Ward, C.H., Tomson, M.B. (1998). Irreversible adsorption of neutral hydrocarbons to sediments. Experimental observations and model predictions. Environmental Science & Technology, 32, 892-902.
Karickhoff, S.W. (1984). Organic pollutant sorption in aquatic systems. J. Hydraul. Eng. Am. Soc. Civil Eng. 110 (6), 707-735.
Kiilerich, O., Arvin, E. (1996). Ground water contamination from creosote contaminated sites. GWMR, 16, Winter, 112-117.
Kile, D.E., Chiou, C.T., Zhou, H.D., Li, H., Xu, O.Y. (1995). Partition of nonpolar organic pollutants from water to soil and sediment organic matters. Environmental Science & Technology, 29, 1401-1406.
Kjeldsen, P., Andersen, L.J., Christiansen, K., Grøn, C., Kirkegaard, C., Lund, U., Olsen, A.N., Segato, H., Wium, M. (1989). Grundvandsprøvetagning og feltmåling. Udredningsrapport U3, Lossepladsprojektet. April 1989.
Kjeldsen, P., Grundtvig, A. (1995). Leaching of organic compounds from industrial waste disposed of at an old municipal landfill. In Christensen, T.H., Cossu, R., Stegemann, R.: Proceedings of Sardinia 95, Fifth International Landfill Symposium, Cagliari, Italy, Bd. III, pp. 201-212.
Kjeldsen, P. (1996). Sorption af organiske stoffer. Kapitel 7. I: Kjeldsen, P., Christensen, T.H. (1996). Kemiske stoffers opførsel i jord og grundvand. Projekt om jord og grundvand fra Miljøstyrelsen, nr. 21. Miljøstyrelsen, København.
Kleineidam, S., Rugner, H., Ligouis, B., Grathwohl, P. (1999). Organic matter facies and equilibrium sorption of phenanthrene. Environmental Science & Technology 33, 1637-1644.
Korsgaard, T., Petersen, C.R., Nielsen, C., Michaelsen, O., Skaarup, J., Borg, D., Andersen, J., Bjerre, B., Henriksen, P. (1989). Forurenede gasværksgrunde. Udredningsrapport U4, Lossepladsprojektet.
Laha, S., Luthy, R.G. (1991). Inhibition of phenanthrene mineralisation by nonionic surfactants in soil water systems. Environmental Science & Technology 25, 1920-1929.
Larsson, L.B. (1996). Lakning av organiska ämnen. Sammanställning av information från litteraturstudie. Rapport, Sveriges geotekniska intstitut, Linköping.
Leboeuf, E.J., Weber, W.J. (1997). A distributed reactivity model for sorption by soils and sediments. 9. Sorbent organic domains: Discovery of a humic acid glass transition and an argument for a polymer-based model. Environmental Science & Technology, 31, 1697-1702.
Leinonen, P.J., Mackay, D. (1973). The multicomponent solubilty of hydrocarbons in water. Canadian Journal of Chemical Engineering, 51, 230-233.
Luthy, R.G., Aiken, G.R., Brusseau, M.L., Cunningham, S.D., Gschwend, P.M., Pignatello, J.J., Reinhard, M., Traina, S.J., Weber, W.J., Westall, J.C. (1997). Sequestration of hydrophobic organic contaminants by geosorbents. Environmental Science & Technology, 31, 3341-3347.
Lyngkilde, J., Christensen, T.H. (1992a). Redox zones of a landfill leachate pollution plume (Vejen, Denmark). J. Contaminant Hydrology, vol. 10, pp. 273-289.
Lyngkilde, J., Christensen, T.H. (1992b). Fate of organic contaminants in the redox zones of a landfill leachate plume (Vejen, Denmark.). J. Contaminant Hydrology, vol. 10, pp. 291-307.
Ma, L., Selim, H.M. (1994) Predicting atrazine adsorption-desorption in soils. A modified second order kinetic model. Water Resources Research, 30, 447-456.
McCall, P.J., Agin, G.L. (1985) Desorption kinetics of picloram as affected by residence time in the soil. Environmental Toxicology and Chemistry. 4, 37-42.
McCarthy, J.F., Degueldre, C. (1993). Sampling and characterization of colloids and particles in groundwater for studying their role in contaminant transport. I: J. Buffle and H.P. Van Leeuwen (Eds.), Environmental Particles, Lewis Publishers, Chelsea, Michigan, 1-59.
McGinley, P.M., Katz L.E., Weber, W.J. (1996). Competitive sorption and displacement of hydrophobic organic contaminants in saturated subsurface soil systems. Water Resources Research, 32, 3571-3577.
Miljøstyrelsen (1995). Toksikologiske kvalitetskriterier for jord og grundvand. Projekt om jord og grundvand fra Miljøstyrelsen no. 12. Miljø- og Energiministeriet.
Miljøstyrelsen (1996). Kemiske stoffers opførsel i jord og grundvand: Bind 1 og 2. Projekt om jord og grundvand fra Miljøstyrelsen no. 20. Miljø- og Energiministeriet.
Miljøstyrelsen (1998a). Oprydning på forurenede lokaliteter – hovedbind. Vejledning fra Miljøstyrelsen nr. 6. Miljø- og Energiministeriet.
Miljøstyrelsen (1998b). Vakuumventileringssager fra Oliebranchens Miljøpulje. Teknologiudviklingsprogrammet for jord- og grundvandsforurening. Miljøprojekt nr. 421, Miljø- og Energiministeriet.
Miljøstyrelsen (1999). Offentlig høring vedr. Bekendtgørelse om genanvendelse af restprodukter og jord til bygge- og anlægsarbejder. Erhvervsaffaldskontoret. J.nr. M3014-0010. 4. marts 1999. Miljø- og Energiministeriet.
Miller, C.T., Pedit, J.A. (1992). Use of a reactive surface-diffusion model to describe apparent sorption desorption hysteresis and abiotic degradation of lindane in a subsurface material. Environmental Science & Technology, 26, 1417-1427.
Miller, C.T., Weber, W.J. Jr. (1986). Sorption of hydrophobic organic pollutants in saturatedsoil system. Journal of Contaminant Hydrology, 1, 243-261.
Müller, M. (1997). Quantum chemical modelling of soil sorption coefficients: Multiple linear regression models. Chemosphere, 35, 365-377.
Nielsen, P.H., Arvin, E., Kjeldsen, P. (1996). Phenoler. Kapitel 14 i: Kjeldsen, P., Christensen, T.H. (1996). Kemiske stoffers opførsel i jord og grundvand. Projekt om jord og grundvand fra Miljøstyrelsen, nr. 21. Miljøstyrelsen, København.
Nkedi-Kizza, P., Brusseau, M.L., Rao, P.S.C., Hornsby, A.G. (1989). Nonequilibrium sorption during diplacement of hydrophobic organic chemicals and 45Ca through soil columns with aqueous and mixed solvents. Environmental Science & Technology, 23, 814-820.
Ong, S.K., L.W. Lion (1991). Mechanisms for trichloroethylene vapor sorption onto soil minerals. Journal of Environmental Quality, 20, 180-188.
Pankow, J.F. og J.A. Cherry (1996). Dense chlorinated solvents and other DNAPLs in groundwater. Waterloo Press.
Pavlostathis, S.G., Jaglal, K. (1991). Desorption behavior of trichloroethylene in contaminated soil. Environmental Science & Technology, 25, 274-279.
Pedit, J.A., Miller, C.T. (1994). Heterogeneous sorption processes in subsurface systems .1. Model formulations and applications. Environmental Science & Technology 28, 2094-2104.
Piatt, J.J., Backhus, D.A., Capel, P.D., Eisenreich, S.J. (1996). Temperature-dependent sorption of naphthalene, phenanthrene and pyrone to low organic carbon aquifer sediments. Environmental Science & Technology, 30, 751-760.
Pignatello, J.J. (1990). Slowly reversible sorption of aliphatic halocarbons in soils. II. Mechanistic aspects. Environmental Toxicology and Chemistry. 9 (9), 1117-1126.
Pignatello, J.J., Ferrandino, F.J. , Huang, L.Q. (1993). Elution of aged and freshly added herbicides from a soil. Environmental Science & Technology, 27, 1563-1571.
Pignatello, J.J., Xing, B.S. (1996). Mechanisms of slow sorption of organic chemicals to natural particles. Environmental Science & Technology, 30, 1-11.
Poulsen, M., Lemon, L., Barker, J.F. (1991). Chemical fate and impact of oxygenates in groundwater: solubility of BTEX from gasoline-oxygenate mixtures. API Project GW-16, Soil/Groundwater Technical Task Force, American Petroleum Institute.
Priester, T., Köster, R., Eberle, S.H. (1996). Charakterisierung kohlenstoffhaltiger Bestandteile in Hausmüllverbrennungsschlacken unter besonderer Berücksichtigung organischer Stoffe. Müll und Abfall 28 (6), pp. 387-398.
Rood, G.A., Broekman, M.H., Aalbers, Th.G. (1994). Investigating a leaching test for PCBs and organochlorine pesticides in waste and building materials. I: Goumans, J.J.J.M., van der Sloot, H.A., Aalbers, Th.G. (Editors): Environmental Aspects of Construction with Waste Materials. 271-280. Elsevier Science B.V.
Rutherford, D.W., Chiou, C.T., Kile, D.E. (1992). Influence of soil organic matter composition on the partition of organic compounds. Environmental Science & Technology, 26, 336-340.
Sabbah, I., Rebhun, M. (1997). Adsorption-desorption of trichlorophenol in water-soil systems. Water Environment Research 69, 1032-1038.
Sabljic, A., Gusten, H., Verhaar, H., Hermens, J. (1995). QSAR modelling of soil sorption. Improvements and systematics of log K-OC vs log K-OW correlations. Chemosphere, 31, 4489-4514.
Sahoo, D., Smith, J.A. (1997). Enhanced trichloroethene desorption from long term contaminated soil using Triton X-100 and pH increases. Environmental Science & Technology, 31, 1910-1915.
Schlutz, B., Rank, P. (1990). Kilder og forekomst med udgangspunkt i undersøgelse af grundvandskvaliteten i Københavns Amt. I: Klorerede opløsningsmidler, forurening og forekomst. ATV-komiteen vedr. grundvandsforurening. SAS Scandinavia Hotel, 4. oktober, 1-16.
Shiu, W., Kou-Chang, M., Varhanícková, D., Mackay, D. (1994). Chlorophenols and alkylphenols: A review and correlation of environmentally relevant properties and fate in an evalutive environment. Chemosphere, 29, (6), 1155-1224.
Skjold-Jørgensen, S., Kolbe, B., Gmehling, J., Rasmussen, P. (1979). Vapor-liquid equilibria by UNIFAC group contribution. Revision and extension. Industrial & Engineering Chemistry. Process, Design & Development., 18, 714-722.
Skúladóttir, B. (1996). Desorptive behavior of volatile nonpolar organic contaminants in soil. Master Thesis Project. Institute of Environmental Science and Engineering. Technical University of Denmark. Lyngby, July 1996.
Steinberg, S.M., Pignatello, J.J., Sawhney, B.L. (1987). Persistence of 1,2-dibromoethane in soils: Entrapment in intraparticle micropores. Environmental Science and Technology,21 (12), 1201-1208.
Stumm, W. (1992). Chemistry of the Solid-Water Interface. Processes at the Mineral-Water and Particle-Water Interface in Natural Systems, 1st edn., John Wiley & Sons Inc., N.Y., Chichester, Brisbane, Toronto, Singapore.
Toride, N., Leij, F.J., van Genuchten, M.Th. (1995). The CXTFIT code for estimating transport parameters from laboratory or field tracer experiments, Version 2.0. Research Report No. 137, U.S. Salinity Laboratory, USDA, ARS, Riverside, California.
US EPA (1991). Toxicity Characteristic Leaching Procedure (TCLP). US EPA method 1311. Rules and Regulations, 55, 11863-11877.
Valsaraj, K.T., Thibodeaux, L.J. (1992). Equilibrium adsorption of chemical vapors onto surface soils: Model predictions and experimental data. I: Schnoor, J.L.(ed.). Fate of pesticides and chemicals in the environment. John Wiley & Sons, London.
Van der Sloot, H.A., Kosson, D.S., Cnubben, P.A.J.P., Hoede, D. Hjelmar, O. (1997). Waste characterization to modify waste quality prior to disposal. In: Proceedings of Sardinia 97, Sith International Landfill Symposium, S. Margherita di Pula, Cagliari, Italy; 13-17 October 1997, Vol. V, pp. 315-328.
van Genuchten, M.T., Wieranga, P.J. (1976). Mass transfer studies in sorbing porous media: analytical solutions. Soil Sci. Soc. Amer. J., 40, 473-479.
van Houdt, R. Wolf, E., Duzijn, R. (1991). Composition and leaching characteristics of road construction materials. I Goumans, J.J.J.M., van der Sloot, H.A., Aalbers, Th.G. (eds.): Waste Materials in Construction. Studies in Environmental Science 48, Elsevier, Amsterdam, pp. 153-160.
Villholth, K.G. (1999). Colloid characterization and colloidal phase partitioning of polycyclic aromatic hydrocarbons in two creosote-contaminated aquifers in Denmark. Environ. Sci. Technol., 33, 691-699.
Villholth, K.G., Brun, A., Christensen, F.D., Hjelmar, O. (1997). Colloid-facilitated transport of pesticides and other sorbing contaminants in soil and groundwater. 40 pp. Miljøstyrelsen, Miljøministeriet.
Wahlström, M., Thomassen, H., Flyvbjerg, J., Veltkamp, A.C., Oscarsson, C., Sundqvist, J.-O., Rood, G.A. (1994). Leaching of organic contaminants from contaminated soils and waste materials. I: Goumans, J.J.J.M., van der Sloot, H.A., Aalbers, Th.G. (Editors). Environmental Aspects of Construction with Waste Materials. pp. 257-270. Elsevier Science B.V.
Weber, W.J. Jr, Miller, C.T. (1988). Modeling the sorption of hydrophobic contaminants by aquifer materials - I. Rates and equilibria. Water Research, 22 (4), 457-464.
Weber, W.J., McGinley, P.M., Katz, L.E. (1992). A distributed reactivity model for sorption by soils and sediments .1. Conceptual basis and equilibrium assessments. Environmental Science & Technology, 26, 1955-1962.
Weber, W.J., Huang, W.L., Yu, H. (1998). Hysteresis in the sorption and desorption of hydrophobic organic contaminants by soils and sediments - 2. Effects of soil organic matter heterogeneity. Journal of Contaminant Hydrology, 31, 149-165.
Werth, C.J., Reinhard, M. (1997a). Effects of temperature on trichloroethylene desorption from silica gel and natural sediments .2. Kinetics. Environmental Science & Technology, 31, 697-703.
Werth, C.J., Reinhard, M. (1997b). Effects of temperature on trichloroethylene desorption from silica gel and natural sediments .1. Isotherms. Environmental Science & Technology, 31, 689-696.
Wu, S.C., Gschwend, P.M. (1986). Sorption kinetics of hydrophobic organic compounds to natural sediments and soils. Environmental Science & Technology, 20, 717-725.
Wu, S., Gschwend, P.M. (1988). Numerical Modeling of Sorption Kinetics of Organic Compounds to Soil and Sediment Particles. Water Resources Research, 24 (8), 1373-1383.
Xing, B.S., Pignatello, J.J., Gigliotti, B. (1996). Competitive sorption between atrazine and other organic compounds in soils and model sorbents. Environmental Science & Technology, 30, 2432-2440.
Xing, B.S. (1997). The effect of the quality of soil organic matter on sorption of naphthalene. Chemosphere, 35, 633-642.
Xing, B.S., Pignatello, J.J. (1998). Competitive sorption between 1,3-dichlorobenzene or 2,4- dichlorophenol and natural aromatic acids in soil organic matter. Environmental Science & Technology, 32, 614-619.
Yiacoumi, S., Tien, C. (1994). A model of organic solute uptake from aqueous-solutions by soils. Water Resources Research, 30, 571-580.
Young, T.M., Weber, W.J. (1995). A distributed reactivity model for sorption by soils and sediments .3. Effects of diagenetic processes on sorption energetics. Environmental Science & Technology, 29, 92-97.
Bilag A Sorptionsligevægte
Sorptionsligevægte
Isotermer
Sorption beskrives ofte ved en isoterm, som viser sammenhængen mellem koncentrationen af stoffet (sorbatet) i vandet og mængden af stof sorberet til partiklerne (sorbenten) ved ligevægt, dvs efter at sorptionsprocessen er løbet til ende. Betragtes i første omgang det simple tilfælde, hvor sorptionen alene er en adsorptionsproces, og hvor sorptionspladserne på overfladen, S, adsorberer sorbatet, A, fra opløsningen:
| 1. |
| hvor | S er adsorptionspladser |
| A er stof i opløsning | |
| SA er sorberet stof |
Koncentrationen af S og SA regnes i mol/l (kunne også regnes i mol/m2 eller mol/g). Ved brug af massevirkningsloven på ligning 1 fås:
| 2. |
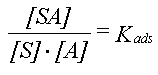
Den maksimale koncentration af sorptionspladser, ST, er:
| 3. |
Fra ligning 2 og 3 fås:
| 4. |
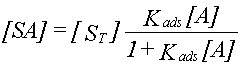
Hvis den sorberede koncentration i stedet opgives i mg/g sorbent og den opløste koncentration i mg/l fås følgende ligning, som kaldes Langmuir adsorptionsisotermen:
| 5. |
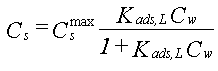
| hvor | Cs er stofkoncentrationen i den faste fase (mg/g) |
| Csmax er den maksimale stofkoncentration i den faste fase (mg/g) | |
| Cw er koncentrationen i vandet (mg/l) | |
| Kads,L er Langmuir adsorptionskonstanten (l/mg) |
I figur 2 er Langmuir isotermen vist. Det ses at den sorberede koncentration stiger asymptotisk til den maksimale værdi med stigende koncentrationer i vandet. Hvis Cw er tilstrækkelig lille er nævneren i ligning 5 approksimativt lig 1, hvilket giver følgende sammenhæng:
| 6. |
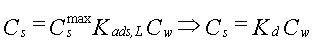
hvor Kd er fordelingskoefficienten (l/kg)
Figur 1
Langmuir adsorptions isotermen. For lave ligevægtskoncentrationer i vandet er Langmuir
adsorptionsisotermen sammenfaldende med den lineære sorptionsisoterm, hvis hældning er
konstant og lig med fordelingskoefficienten, Kd.
Ved lave koncentrationer fås altså en simpel lineær isoterm, hvis hældning er lig fordelingskoefficienten, Kd (se figur 1).
Langmuir adsorptionsisotermen er udviklet under den forudsætning at alle sorptionspladser har den samme aktivitet. (har samme adsorptionskonstant). For bl.a. jorde vil dette sjældent være tilfældet, idet naturlige jorde er sammensat af lermineraler, hydroxider og organiske overflader som vil reagere forskelligt overfor et givent stof. På grund af denne sammensathed benyttes en mere empirisk isoterm, Freundlich isotermen, til beskrivelse af egentlige forsøgsresultater:
| 7. |
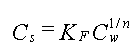
| hvor | KF er Freundlich konstanten |
| 1/n er potensen som Cw opløftes til. |
I figur 2 er vist tre eksempler på Freundlich isotermer. Tilfælde I hvor 1/n<1 opnås en mindre fordelingskoefficient (=hældning på kurven) med stigende koncentration, et forhold tilsvarende til det af Langmuir isotermen beskrevne, hvor de tilrådighed værende sorptionspladser fyldes op. Tilfælde III hvor 1/n>1 beskriver den modsatte situation, hvor der via de allerede sorberede molekyler sker en modifikation af partiklerne, som favoriserer yderligere sorption. I tilfælde II er 1/n=1, hvorved man opnår den lineære isoterm som allerede beskrevet. Som det ses på figur 2 er de tre isotermer tilnærmelsesvis sammenfaldende ved lave værdier af Cw, hvilket indikere at for tilfælde, hvor man kun interesserer sig i lave koncentrationer kan sorptionen beskrives tilstrækkeligt nøjagtigt med den lineære isoterm.
Figur 2
Tre eksempler på Freundlich isotermer, nemlig hvor 1/n<1, 1/n=1 og 1/n>1 i
Freundlich isotermudtrykket (formel 7) (efter Schwarzenbach et al. 1993).
Referencer
Schwarzenbach, R.P., Gschwend, P.M., Imboden, D.M. (1993). Environmental organic chemistry. John Wiley & Sons, New York.
Bilag B Fasefordelingsberegninger i et luft-/vand-/jordsystem
Eksempel på fasefordelingsberegning
Hvis man kender begyndelsesværdien i jorden, som det er tilfældet i beregning nr. 1, 2 og 4, forløber beregninger som vist i det følgende.
Den totale koncentration til start fordeler sig efter følgende massebalance, hvis der ikke er fri fase:
![]()
hvor
CT er den oprindelig start koncentrationen i jorden (mg/kg TS)
C med indeks L, V eller J er henholdsvis koncentrationen i luften, vandet og jorden efter
ligevægt
VL er luftvolumet i systemet
VV er væskevolumet i systemet
M er jordmængden i systemet.
Ligevægt med luft beskrives med Henrys lov:
hvor
H er Henrys konstant (fordelingen mellem luft og vand) (-).
Fordelingen mellem vand og jord beskrives med lineær sorptionsisoterm:
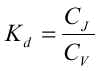
hvor
Kd er den lineære fordelingskoefficient mellem jord og vand (l/kg)
Ud fra en sammenhæng mellem henholdsvis Koc og Kow og henholdsvis Kd, foc og Koc er følgende sammenhæng anvendt (Miljøstyrelsen 1996):
![]()
![]()
hvor
Koc er fordelingskoefficienten mellem det organiske stof i jorden og vand
Kow er fordelingskoefficienten mellem octanol og vand
foc udtrykker indeholdet af naturligt organisk stof i jorden.
Indsættes fordelingen mellem luft og vand og mellem jord og vand i den overordnede massebalance fås:
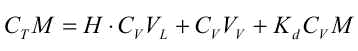
Isoleres CV fås
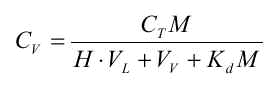
I standardudvaskningssystemet er
VL = 0,050 l
VV = 1,88 l
M = 0,94 kg TS
Som eksempel regnes på benzen, der har følgende egenskaber
H = 0,22
log(KOW) = 2,1
Jorden har følgende egenskaber
fOC = 0,001
CT =1,5 mg/kg TS
KOC beregnes til
![]()
dvs
![]()
og Kd beregnes til
![]()
Væskekoncentrationen efter ligevægt kan nu beregens
![]()
Hvis der er fri fase vil den beregnede væskekoncentration efter ligevægt være større end opløseligeheden for stoffet. Hvis dette er tilfældet sættes væskekoncentrationen til opløseligheden og beregningerne forløber som normalt.
Koncentrationen i luften efter ligevægt beregnes vha. Henrys lov:
![]()
Koncentration på jorden efter ligevægt beregnes udefra ligevægten mellem jord og vand:
![]()
Den fordeling mellem faserne, der er vist i figur 7.1, beregnes udfra mængderne i de enkelte faser. Mængden, MV, i væskefasen er:
![]()
Mængden i luften, ML, er:
![]()
Mængden på jorden, MJ, er:
![]()
I alt er der:
![]()
så andelen, der er i væskefasen, er:
og i luften:
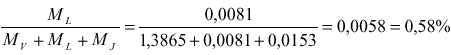
og på jorden:
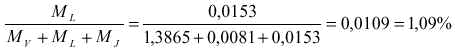
Hvis der er fri fase beregnes mængden af fri fase, MF, som følger:
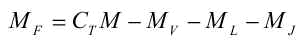
hvor mængderne i væsken, i luften og på jorden er fundet ved at følgende ovenstående beregninger og bruge opløseligheden som væskekoncentration. Mængden af fri fase skal selvfølgelig også medtages når andelene i de enkelte faser beregnes.
Hvis istedet væskekoncentration ved ligevægt er kendt, som det er tilfældet i beregning nr. 3, forløber beregninger således:
For benzen er
CV = 0,00001 mg/l
Koncentrationen i luften efter ligevægt beregnes vha. Henrys lov:
![]()
Koncentration på jorden efter ligevægt beregnes udefra ligevægten mellem jord og vand:
![]()
Mængden i væskefasen er:
![]()
Mængden i luften er:
![]()
Mængden på jorden er:
![]()
I alt er der:
![]()
Koncentrationen i jorden, der svarer til detektionsgrænsen for benzen i vand beregnes som:
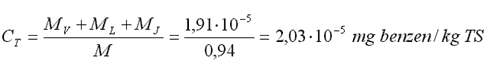
Fasefordelingsberegning til Figur 7.1 Se her!
Beregning af den relative fordeling af stof mellem de tre faser (jord, vand og luft) udfra en arbitrær jordkoncentration, der ikke resulterer i fri fase. Kolonnen "Kor. konc i vand" indeholder en kontrol af om den beregnede koncentration i vandet er højere end opløseligheden, og hvis det er tilfælde benyttes opløseligheden i de resterende beregninger. Værdier i fed er enten estimeret/vurderet eller sat til 0, fordi de er ubetydelige.
Fasefordelingsberegning til Figur 7.2 Se her!
Beregning af den eluatkoncentrationen, der svarer til jordkvalitetskriterierne for de enkelte stoffer. Kolonnen "Kor. konc i vand" indeholder en kontrol af om den beregnede koncentration i vandet er højere end opløseligheden, og hvis det er tilfælde benyttes opløseligheden i de resterende beregninger.
Fasefordelingsberegning til Figur 7.3 Se her!
Beregning af den jordkoncentration, der svarer til detektionsgrænserne for de enkelte stoffer.
Fasefordelingsberegning til Figur 7.4 Se her!
Beregning af den eluatkoncentrationen, der svarer til jordkvalitetskriterierne for de enkelte stoffer. Her er benyttet et LS-forhold på 10 l/kg i modsætning til de øvrige beregninger, hvor der er benyttet et LS-forhold på 2 l/kg. Kolonnen "Kor. konc i vand" indeholder en kontrol af om den beregnede koncentration i vandet er højere end opløseligheden, og hvis det er tilfælde benyttes opløseligheden i de resterende beregninger.
Bilag C Anvendte analysemetoder
BESTEMMELSE AF TØRSTOF OG GLØDETAB
PRINCIP: Tørstofindholdet bestemes ved at en kendt prøvemængde tørrres ved
105°C, og den tilbageværende rest vejes. Glødetabet bestemmes ved, at prøvens tørstof
glødes ved 550°C, og den tilbageværende rest vejes. Glødetabet er differencen mellem
tørstofmængden og gløderesten.
REFERENCE: DS 204: 1980.
DETEKTIONSGRÆNSE: Tørstof: 20 mg/kg, glødetab: 200 mg/kg.
INTERN KVALITETSKONTROL: Metoden kontrolleres ved dobbeltbestemmelse af en naturlig
prøve i hver analyseserie.
USIKKERHED: Ved dobbeltbestemmelse af en naturlig prøve er der en
analyseusikkerhed indenfor dagen på , CVW, tørstof, 0,1-1% og , CVW,
glødetab,5-10%.
SIGTEANALYSE
PRINCIP: Prøven tørres. Sedimentet overføres til en beholder med vand og
sedimentet-vand suspensionen tilsættes natriumpylophosphat. Efter henstand vaskes
sedimentet igennem en 63 µm sigte, hvorefter silt (>63µm) og sedimentet tørres.
Efter tørring sigtes sedimentet igennem en stak af nedenstående sigter.
Sigter: 2000, 1000, 500, 250, 125, 63 µm.
Ved fjernelse af organisk stof tilsættes en brintperoxidopløsning efter tørring og før tilsætning af natriumpolyphosphat. Derefter filtreres prøven og sedimentet skylles med vand. Herefter tilsættes natriumpolyphosphat og analysen fortsættes efter ovenstående beskrivelse.
REFERENCE: Buchanan, J.B. (1984) Sediment Analysis. In "Metods for the study of Marine Benthos", (eds.) N.A. Holme and A.D. MacIntyre, Blackwell Scientific (Oxford), pp. 41-65.
BESTEMMELSE AF IKKE-FLYGTIGT ORGANISK KULSTOF (NVOC) I VAND
PRINCIP: Prøven tilsættes syre til pH < 2, hvorefter prøven blæses
fri for CO2 og flygtige organiske forbindelser med N2. Prøvens
indhold af organiske forbindelser oxideres katalytisk ved 680°C og dannet CO2
kvantificeres ved IR-spektrofotometri. Partikler indtil 0,5 mm i diameter medbestemmes.
Der benyttes et Dohrmann DC-190 apparat ved analysen. Der foretages dobbeltbestemmelser.
Faste prøver kan bestemmes efter opslemning i vand.
REFERENCE: Standard Methods 5310 A + B.
DETEKTIONSGRÆNSE: 1 mg/L C.
INTERN KVALITETSKONTROL: Metoden kontrolleres ved samtidig analyse af certificeret
kontrolprøve.
USIKKERHED: Ved kontrolanalyse af syntetiske kontrolprøver er der en
analyseusikkerhed indenfor dagen, CVW, 1,4% og mellem dagene, CVB,
1,9%.
BESTEMMELSE AF PENTANEKSTRAHERBARE STOFFER I VAND OG JORD
PRINCIP: Vandprøve (1 l) ekstraheres på magnetomrører med pentan. Jord (50 g)
tilsat vandig pyrophosphatopløsning ekstraheres med pentan ved ultralydsbehandling og
ekstraktion på rysteapparat. Pentanekstraktet analyseres ved gaschromatografi med
kapillarkolonne, temperaturprogrammering og med flammeionisationsdetektion. Der
enkeltorrbindelser kalibreres over for aktuelle analytiske standarder under anvendelse af
chlorbenzen som intern standard, mens totalindholdet bestemmes over for udvalgte
n-alkaner. Der korrigeres med resultatet af de gennemsnitlige genfindelse for
enkeltkomponenter. Metoden medbestemmer rutinemæssigt benzen, toluen, xylener og
naphthalen samt benzin, gasolie eller stenkulstjære og kan benyttes til andre
forbindelser eller produkter. Identifikationen foretages ud fra retentionstiden.
REFERENCE: ISO/CD 11423-part 2.
DETEKTIONSGRÆNSER: For vandprøver er den 0,1 µg/L for enkeltkomponenter og 5 -
25 µg/L for total kulbrinter afhængigt af sammensætningen og evt. nedbrydning. For
jordprøver er detektionsgrænsen 0,01 - 0,1 mg/kg for enkeltkomponenter og 5 mg/kg for
total kulbrinter.
INTERN KVALITETSKONTROL: Resultaterne er kontrolleret ved samtidig analyse af
spikede, naturlige jord- henholdsvis vandprøver.
USIKKERHED: Ved kontrolanalyse af spikede, naturlige prøver er der en
analyseusikkerhed, CVTotal, på 15 - 20 %.
BESTEMMELSE AF PAH I VAND OG JORD VED GC-MS
PRINCIP: Jordprøver (10-50 g) tilsættes vand og ekstraheres med to gange
isopropanol/cyclohexan (1:2) ved behandling på ultralydsbad og rysteapparat. De samlede
ekstrakter vaskes to gange med vand. Vandprøve (1 l) ekstraheres to gange med cyclohexan
ved omrøring på magnetomrører. Ekstrakterne tørres med vandfrit natriumsulfat og
koncentreres ved inddampning til 1 - 5 mL. Cyclohexanekstraktet analyseres ved
gaschromatografi med kapillarkolonne, temperaturprogrammering og med masse selektiv
detektor (MS-SIM). Der kalibreres over for aktuelle analytiske standarder under anvendelse
af deuterium mærket anthracen som intern standard.
Metoden kan anvendes til bestemmelse af følgende PAH’er: acenaphthen, acenaphthylen,
anthracen, benzo(a)anthracen, benzo(a)pyren, benzo(b+j)fluoranthen, , benzo(k)fluoranthen,
benzo(ghi)perylen, chrysen, dibenzo(a,h)anthracen, fluoranthen, fluoren, indenopyren,
naphthalen, phenanthren og pyren.
DETEKTIONSGRÆNSER: 0,01-0,05 µg/L for vandprøver og 0,002-0,005 mg/kg ts for
jordprøver.
INTERN KVALITETSKONTROL: Resultaterne er kontrolleret ved samtidig analyse af
spikede, naturlige jord- henholdsvis vandprøver.
USIKKERHED: Ved kontrol af spikede, naturlige prøver er der en analyseusikkerhed,
CVTotal, på under 15 %.
BESTEMMELSE AF CHLOREREDE OPLØSNINGSMIDLER I VAND OG JORD
PRINCIP: Vandprøve (100 mL) ekstraheres med pentan. Jord (50 g) tilsat vand
ekstraheres med pentan ved ultralydsbehandling og ekstraktion på rysteapparat.
Pentanekstraktet analyseres ved gaschromatografi med kapillarkolonne og electron capture
detektion, GC-ECD. Der kalibreres over for aktuelle analytiske standarder. Der korrigeres
med resultatet af de gennemsnitlige genfindelser for enkeltkomponenter. Metoden
medbestemmer rutinemæssigt chloroform, 1,1,1-trichlorethan, tetrachlorethylen,
trichlorethylen, tetrachlormethan samt trihalomethaner. Identifikationen foretages ud fra
retentionstiden.
DETEKTIONSGRÆNSER: 0,01 µg/L for vandprøver, dog 0,03 µg/L for chloroform og
0,1 - 1 µg/kg for jordprøver.
INTERN KVALITETSKONTROL: Resultaterne er kontrolleret ved samtidig analyse af
spikede, naturlige jord- henholdsvis vandprøver.
USIKKERHED: Ved kontrolanalyse af spikede, naturlige prøver er der en
analyseusikkerhed, CVTotal, på 15-20 %.
BESTEMMELSE AF MTBE I VAND OG JORD
PRINCIP: Vandprøve (0,5 L) ekstraheres på magnetomrører med dichlormethan.
Ekstraktet analyseres ved gaschromatografi med kapillarkolonne, temperaturprogrammering og
med massespectrometrisk detektion ved enkelt ion bestemmelse (GC-MS-SIM). Der kalibreres
over for aktuel analytisk standard under anvendelse af fluorbenzen som intern standard.
Metoden anvendes til bestemmelse af MTBE, methyl-tertiær-butyl-ether. Der
korrigeres med den gennemsnitlige genfindelse bestemt udfra den løbende kvalitetskontrol.
Identifikationen foretages ud fra retentionstiden og forholdet mellem to karakteristiske
ioner.
Metoden kan også benyttes til jordprøver efter opslæmning i vand.
REFERENCE: VKI O-45
DETEKTIONSGRÆNSER: 0,1 µg/L henholdsvis 1 µg/kg.
INTERN KVALITETSKONTROL: Resultaterne er kontrolleret ved samtidig analyse af
spikede, naturlige prøver.
USIKKERHED: Ved kontrolanalyse af spikede, naturlige prøver er der en
analyseusikkerhed, CVTotal, på 10 %.
Bilag D Resultater af fugacitetsberegninger
Beregning af koncentrationen i porevandet i et naturligt system udfra de målte jordkoncentrationer. Værdier i fed er enten estimeret/vurderet eller sat til 0, fordi de er ubetydelige.
Bilag E Indledende forslag til testprotokol forudførelse af udvaskningstests med jord forurenet med ikke-flygtige organiske stoffer
Indledende forslag til testprotokol for udførelse af udvaskningstest med jord forurenet med ikke-flygtige organiske stoffer
Denne protokol skal kun benyttes ved testning af udvaskningen fra jord forurenet med ikke-flygtige organiske stoffer som olieprodukter (undtagen benzin) eller PAH`er.
Nedenstående fremgangsmåde beskriver en udvaskningstest udført ved et L/S-forhold på 2 l/kg TS og separering ved centrifugering. Testen udføres ved 20 ° C +/- 5 ° C.
Forsøget udføres i en 2 l Pyrex glasflaske med Teflon coated septum (Red Cap flakse). Glasflasken varmebehandles ved 550°C i minimum 2 timer.
| 1880 ml af en 0,001 M CaCl2 opløsning i demineraliseret vand hældes i flasken. |
| Vandet gøres det iltfrit ved at gennemblæse det med kvælstof i 24 timer. |
| 940 g TS af den jord, som skal undersøges, anbringes i flasken |
| Til sidst tilsættes 200 mg Na2SO3/l til flasken, som derefter lukkes. |
| Flasken agiteres ved rotation om sin tværakse ved 5-10 rpm i mørke i 1 eller 5 døgn. |
| Flasken henstår i 1 time, hvorved de største partikler vil sedimentere. |
| Herefter overføres vandfasen til centriufugebeholdere (af glas eller stål), som herefter centrifugeres med en hastighed og tid, så der sker en partikelafskæring ved 0,7 µm. |
| Efter centrifugeringen overføres eluatet forsigtigt med glaspipette til en analysebeholder. |